
Chemoprevention of Lung Cancer with a Combination of Mitochondria-Targeted Compounds


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Reagents and Animals
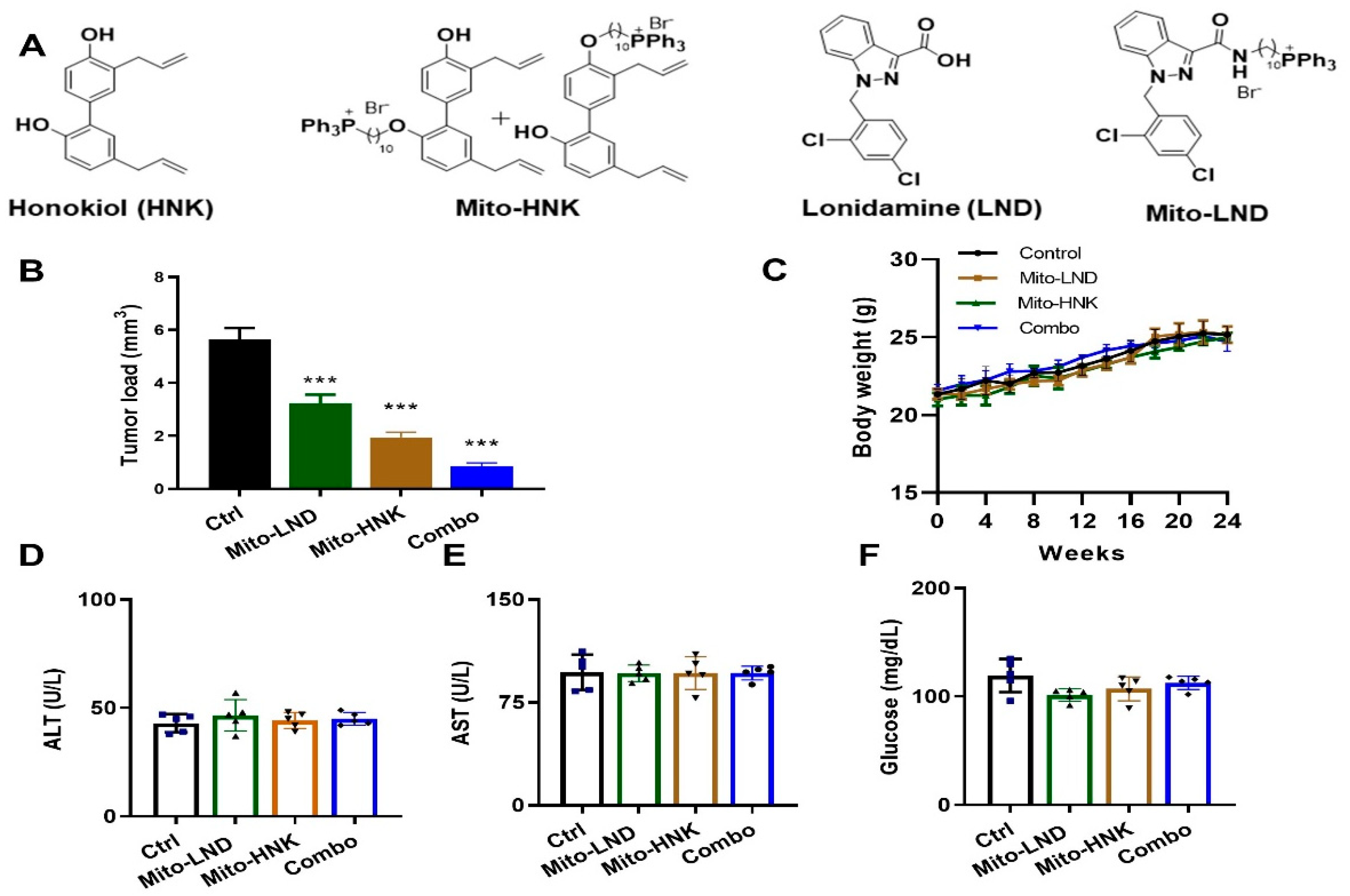
2.2. Chemopreventive Efficacy of Mito-HNK on Lung Tumorigenesis
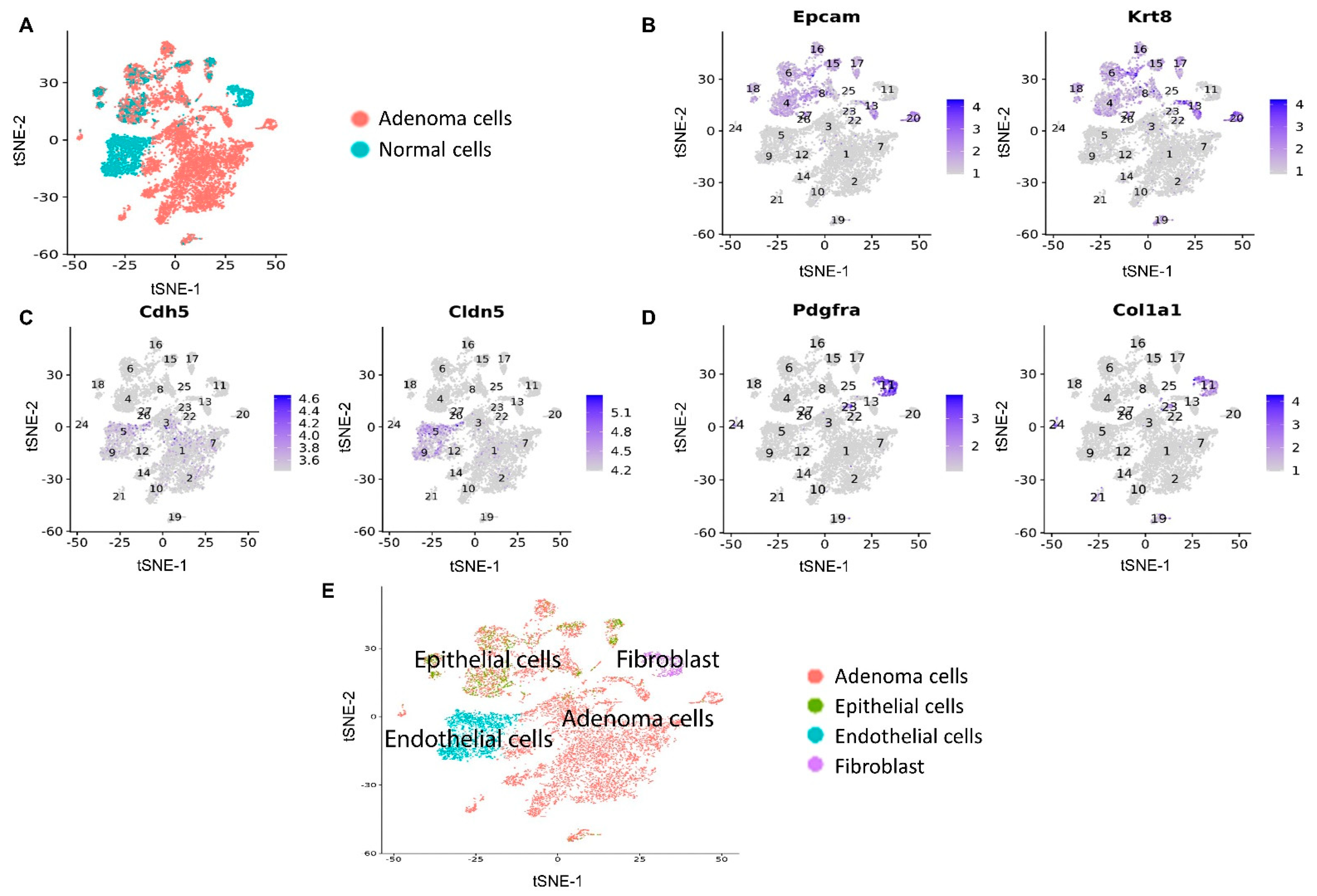
2.3. scRNA-seq Experiment of Mice Lung Tumors
2.4. scRNA-seq Data Analysis
2.5. Statistical Analysis
3. Results
3.1. Efficacy of Mito-HNK in Inhibiting Lung Tumor Progression
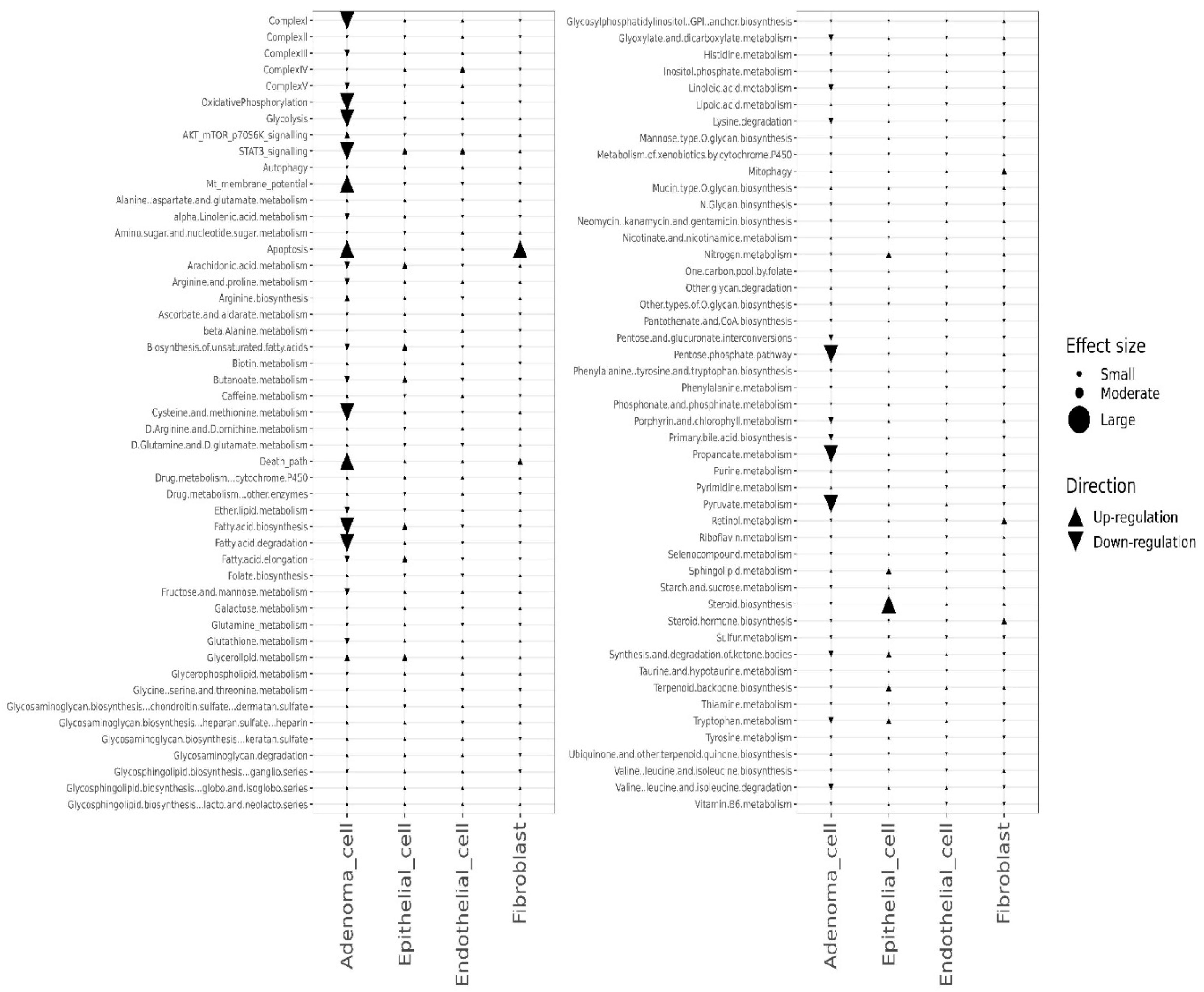
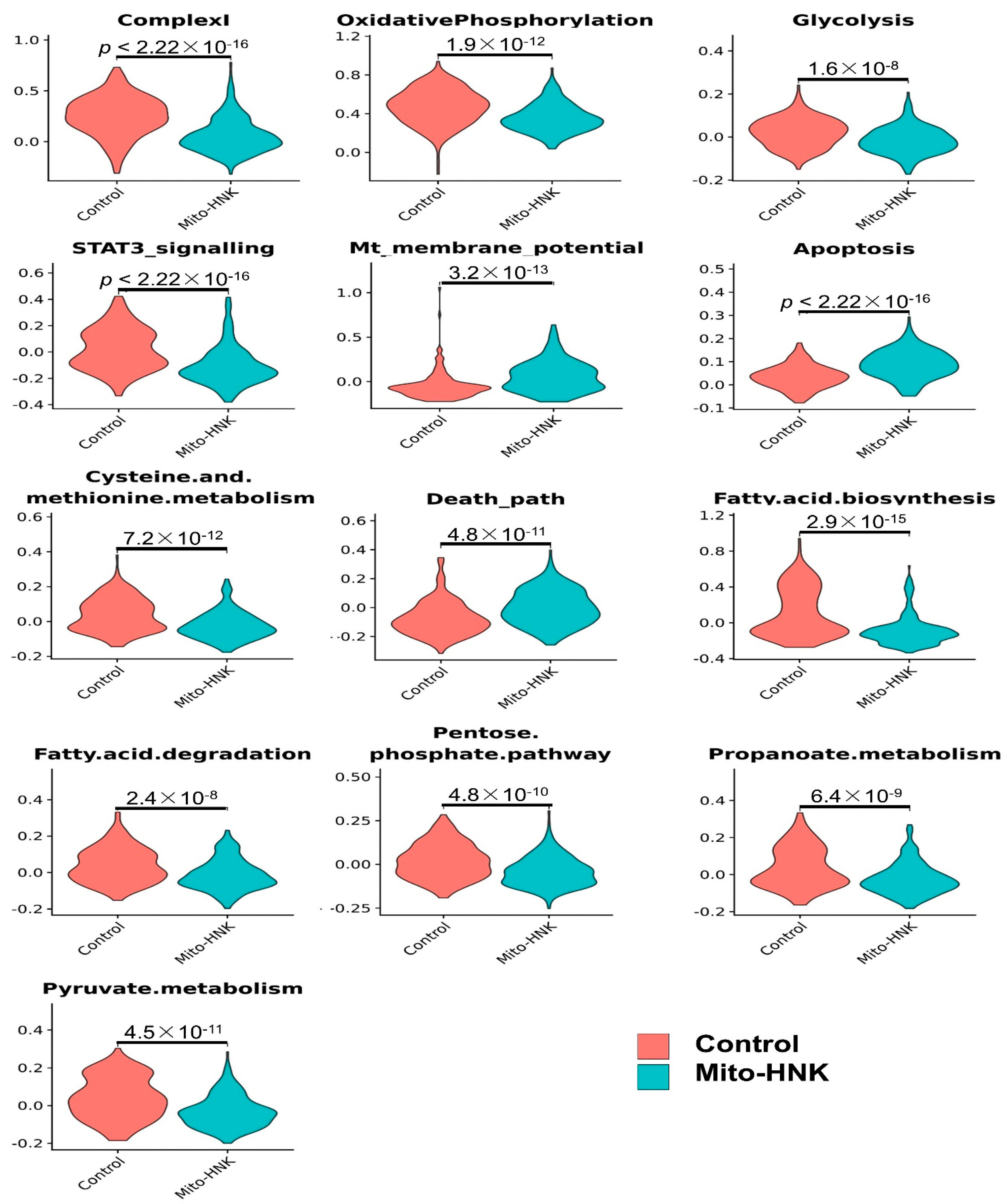
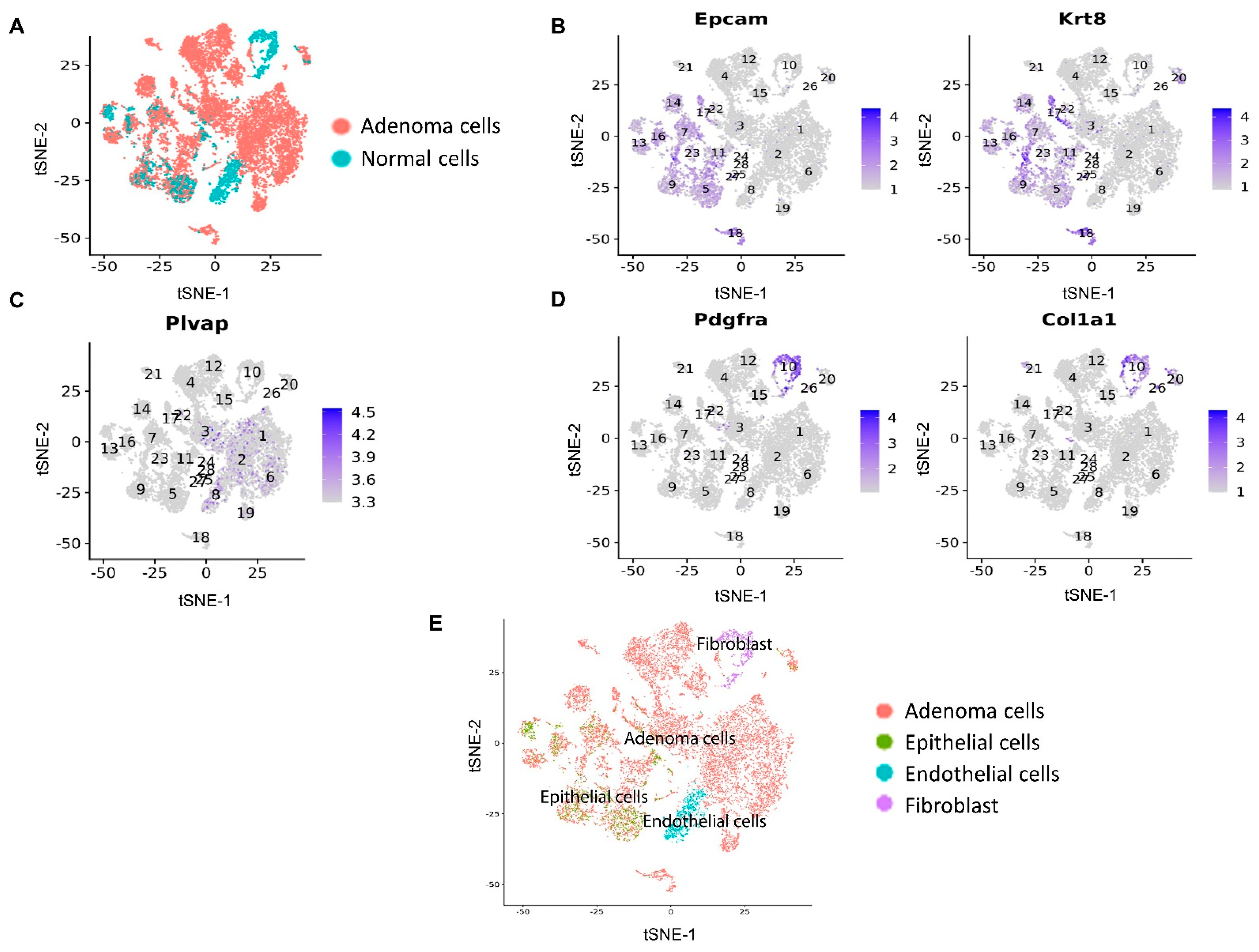
3.2. scRNA-seq Identified the Pathway Changes in Mouse Lung Adenoma Cells Associated with Mito-HNK Treatment
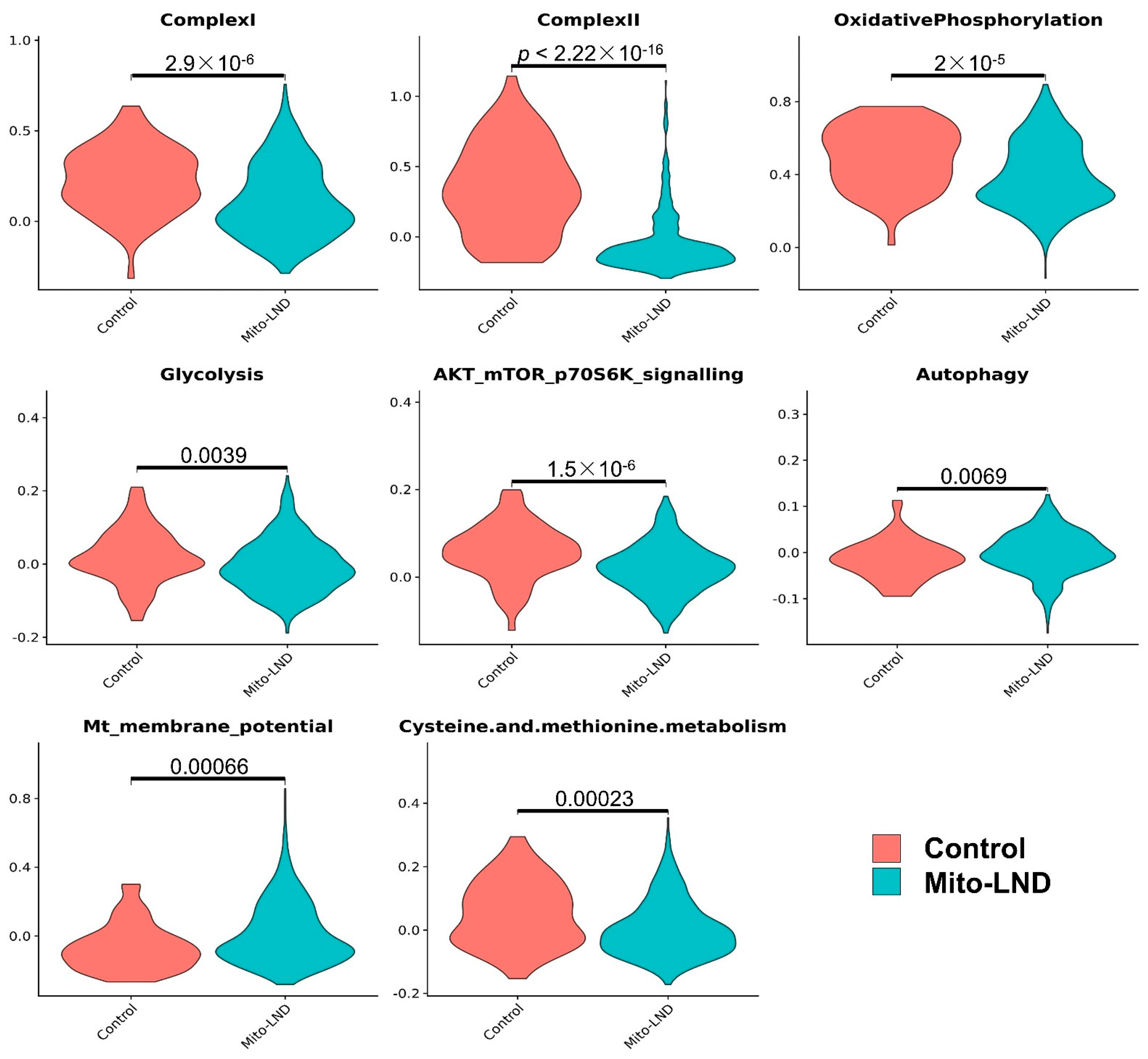
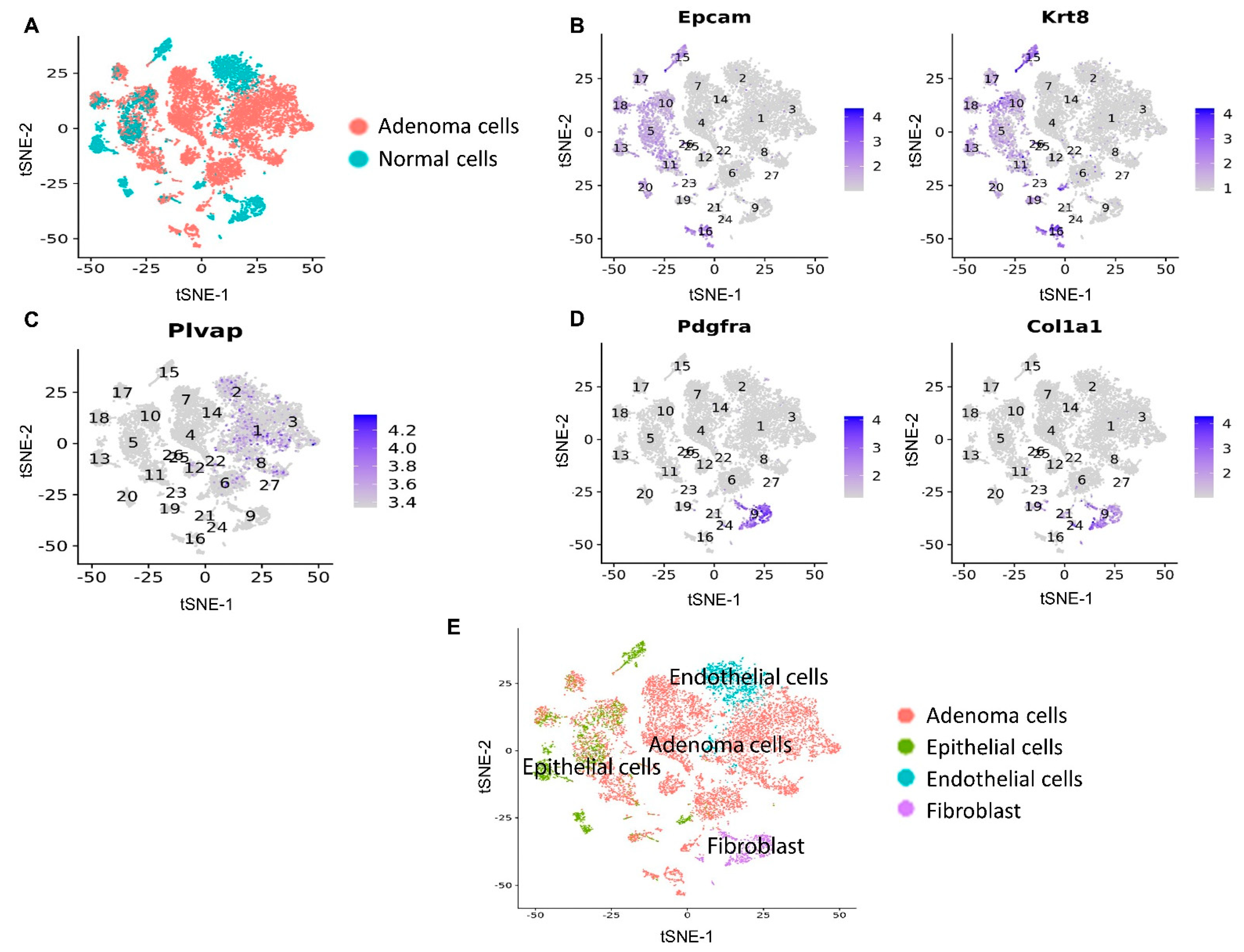
3.3. Mito-LND Treatment Altered Multiple Pathways in Mouse Lung Adenoma Cells
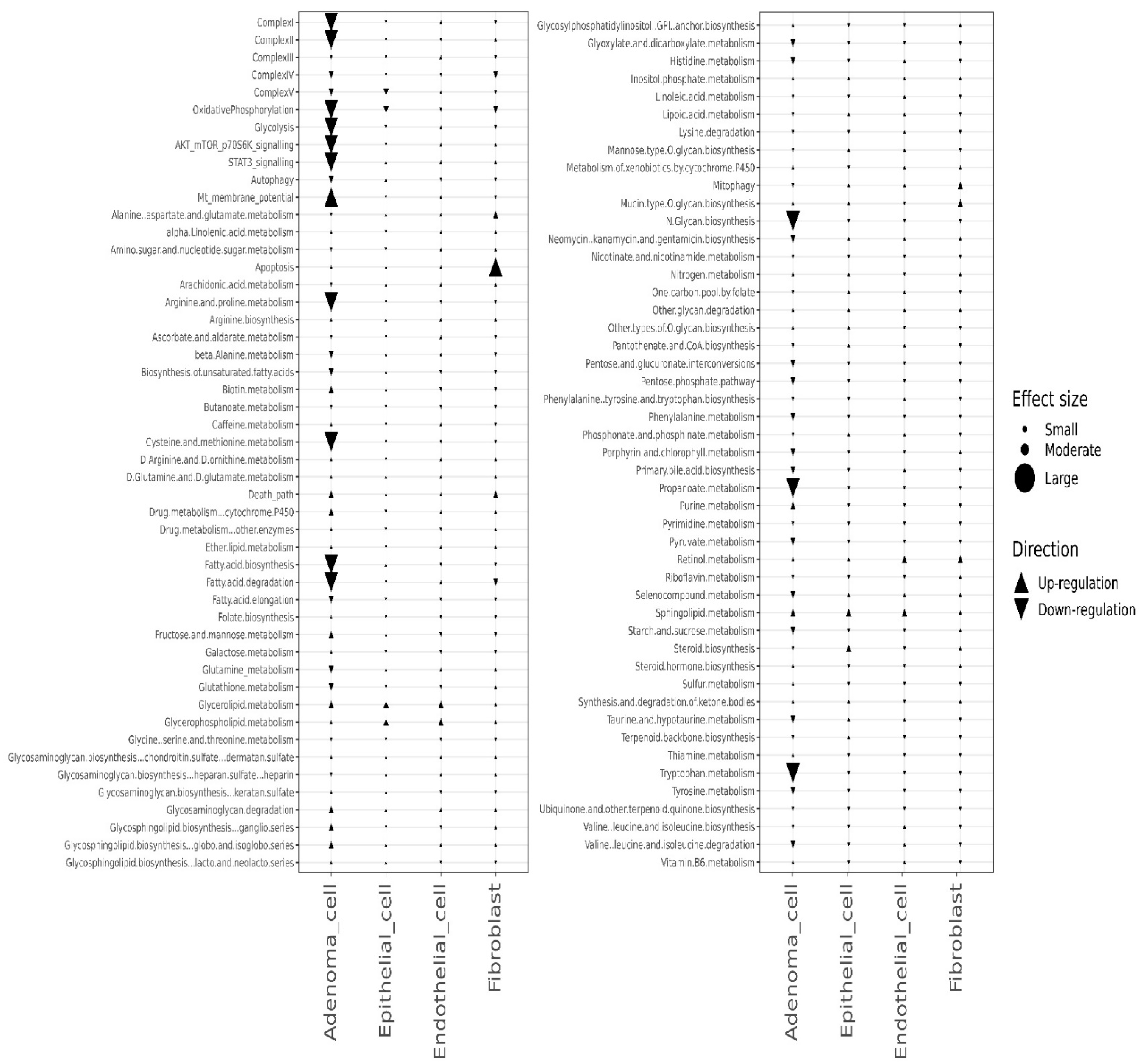
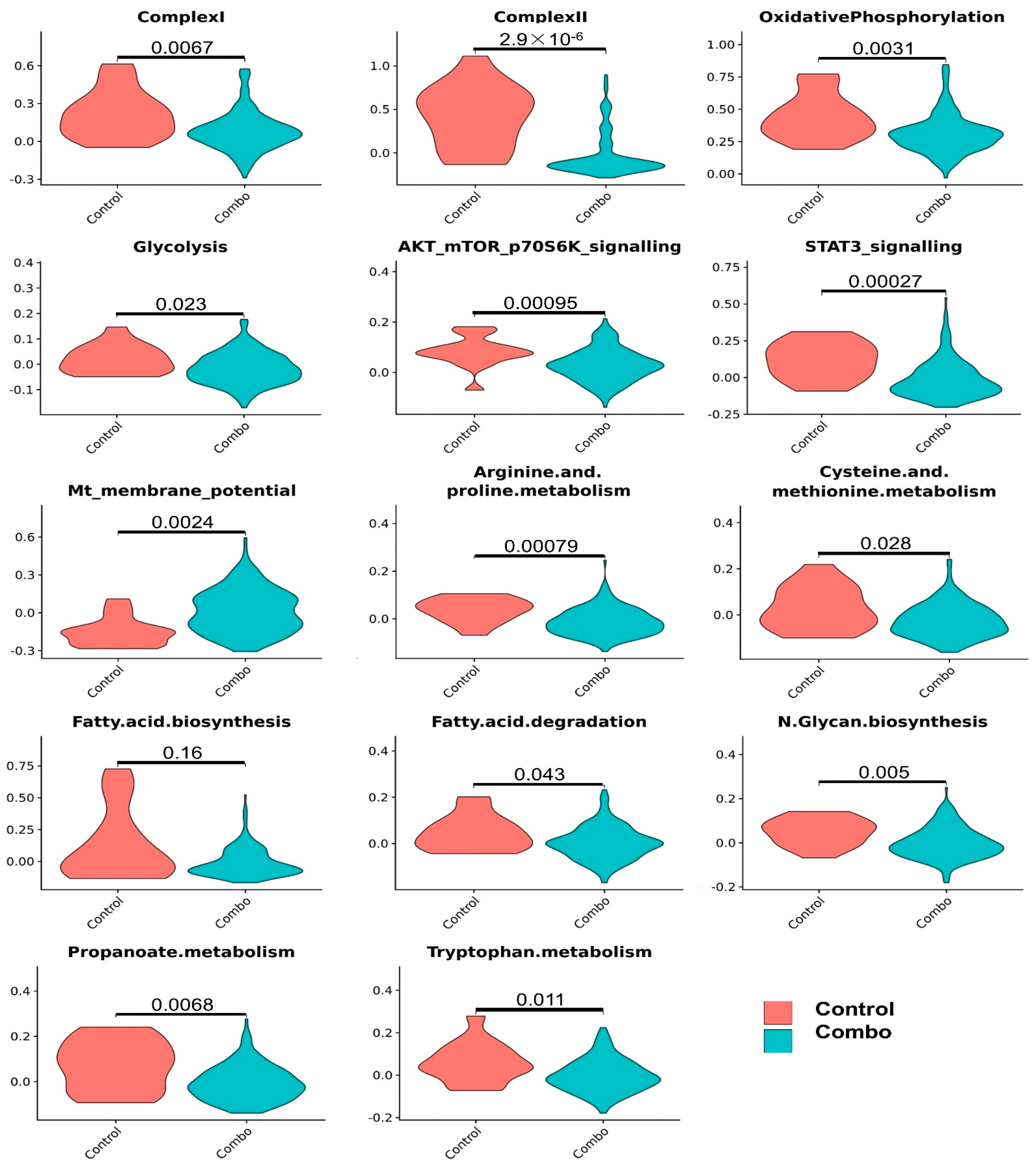
3.4. Effects of the Combined Treatment (Mito-HNK and Mito-LND) on Pathway Activities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Lippman, S.M.; Hawk, E.T. Cancer prevention: From 1727 to milestones of the past 100 years. Cancer Res. 2009, 69, 5269–5284. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Zhang, Q.; Liu, Q.; Komas, S.M.; Kalyanaraman, B.; Lubet, R.A.; Wang, Y.; You, M. Honokiol inhibits lung tumorigenesis through inhibition of mitochondrial function. Cancer Prev. Res. 2014, 7, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Lee, Y.; Cheng, G.; Zielonka, J.; Zhang, Q.; Bajzikova, M.; Xiong, D.; Tsaih, S.W.; Hardy, M.; Flister, M.; et al. Mitochondria-targeted honokiol confers a striking inhibitory effect on lung cancer via inhibiting complex I activity. iScience 2018, 3, 192–207. [Google Scholar] [CrossRef] [Green Version]
- De Lena, M.; Lorusso, V.; Latorre, A.; Fanizza, G.; Gargano, G.; Caporusso, L.; Guida, M.; Catino, A.; Crucitta, E.; Sambiasi, D.A.; et al. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur. J. Cancer 2001, 37, 364–368. [Google Scholar] [CrossRef]
- Cervantes-Madrid, D.; Romero, Y.; Duenas-Gonzalez, A. Reviving lonidamine and 6-diazo-5-oxo-L-norleucine to be used in combination for metabolic cancer therapy. Biomed. Res. Int. 2015, 2015, 690492. [Google Scholar] [CrossRef] [Green Version]
- Nancolas, B.; Guo, L.; Zhou, R.; Nath, K.; Nelson, D.S.; Leeper, D.B.; Blair, I.A.; Glickson, J.D.; Halestrap, A.P. The anti-tumour agent lonidamine is a potent inhibitor of the mitochondrial pyruvate carrier and plasma membrane monocarboxylate transporters. Biochem. J. 2016, 473, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Zhang, Q.; Pan, J.; Lee, Y.; Ouari, O.; Hardy, M.; Zielonka, M.; Myers, C.R.; Zielonka, J.; Weh, K.; et al. Targeting lonidamine to mitochondria mitigates lung tumorigenesis and brain metastasis. Nat. Commun. 2019, 10, 2205. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Chen, Y.; Zhang, Q.; Khatun, A.; Palen, K.; Xin, G.; Wang, L.; Yang, C.; Johnson, B.D.; Myers, C.R.; et al. Inhibition of lung tumorigenesis by a small molecule CA170 targeting the immune checkpoint protein VISTA. Commun. Biol. 2021, 4, 906. [Google Scholar] [CrossRef]
- Chen, Y.; Zander, R.A.; Wu, X.; Schauder, D.M.; Kasmani, M.Y.; Shen, J.; Zheng, S.; Burns, R.; Taparowsky, E.J.; Cui, W. BATF regulates progenitor to cytolytic effector CD8(+) T cell transition during chronic viral infection. Nat. Immunol. 2021, 22, 996–1007. [Google Scholar] [CrossRef]
- Khatun, A.; Kasmani, M.Y.; Zander, R.; Schauder, D.M.; Snook, J.P.; Shen, J.; Wu, X.; Burns, R.; Chen, Y.G.; Lin, C.W.; et al. Single-cell lineage mapping of a diverse virus-specific naive CD4 T cell repertoire. J. Exp. Med. 2021, 218, e20200650. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, J.; Lubet, R.A.; Komas, S.M.; Kalyanaraman, B.; Wang, Y.; You, M. Enhanced antitumor activity of 3-bromopyruvate in combination with rapamycin in vivo and in vitro. Cancer Prev. Res. 2015, 8, 318–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Wang, D.; Wang, S.; Shan, Y.; Liu, C.; Gu, J. scCancer: A package for automated processing of single-cell RNA-seq data in cancer. Brief. Bioinform. 2021, 22, bbaa127. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox. Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef]
- Huang, M.; Myers, C.R.; Wang, Y.; You, M. Mitochondria as a novel target for cancer chemoprevention: Emergence of mitochondrial-targeting agents. Cancer Prev. Res. 2021, 14, 285–306. [Google Scholar] [CrossRef]
- Wanders, D.; Hobson, K.; Ji, X. Methionine restriction and cancer biology. Nutrients 2020, 12, 684. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yip, L.Y.; Lee, J.H.J.; Wu, Z.; Chew, H.Y.; Chong, P.K.W.; Teo, C.C.; Ang, H.Y.K.; Peh, K.L.E.; Yuan, J.; et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat. Med. 2019, 25, 825–837. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Li, Y.; Su, Y.; Guo, Z.; Wang, X.; Wang, S.; Zhang, F.; Zhang, Z.; Shao, J.; Zheng, S. Novel copper complex CTB regulates methionine cycle induced TERT hypomethylation to promote HCC cells senescence via mitochondrial SLC25A26. Cell Death Dis. 2020, 11, 844. [Google Scholar] [CrossRef]
- Li, M.; Shao, J.; Guo, Z.; Jin, C.; Wang, L.; Wang, F.; Jia, Y.; Zhu, Z.; Zhang, Z.; Zhang, F.; et al. Novel mitochondrion-targeting copper(II) complex induces HK2 malfunction and inhibits glycolysis via Drp1-mediating mitophagy in HCC. J. Cell. Mol. Med. 2020, 24, 3091–3107. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Li, W.; Kremer, D.M.; Sajjakulnukit, P.; Li, S.; Crespo, J.; Nwosu, Z.C.; Zhang, L.; Czerwonka, A.; Pawłowska, A.; et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 2020, 585, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Development of cancer metabolism as a therapeutic target: New pathways, patient studies, stratification and combination therapy. Br. J. Cancer 2020, 122, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Salunkhe, S.; Mishra, S.V.; Ghorai, A.; Hole, A.; Chandrani, P.; Dutt, A.; Chilakapati, M.; Dutt, S. Metabolic rewiring in drug resistant cells exhibit higher OXPHOS and fatty acids as preferred major source to cellular energetics. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148300. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H.W.; Feun, L.G.; Savaraj, N. Targeting the proline-glutamine-asparagine-arginine metabolic axis in amino acid starvation cancer therapy. Pharmaceuticals 2021, 14, 72. [Google Scholar] [CrossRef]
- Geck, R.C.; Toker, A. Nonessential amino acid metabolism in breast cancer. Adv. Biol. Regul. 2016, 62, 11–17. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsu, S.C.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine signaling and cancer metabolism. Cancers 2021, 13, 3541. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [Green Version]
- Helenius, I.T.; Madala, H.R.; Yeh, J.J. An asp to strike out cancer? Therapeutic possibilities arising from aspartate’s emerging roles in cell proliferation and survival. Biomolecules 2021, 11, 1666. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.T.; Qi, Y.; Wang, Y.C.; Chi, K.K.; Chung, Y.; Ouyang, C.; Chen, Y.R.; Oh, M.E.; Sheng, X.; Tang, Y.; et al. Arginine starvation kills tumor cells through aspartate exhaustion and mitochondrial dysfunction. Commun. Biol. 2018, 1, 178. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.C.; Falcone, M.; Uribe, A.H.; Zhang, T.; Athineos, D.; Pietzke, M.; Vazquez, A.; Blyth, K.; Maddocks, O.D.K. Immune-regulated IDO1-dependent tryptophan metabolism is source of one-carbon units for pancreatic cancer and stellate cells. Mol. Cell 2021, 81, 2290–2302.e7. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Nollen, E.A.A.; Rohrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lee, J.; He, C.; Zou, M.H.; Xie, Z. Suppression of the mTORC1/STAT3/Notch1 pathway by activated AMPK prevents hepatic insulin resistance induced by excess amino acids. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E197–E209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silsirivanit, A. Glycosylation markers in cancer. Adv. Clin. Chem. 2019, 89, 189–213. [Google Scholar] [CrossRef] [PubMed]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the era of cancer-targeted therapy: Where are we heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.N.; Lee, H.H.; Hsu, J.L.; Yu, D.; Hung, M.C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef]
- Greco, B.; Malacarne, V.; De Girardi, F.; Scotti, G.M.; Manfredi, F.; Angelino, E.; Sirini, C.; Camisa, B.; Falcone, L.; Moresco, M.A.; et al. Disrupting N-glycan expression on tumor cells boosts chimeric antigen receptor T cell efficacy against solid malignancies. Sci. Transl. Med. 2022, 14, eabg3072. [Google Scholar] [CrossRef]
- Zegeye, M.M.; Lindkvist, M.; Fälker, K.; Kumawat, A.K.; Paramel, G.; Grenegård, M.; Sirsjö, A.; Ljungberg, L.U. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun. Signal. 2018, 16, 55. [Google Scholar] [CrossRef]
- Riethmueller, S.; Somasundaram, P.; Ehlers, J.C.; Hung, C.W.; Flynn, C.M.; Lokau, J.; Agthe, M.; Düsterhöft, S.; Zhu, Y.; Grötzinger, J.; et al. Proteolytic origin of the soluble human IL-6R in vivo and a decisive role of N-glycosylation. PLoS Biol. 2017, 15, e2000080. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Schild, L.; Kunz, W. Long-chain fatty acids act as protonophoric uncouplers of oxidative phosphorylation in rat liver mitochondria. Biochim. Biophys. Acta 1989, 977, 266–272. [Google Scholar] [CrossRef]
- Reed, L.; Arlt, V.M.; Phillips, D.H. The role of cytochrome P450 enzymes in carcinogen activation and detoxication: An in vivo-in vitro paradox. Carcinogenesis 2018, 39, 851–859. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Xiong, D.; Pan, J.; Wang, Y.; Hardy, M.; Kalyanaraman, B.; You, M. Chemoprevention of Lung Cancer with a Combination of Mitochondria-Targeted Compounds. Cancers 2022, 14, 2538. https://doi.org/10.3390/cancers14102538
Zhang Q, Xiong D, Pan J, Wang Y, Hardy M, Kalyanaraman B, You M. Chemoprevention of Lung Cancer with a Combination of Mitochondria-Targeted Compounds. Cancers. 2022; 14(10):2538. https://doi.org/10.3390/cancers14102538
Chicago/Turabian StyleZhang, Qi, Donghai Xiong, Jing Pan, Yian Wang, Micael Hardy, Balaraman Kalyanaraman, and Ming You. 2022. "Chemoprevention of Lung Cancer with a Combination of Mitochondria-Targeted Compounds" Cancers 14, no. 10: 2538. https://doi.org/10.3390/cancers14102538
APA StyleZhang, Q., Xiong, D., Pan, J., Wang, Y., Hardy, M., Kalyanaraman, B., & You, M. (2022). Chemoprevention of Lung Cancer with a Combination of Mitochondria-Targeted Compounds. Cancers, 14(10), 2538. https://doi.org/10.3390/cancers14102538