
A Real-World Systematic Analysis of Driver Mutations’ Prevalence in Early- and Advanced-Stage NSCLC: Implications for Targeted Therapies in the Adjuvant Setting

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Specimens
2.2. Immunohistochemistry (IHC)
2.3. Fluorescent In Situ Hybridization
2.4. Nucleic Acid Extraction and Quantification
2.5. Next-Generation Sequencing (NGS)
2.6. Statistical Analysis
3. Results
3.1. Stage-Specific Analysis of NSCLC Actionable Alterations in a Single-Institution Cohort
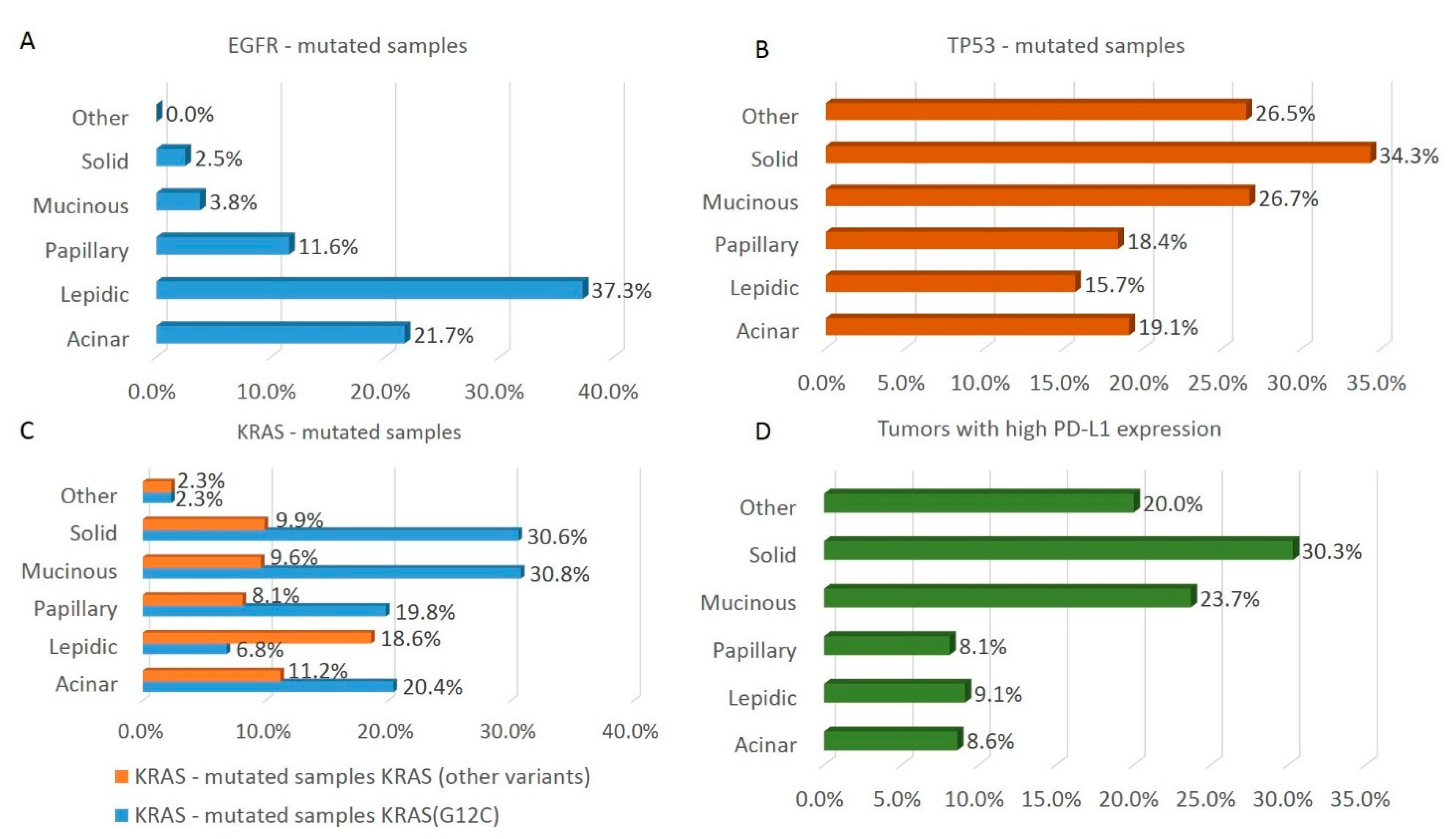
3.2. Distribution of EGFR Mutation Classes in Early Stage NSCLC
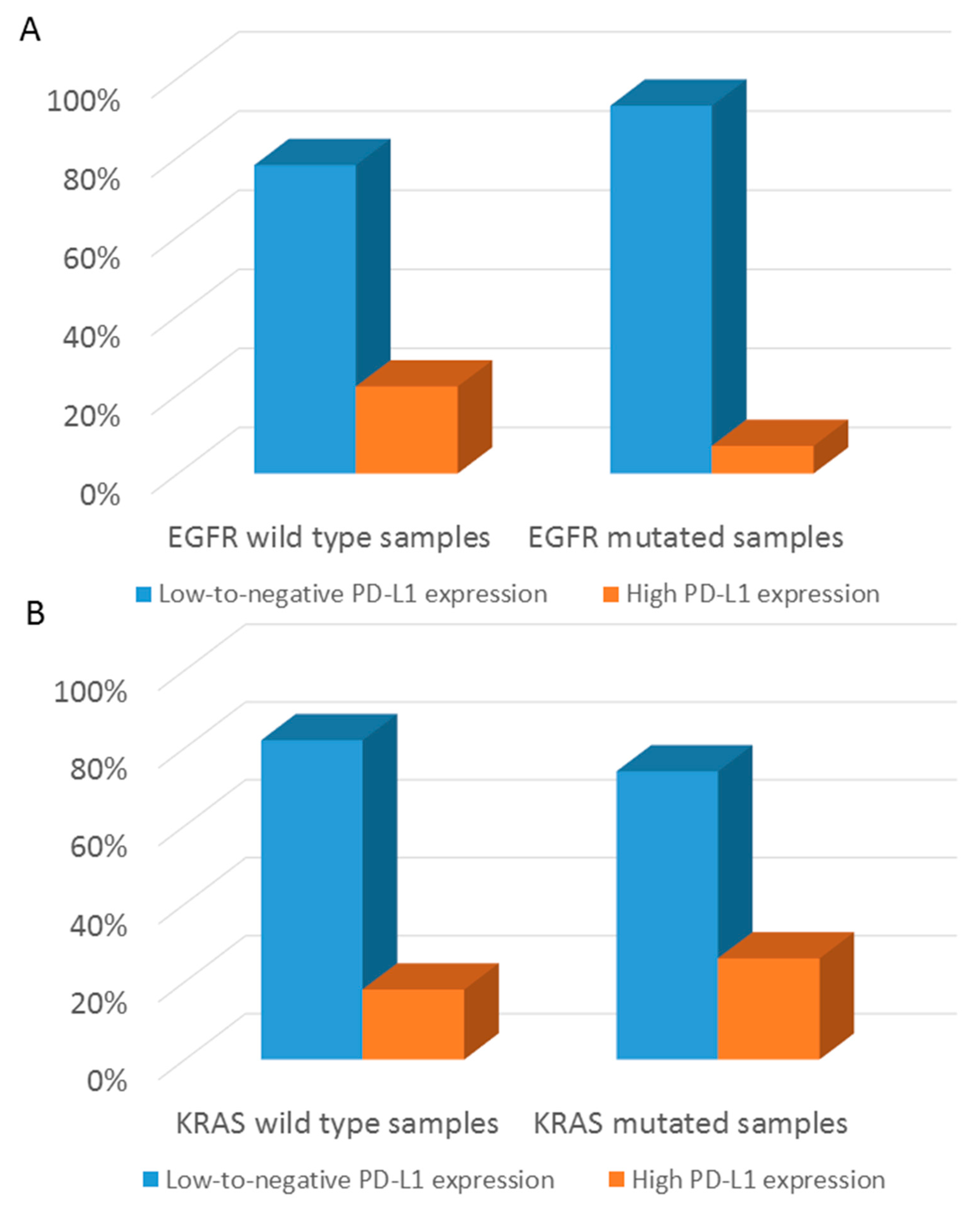
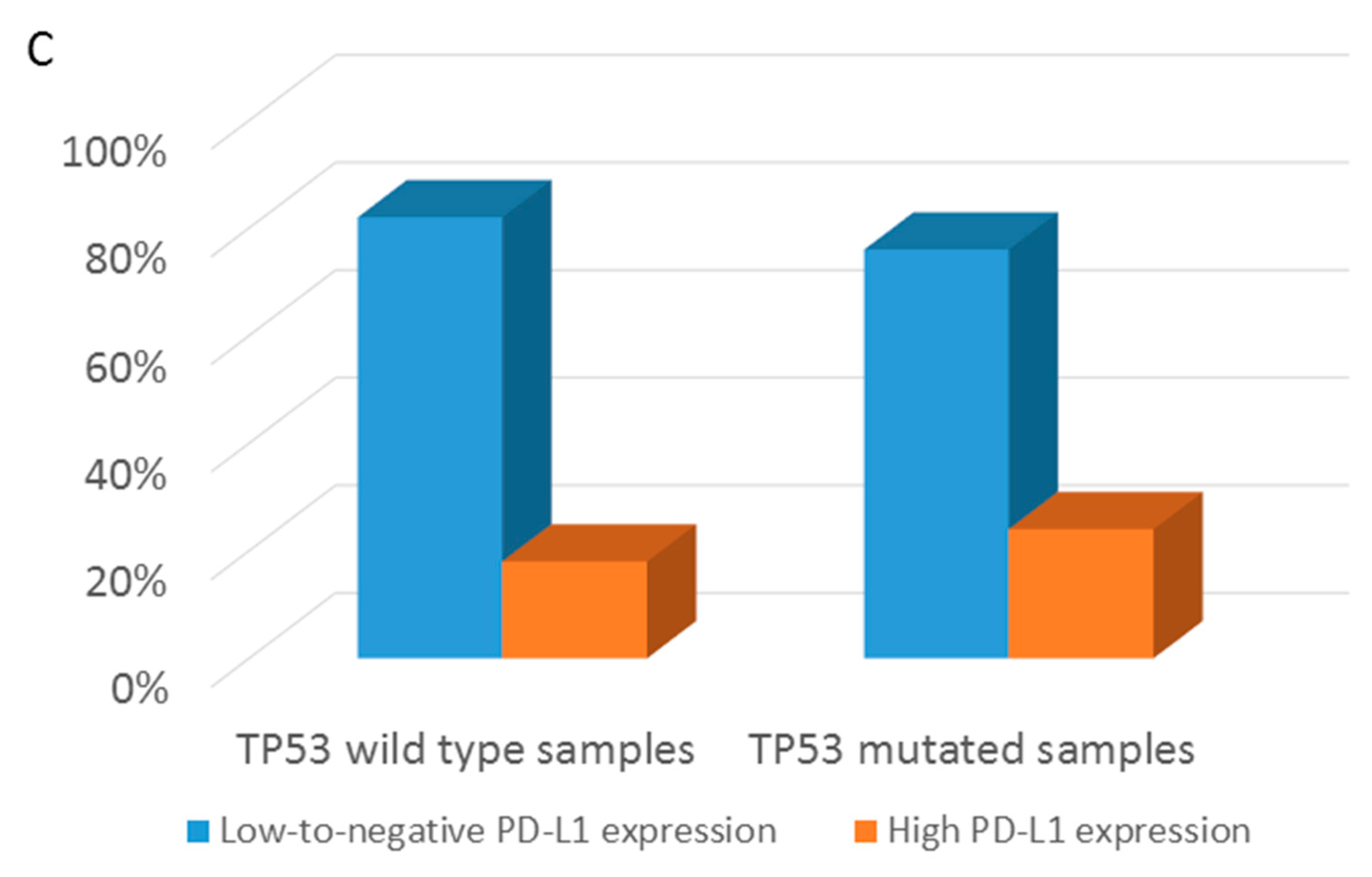
3.3. Distribution of Targetable Alterations in Early-Stage NSCLC Based on Histological Pattern
3.4. Overall Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S.; et al. Global, regional, and National Cancer Incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: A systematic analysis for the global burden of disease study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar]
- AIOM-Associazione Italiana Oncologia Medica. Linee Guida Neoplasie del Polmone Ottobre 2021. Available online: https://snlg.iss.it/wp-content/uploads/2021/11/LG-149_Polmone_agg2021.pdf (accessed on 31 December 2021).
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [Green Version]
- Thoracic Tumours. WHO Classification of Tumours, 5th ed.; WHO Classification of Tumours Editorial Board: Geneva, Switzerland, 2021; Volume 5, ISBN 978-92-832-4506-3. [Google Scholar]
- Le Chevalier, T. Adjuvant chemotherapy for resectable non-small-cell lung cancer: Where is it going. Ann. Oncol. 2010, 21 (Suppl. 7), vii196–vii198. [Google Scholar] [CrossRef]
- Cagle, P.T.; Allen, T.C.; Olsen, R.J. Lung cancer biomarkers: Present status and future developments. Arch. Pathol. Lab. Med. 2013, 137, 1191–1198. [Google Scholar] [CrossRef]
- Nagasaka, M.; Gadgeel, S.M. Role of chemotherapy and targeted therapy in early stage non-small cell lung cancer. Expert Rev. Anticancer Ther. 2018, 18, 63–70. [Google Scholar] [CrossRef]
- Pignon, J.P.; Tribodet, H.; Scagliotti, G.V.; Douillard, J.Y.; Shepherd, F.A.; Stephens, R.J.; Dunant, A.; Torri, V.; Rosell, R.; Seymour, L.; et al. Lung adjuvant cisplatin evaluation: A pooled analysis by the LACE collaborative group. J. Clin. Oncol. 2008, 26, 3552–3559. [Google Scholar] [CrossRef]
- Kris, M.G.; Gaspar, L.E.; Chaft, J.E.; Kennedy, E.B.; Azzoli, C.G.; Ellis, P.M.; Lin, S.H.; Pass, H.; Seth, R.; Shepherd, F.A.; et al. Adjuvant systemic therapy and adjuvant radiation therapy for stage I to IIIA completely resected non-small-cell lung cancers: American Society of Clinical Oncology/Cancer Care Ontario clinical practice guideline update. J. Clin. Oncol. 2017, 35, 2960–2974. [Google Scholar] [CrossRef] [Green Version]
- Uramoto, H.; Nakanishi, R.; Nagashima, A.; Uchiyama, A.; Inoue, M.; Osaki, T.; Yoshimatsu, T.; Sakata, H.; Nakanishi, K.; Yasumoto, K. A randomized phase II trial of adjuvant chemotherapy with bi-weekly carboplatin plus paclitaxel versus carboplatin plus gemcitabine in patients with completely resected non-small cell lung cancer. Anticancer Res. 2010, 30, 4695–4699. [Google Scholar]
- Kreuter, M.; Vansteenkiste, J.; Fischer, J.R.; Eberhardt, W.; Zabeck, H.; Kollmeier, J.; Serke, M.; Frickhofen, N.; Reck, M.; Engel-Riedel, W.; et al. Randomized phase 2 trial on refinement of early-stage NSCLC adjuvant chemotherapy with cisplatin and pemetrexed versus cisplatin and vinorelbine: The TREAT study. Ann. Oncol. 2013, 24, 986–9892. [Google Scholar] [CrossRef]
- Majeed, U.; Manochakian, R.; Zhao, Y.; Lou, Y. Targeted therapy in advanced non-small cell lung cancer: Current advances and future trends. J. Hematol. Oncol. 2021, 14, 108. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Han, J.-Y.; Katakami, N.; Kim, H.R.; Hodge, R.; Kaur, P.; Brown, A.P.; Ghiorghiu, D.; et al. CNS efficacy of osimertinib in patients with T790M-positive advanced non-small-cell lung cancer: Data from a randomized phase III trial (AURA3). J. Clin. Oncol. 2018, 36, 2702–2709. [Google Scholar] [CrossRef] [PubMed]
- Reungwetwattana, T.; Nakagawa, K.; Cho, B.C.; Cobo, M.; Cho, E.K.; Bertolini, A.; Bohnet, S.; Zhou, C.; Lee, K.H.; Nogami, N.; et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J. Clin. Oncol. 2018, 36, 3290–3297. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. Osimertinib in Resected EGFR-Mutated Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef]
- Koch, A.L.; Vellanki, P.J.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Shen, Y.L.; Xia, H.; Li, Y.; Liu, J.; Zirkelbach, J.F.; et al. FDA Approval Summary: Osimertinib for adjuvant treatment of surgically resected non-small cell lung cancer, a collaborative Project Orbis review. Clin. Cancer Res. 2021, 27, 6638–6643. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.B.; Spranger, S. Modulation of the immune microenvironment by tumor-intrinsic oncogenic signaling. J. Cell Biol. 2020, 219, e201908224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.L.; Walsh, K.; Diamond, A.; Oniscu, A.; Deans, Z.C. Validation of the Oncomine™ focus panel for next-generation sequencing of clinical tumour samples. Virchows Arch. 2018, 473, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Kerr, K.M.; Bibeau, F.; Thunnissen, E.; Botling, J.; Ryška, A.; Wolf, J.; Öhrling, K.; Burdon, P.; Malapelle, U.; Büttner, R. The evolving landscape of biomarker testing for non-small cell lung cancer in Europe. Lung Cancer 2021, 154, 161–175. [Google Scholar] [CrossRef]
- Thai, A.A.; Solomon, B.J.; Sequist, L.V.; Gainor, J.F.; Heist, R.S. Seminar Lung Cancer. Lancet 2021, 398, 535–554. [Google Scholar] [CrossRef]
- Liu, B.; Quan, X.; Xu, C.; Lv, J.; Li, C.; Dong, L.; Liu, M. Lung cancer in young adults aged 35 years or younger: A full-scale analysis and review. J. Cancer 2019, 10, 3553–3559. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.; O’Sullivan, B.; Hughes, F.; Mullis, T.; Smith, M.; Trim, N.; Taniere, P. The Clinicopathological and Molecular Associations of PD-L1 Expression in Non-small Cell Lung Cancer: Analysis of a Series of 10,005 Cases Tested with the 22C3 Assay. Pathol. Oncol. Res. 2020, 26, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gristina, V.; Malapelle, U.; Galvano, A.; Pisapia, P.; Pepe, F.; Rolfo, C.; Tortorici, S.; Bazan, V.; Troncone, G.; Russo, A. The significance of epidermal growth factor receptor uncommon mutations in non-small cell lung cancer: A systematic review and critical appraisal. Cancer Treat. Rev. 2020, 85, 101994. [Google Scholar] [CrossRef] [PubMed]
- Passaro, A.; Mok, T.; Peters, S.; Popat, S.; Ahn, M.-J.; de Marinis, F. Recent Advances on the Role of EGFR Tyrosine Kinase Inhibitors in the Management of NSCLC With Uncommon, Non Exon 20 Insertions, EGFR Mutations. J. Thorac. Oncol. 2021, 16, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Lim, S.H.; An, H.J.; Kim, K.H.; Park, K.U.; Kang, E.J.; Choi, Y.H.; Ahn, M.S.; Lee, M.H.; Sun, J.M.; et al. Osimertinib for Patients With Non-Small-Cell Lung Cancer Harboring Uncommon EGFR Mutations: A Multicenter, Open-Label, Phase II Trial (KCSG-LU15-09). J. Clin. Oncol. 2020, 38, 488–495. [Google Scholar] [CrossRef]
- Xu, Y.; Tong, X.; Yan, J.; Wu, X.; Shao, Y.W.; Fan, Y. Short-Term Responders of Non-Small Cell Lung Cancer Patients to EGFR Tyrosine Kinase Inhibitors Display High Prevalence of TP53 Mutations and Primary Resistance Mechanisms. Transl. Oncol. 2018, 11, 1364–1369. [Google Scholar] [CrossRef]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Zhang, S.M.; Zhu, Q.G.; Ding, X.X.; Lin, S.; Zhao, J.; Guan, L.; Li, T.; He, B.; Zhang, H.Q. Prognostic value of EGFR and KRAS in resected non-small cell lung cancer: A systematic review and meta-analysis. Cancer Manag. Res. 2018, 10, 3393–3404. [Google Scholar] [CrossRef] [Green Version]
- Koopman, B.; Garcia, B.C.; Kuijpers, C.; Damhuis, R.; van der Wekken, A.; Groen, H.; Schuuring, E.; Willems, S.; van Kempen, L. A Nationwide Study on the Impact of Routine Testing for EGFR Mutations in Advanced NSCLC Reveals Distinct Survival Patterns Based on EGFR Mutation Subclasses. Cancers 2021, 13, 3641. [Google Scholar] [CrossRef]
- Gu, J.; Zhou, Y.; Huang, L.; Ou, W.; Wu, J.; Li, S.; Xu, J.; Feng, J.; Liu, B. TP53 mutation is associated with a poor clinical outcome for non-small cell lung cancer: Evidence from a meta-analysis. Mol. Clin. Oncol. 2016, 5, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Tuminello, S.; Sikavi, D.; Veluswamy, R.; Gamarra, C.; Lieberman-Cribbin, W.; Flores, R.; Taioli, E. PD-L1 as a prognostic biomarker in surgically resectable non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2020, 9, 1343–1360. [Google Scholar] [CrossRef]
- Guo, Y.; Song, J.; Wang, Y.; Huang, L.; Sun, L.; Zhao, J.; Zhang, S.; Jing, W.; Ma, J.; Han, C. Concurrent Genetic Alterations and Other Biomarkers Predict Treatment Efficacy of EGFR-TKIs in EGFR-Mutant Non-Small Cell Lung Cancer: A Review. Front. Oncol. 2020, 10, 610923. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | N = 1961 | % |
|---|---|---|
| Gender | ||
| Female | 826 | 42% |
| Male | 1135 | 58% |
| Age at molecular diagnosis | ||
| Median (min–max) | 69 (23–90) | |
| Stage | ||
| Early (IA–IIIA) | 513 | 26% |
| Advanced (IIIB–IV) | 1448 | 74% |
| Type of panel | ||
| 22 Genes | 1745 | 89% |
| Focus | 202 | 10% |
| Both | 14 | 1% |
| EGFR | 270/1961 | 13.8% |
| ALK | 107/1951 | 5.5% |
| ROS1 | 9/1956 | 0.5% |
| BRAF | 71/1890 | 3.8% |
| V600E | 24/1890 | 1.3% |
| Other variant | 47/1890 | 2.7% |
| KRAS | 536/1961 | 27.3% |
| G12C | 199/1961 | 10.1% |
| Other variant | 337/1961 | 17.2% |
| MET exon 14 skipping | 13/207 | 6.3% |
| RET fusions | 7/207 | 3.4% |
| TP53 | 454/1759 | 25.8% |
| PIK3CA | 75/1961 | 3.8% |
| ERBB2 | 17/1961 | 0.9% |
| PD-L1 | ||
| score 0 (<1%) | 568/1351 | 42.0% |
| score 1 (1–49%) | 512/1351 | 37.8% |
| score 2 (≥50%) | 271/1351 | 20.2% |
| Variable | Early Stage | Advanced Stage | p-Value |
|---|---|---|---|
| N (%) | N (%) | ||
| 513 | 1448 | ||
| Gender | 0.078 | ||
| Female | 233 (45%) | 593 (41%) | |
| Male | 280 (55%) | 855 (59%) | |
| Age at molecular diagnosis | 0.023 | ||
| Median (min–max) | 70 (27–86) | 68 (23–90) | |
| EGFR | 70/513 (13.6%) | 200/1448 (13.8%) | 0.925 |
| ALK | 25/513 (4.8%) | 82/1438 (5.7%) | 0.479 |
| ROS1 | 2/513 (0.39%) | 7/1443 (0.48%) | 0.784 |
| BRAF | 18/513 (3.5%) | 53/1448 (3.6%) | 0.543 |
| V600E | 10 (1.9%) | 37 (2.5%) | |
| Other variant | 8 (1.5%) | 16 (1.1%) | |
| KRAS | 159/513 (30.9%) | 377/1448 (26%) | 0.045 |
| G12C | 53/513 (10.3%) | 146/1448 (10.1%) | |
| Other variant | 106/513 (20.6%) | 231/1448 (15.9%) | |
| MET exon 14 skipping | 3/58 (5.2%) | 10/149 (6.7%) | 0.682 |
| RET | 2/58 (3.4%) | 5/149 (3.3%) | 0.974 |
| TP53 | 107/455 (23.5%) | 347/1304 (26.6%) | 0.194 |
| PIK3CA | 16/513 (3.1%) | 59/1448 (4.1%) | 0.332 |
| ERBB2 | 4/513 (0.8%) | 13/1448 (0.9%) | 1.000 |
| PD-L1 | |||
| score 0 (<1%) | 163/357(45.7%) | 405/994 (40.7%) | 0.104 |
| score 1 (1–49%) | 135/357 (37.8%) | 377/994 (37.9%) | |
| score 2 (≥50%) | 59/357 (16.5%) | 212/994 (21.4%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terrenato, I.; Ercolani, C.; Di Benedetto, A.; Gallo, E.; Melucci, E.; Casini, B.; Rollo, F.; Palange, A.; Visca, P.; Pescarmona, E.; et al. A Real-World Systematic Analysis of Driver Mutations’ Prevalence in Early- and Advanced-Stage NSCLC: Implications for Targeted Therapies in the Adjuvant Setting. Cancers 2022, 14, 2971. https://doi.org/10.3390/cancers14122971
Terrenato I, Ercolani C, Di Benedetto A, Gallo E, Melucci E, Casini B, Rollo F, Palange A, Visca P, Pescarmona E, et al. A Real-World Systematic Analysis of Driver Mutations’ Prevalence in Early- and Advanced-Stage NSCLC: Implications for Targeted Therapies in the Adjuvant Setting. Cancers. 2022; 14(12):2971. https://doi.org/10.3390/cancers14122971
Chicago/Turabian StyleTerrenato, Irene, Cristiana Ercolani, Anna Di Benedetto, Enzo Gallo, Elisa Melucci, Beatrice Casini, Francesca Rollo, Aldo Palange, Paolo Visca, Edoardo Pescarmona, and et al. 2022. "A Real-World Systematic Analysis of Driver Mutations’ Prevalence in Early- and Advanced-Stage NSCLC: Implications for Targeted Therapies in the Adjuvant Setting" Cancers 14, no. 12: 2971. https://doi.org/10.3390/cancers14122971
APA StyleTerrenato, I., Ercolani, C., Di Benedetto, A., Gallo, E., Melucci, E., Casini, B., Rollo, F., Palange, A., Visca, P., Pescarmona, E., Melis, E., Gallina, F., Sacconi, A., Cecere, F. L., Landi, L., Cappuzzo, F., Ciliberto, G., & Buglioni, S. (2022). A Real-World Systematic Analysis of Driver Mutations’ Prevalence in Early- and Advanced-Stage NSCLC: Implications for Targeted Therapies in the Adjuvant Setting. Cancers, 14(12), 2971. https://doi.org/10.3390/cancers14122971