
α-Ketoglutarate-Mediated DNA Demethylation Sustains T-Acute Lymphoblastic Leukemia upon TCA Cycle Targeting

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Metabolomics Profiling and Analysis
2.2. Cell Culture and α-KG Treatment
2.3. Protein Extraction and Western Blotting
2.4. Lentivirus Infection
2.5. Immunofluorescence Staining and Imaging
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. Zebrafish Xenograft Assays
2.8. Statistical Analysis
2.9. Timeline Proposed to Complete Future Experiments
3. Results
3.1. DLST Knockdown in Human T-ALL Cells Enhances Reductive Carboxylation While Decreasing Glutamine-Derived TCA Cycle Flux
3.2. DLST Knockdown Leads to Upregulation of IDH1 and IDH2 in Human T-ALL Cells
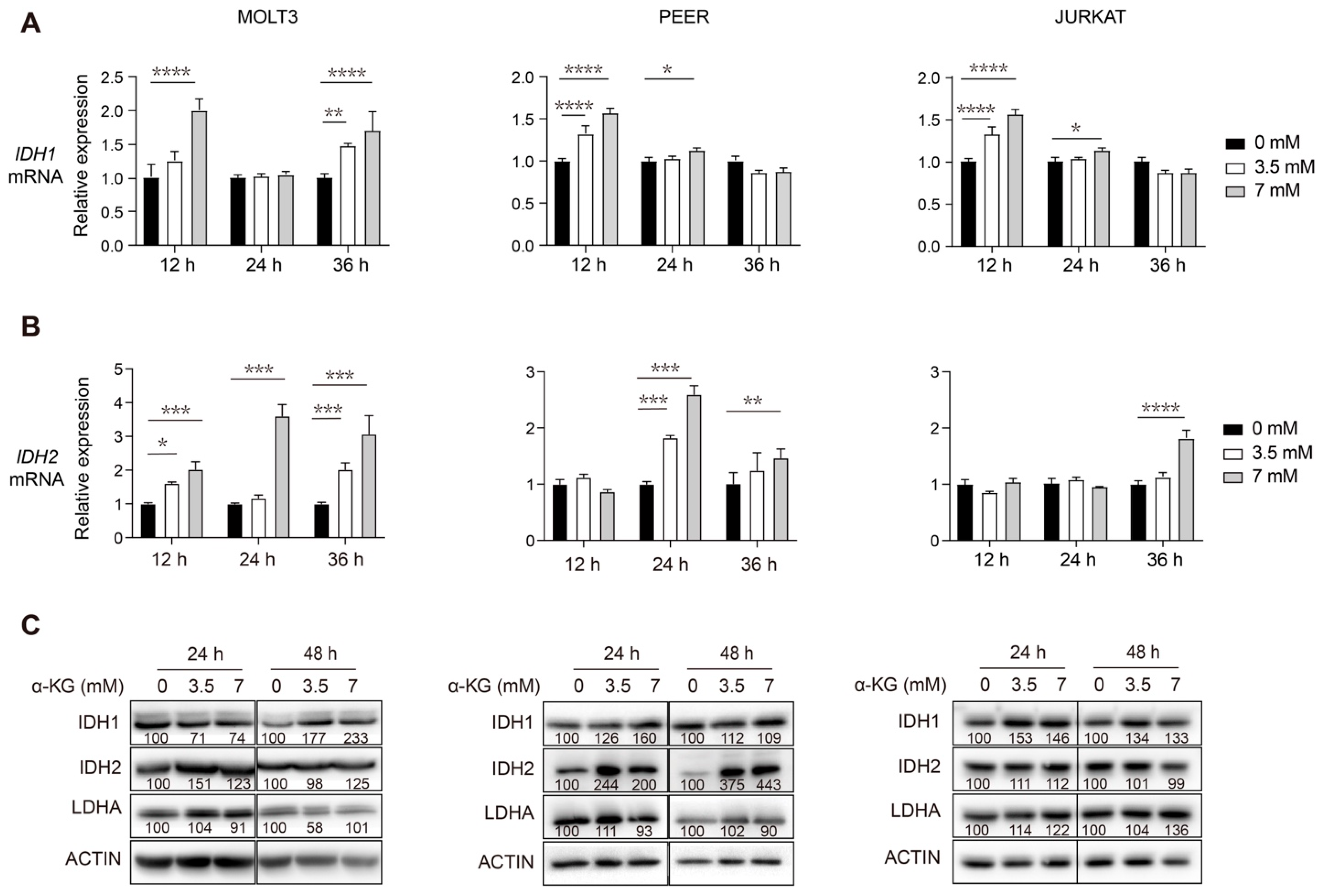
3.3. Supplementation of Exogenous α-KG Induces Upregulation of IDH1 and IDH2 in Human T-ALL Cells
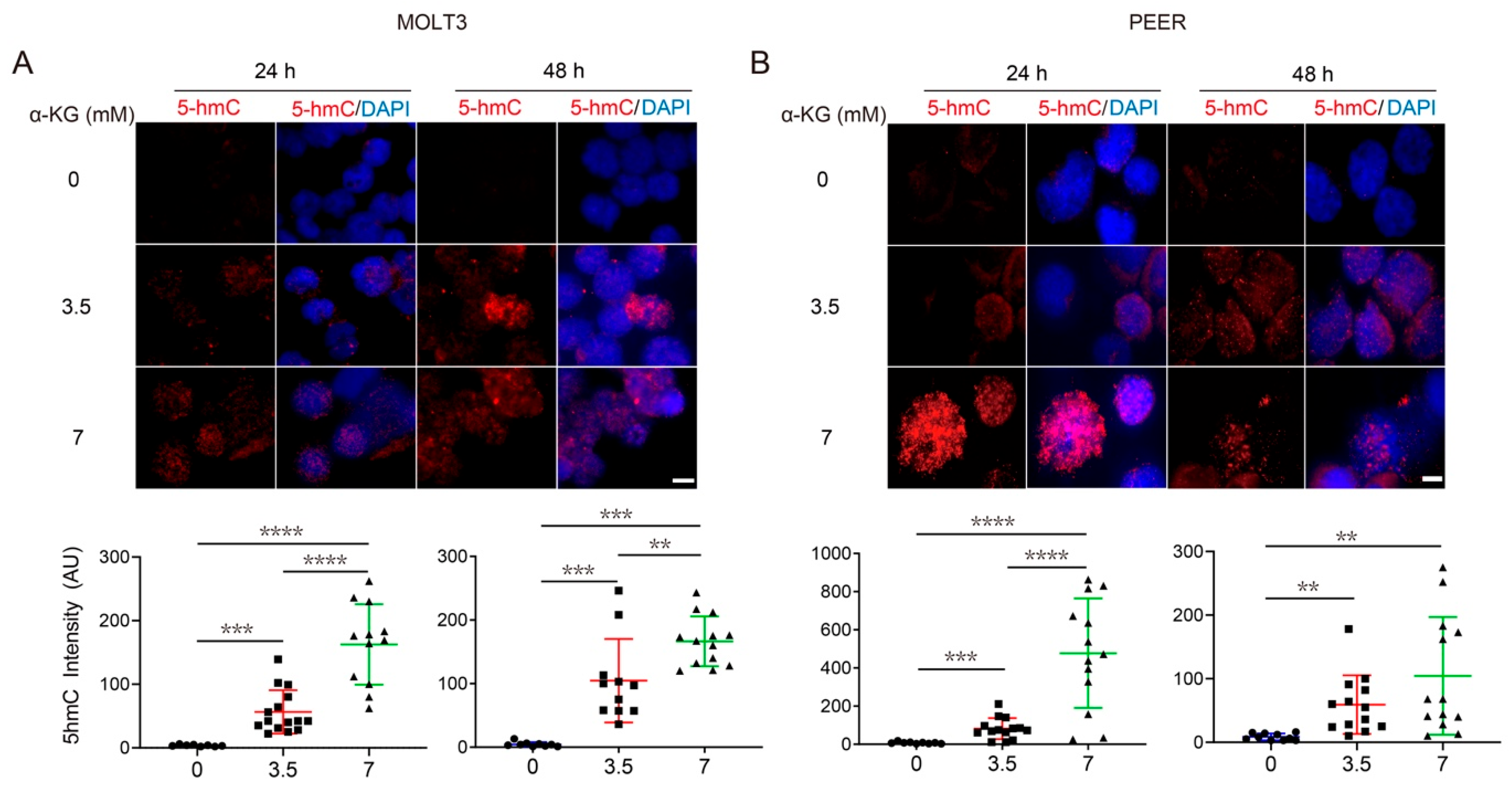
3.4. Both α-KG Treatment and DLST Depletion Lead to Increased DNA Demethylation in Human T-ALL Cells
3.5. Combined Treatment with CPI-613 and Bobcat339 Significantly Decreases Tumor Burden in Zebrafish Xenografts of Human T-ALL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2021, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2017, 9, 216–237. [Google Scholar] [CrossRef]
- Wahlström, T.; Henriksson, M.A. Impact of MYC in regulation of tumor cell metabolism. Biochim. Biophys. Acta 2015, 1849, 563–569. [Google Scholar] [CrossRef]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC Activation Is a Hallmark of Cancer Initiation and Maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [Green Version]
- Rivera, G.O.R.; Knochelmann, H.M.; Dwyer, C.J.; Smith, A.S.; Wyatt, M.M.; Rivera-Reyes, A.M.; Thaxton, J.E.; Paulos, C.M. Fundamentals of T Cell Metabolism and Strategies to Enhance Cancer Immunotherapy. Front. Immunol. 2021, 12, 782. [Google Scholar] [CrossRef]
- Anderson, N.M.; Li, D.; Peng, H.L.; Laroche, F.J.F.; Mansour, M.R.; Gjini, E.; Aioub, M.; Helman, D.J.; Roderick, J.E.; Cheng, T.; et al. The TCA cycle transferase DLST is important for MYC-mediated leukemogenesis. Leukemia 2016, 30, 1365–1374. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-K.; Park, K.-G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Hu, Z.; Shi, X.; Jiang, L.; Boroughs, L.K.; Kovacs, Z.; Boriack, R.; Rakheja, D.; Sullivan, L.B.; Linehan, W.M.; et al. Oxidation of Alpha-Ketoglutarate Is Required for Reductive Carboxylation in Cancer Cells with Mitochondrial Defects. Cell Rep. 2014, 7, 1679–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re Not “DON” Yet: Optimal Dosing and Prodrug Delivery of 6-Diazo-5-oxo-L-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Per-spectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 2321. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wang, S.; Zaal, E.A.; Wang, C.; Wu, H.; Bosma, A.; Jochems, F.; Isima, N.; Jin, G.; Lieftink, C.; et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. Elife 2020, 9, e56749. [Google Scholar] [CrossRef] [PubMed]
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; DiPaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase I dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.-A.; Chen, C.-L.; Huang, Y.-H.; Evans, E.E.; Cheng, C.-C.; Chuang, Y.-J.; Zhang, C.; Le, A. Inhibition of glutaminolysis in combination with other therapies to improve cancer treatment. Curr. Opin. Chem. Biol. 2021, 62, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, T.N.; Patwardhan, P.P.; Cremers, S.; Schwartz, G.K. Targeted inhibition of glutaminase as a potential new approach for the treatment of NF1 associated soft tissue malignancies. Oncotarget 2017, 8, 94054–94068. [Google Scholar] [CrossRef] [Green Version]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.; Siska, P.J.; Macintyre, A.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Anderson, N.M.; Qin, X.; Finan, J.M.; Lam, A.; Athoe, J.; Missiaen, R.; Skuli, N.; Kennedy, A.; Saini, A.S.; Tao, T.; et al. Metabolic Enzyme DLST Promotes Tumor Aggression and Reveals a Vulnerability to OXPHOS Inhibition in High-Risk Neuroblastoma. Cancer Res. 2021, 81, 4417–4430. [Google Scholar] [CrossRef]
- Shen, N.; Korm, S.; Karantanos, T.; Li, D.; Zhang, X.; Ritou, E.; Xu, H.; Lam, A.; English, J.; Zong, W.-X.; et al. DLST-dependence dictates metabolic heterogeneity in TCA-cycle usage among triple-negative breast cancer. Commun. Biol. 2021, 4, 1289. [Google Scholar] [CrossRef]
- Baran, N.; Lodi, A.; Sweeney, S.R.; Renu, P.; Kuruvilla, V.M.; Cavazos, A.; Herranz, D.; Skwarska, A.; Warmoes, M.; Davis, R.E.; et al. Mitochondrial Complex I Inhibitor Iacs-010759 Reverses the NOTCH1-Driven Metabolic Reprogramming in T-ALL via Blockade of Oxidative Phosphorylation: Synergy with Chemotherapy and Glutaminase Inhibition. Blood 2018, 132, 4020. [Google Scholar] [CrossRef]
- Pardee, T.; DeFord-Watts, L.M.; Peronto, E.; Levitan, D.A.; Hurd, D.D.; Kridel, S. Altered Lipid and Mitochondrial Metabolism Are Viable Targets in Acute Leukemia. Blood 2011, 118, 3618. [Google Scholar] [CrossRef]
- Lycan, T.W.; Pardee, T.S.; Petty, W.J.; Bonomi, M.; Alistar, A.; Lamar, Z.S.; Isom, S.; Chan, M.D.; Miller, A.A.; Ruiz, J. A phase II clinical trial of CPI-613 in patients with relapsed or refractory small cell lung carcinoma. PLoS ONE 2016, 11, e0164244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, L.; Wang, F.; Ding, Z.; Zhao, X.; Loveless, R.; Xie, J.; Shay, C.; Qiu, P.; Ke, Y.; Saba, N.F.; et al. Blockade of gluta-mine-dependent cell survival augments antitumor efficacy of CPI-613 in head and neck cancer. J. Exp. Clin. Cancer Res. 2021, 40, 393. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.N.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Zdzisińska, B.; Żurek, A.; Kandefer-Szerszeń, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Ther. Exp. 2016, 65, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of alpha-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2021, 1–43. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W. Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic plasticity underpins innate and acquired resistance to LDHA inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, D.; Ambesi-Impiombato, A.; Sudderth, J.; Sánchez-Martín, M.; Belver, L.; Tosello, V.; Xu, L.; Wendorff, A.A.; Castillo, M.; Haydu, J.E.; et al. Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 2015, 21, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.; Wang, Q.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 532, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Van Dyck, L.; Boeckx, B.; Schoonjans, L.; et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.-W.J.; Yuita, H.; Rao, A. Dysregulation of the TET family of epigenetic regulators in lymphoid and myeloid malignancies. Blood 2019, 134, 1487–1497. [Google Scholar] [CrossRef]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; VasanthaKumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- López-Moyado, I.F.; Tsagaratou, A.; Yuita, H.; Seo, H.; Delatte, B.; Heinz, S.; Benner, C.; Rao, A. Paradoxical association of TET loss of function with genome-wide DNA hypomethylation. Proc. Natl. Acad. Sci. USA 2019, 116, 16933–16942. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Shen, N.; Spurlin, G.; Korm, S.; Huang, S.; Anderson, N.M.; Huiting, L.N.; Liu, H.; Feng, H. α-Ketoglutarate-Mediated DNA Demethylation Sustains T-Acute Lymphoblastic Leukemia upon TCA Cycle Targeting. Cancers 2022, 14, 2983. https://doi.org/10.3390/cancers14122983
Wang Y, Shen N, Spurlin G, Korm S, Huang S, Anderson NM, Huiting LN, Liu H, Feng H. α-Ketoglutarate-Mediated DNA Demethylation Sustains T-Acute Lymphoblastic Leukemia upon TCA Cycle Targeting. Cancers. 2022; 14(12):2983. https://doi.org/10.3390/cancers14122983
Chicago/Turabian StyleWang, Yanwu, Ning Shen, Gervase Spurlin, Sovannarith Korm, Sarah Huang, Nicole M. Anderson, Leah N. Huiting, Hudan Liu, and Hui Feng. 2022. "α-Ketoglutarate-Mediated DNA Demethylation Sustains T-Acute Lymphoblastic Leukemia upon TCA Cycle Targeting" Cancers 14, no. 12: 2983. https://doi.org/10.3390/cancers14122983
APA StyleWang, Y., Shen, N., Spurlin, G., Korm, S., Huang, S., Anderson, N. M., Huiting, L. N., Liu, H., & Feng, H. (2022). α-Ketoglutarate-Mediated DNA Demethylation Sustains T-Acute Lymphoblastic Leukemia upon TCA Cycle Targeting. Cancers, 14(12), 2983. https://doi.org/10.3390/cancers14122983