atm Mutation and Oxidative Stress Enhance the Pre-Cancerous Effects of UHRF1 Overexpression in Zebrafish Livers

 , and
, and 
Abstract
:Simple Summary
Abstract
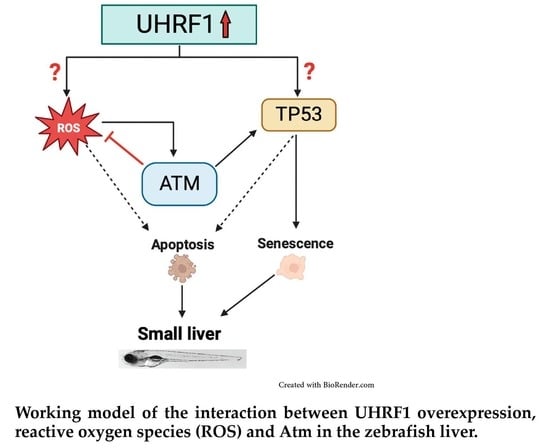
1. Introduction
2. Materials and Methods
2.1. Zebrafish Husbandry, Genotyping and Exposure
2.2. Generation of atm Mutant Zebrafish
2.3. Sanger Sequencing
2.4. RNA Extraction
2.5. cDNA Production and qPCR
2.6. RNAseq and Bioinformatic Analysis
2.7. Cy3 Streptavidin (CY3-SA) Staining and Liver Size Measurement
2.8. Statistical Analysis
3. Results
3.1. hUHRF1 Overexpression in Zebrafish Hepatocytes Activates the tp53 Pathway at 5 dpf

3.2. Deletion of atm Is Well Tolerated in Zebrafish Embryos

3.3. atm Mutation Suppresses DNA Damage and Oxidative Stress Induced Phenotypes in Zebrafish Embryos

3.4. atm Mutation and H2O2 Exposure Enhances the Small Liver Phenotype Caused by hUHRF1 Overexpression in Hepatocytes

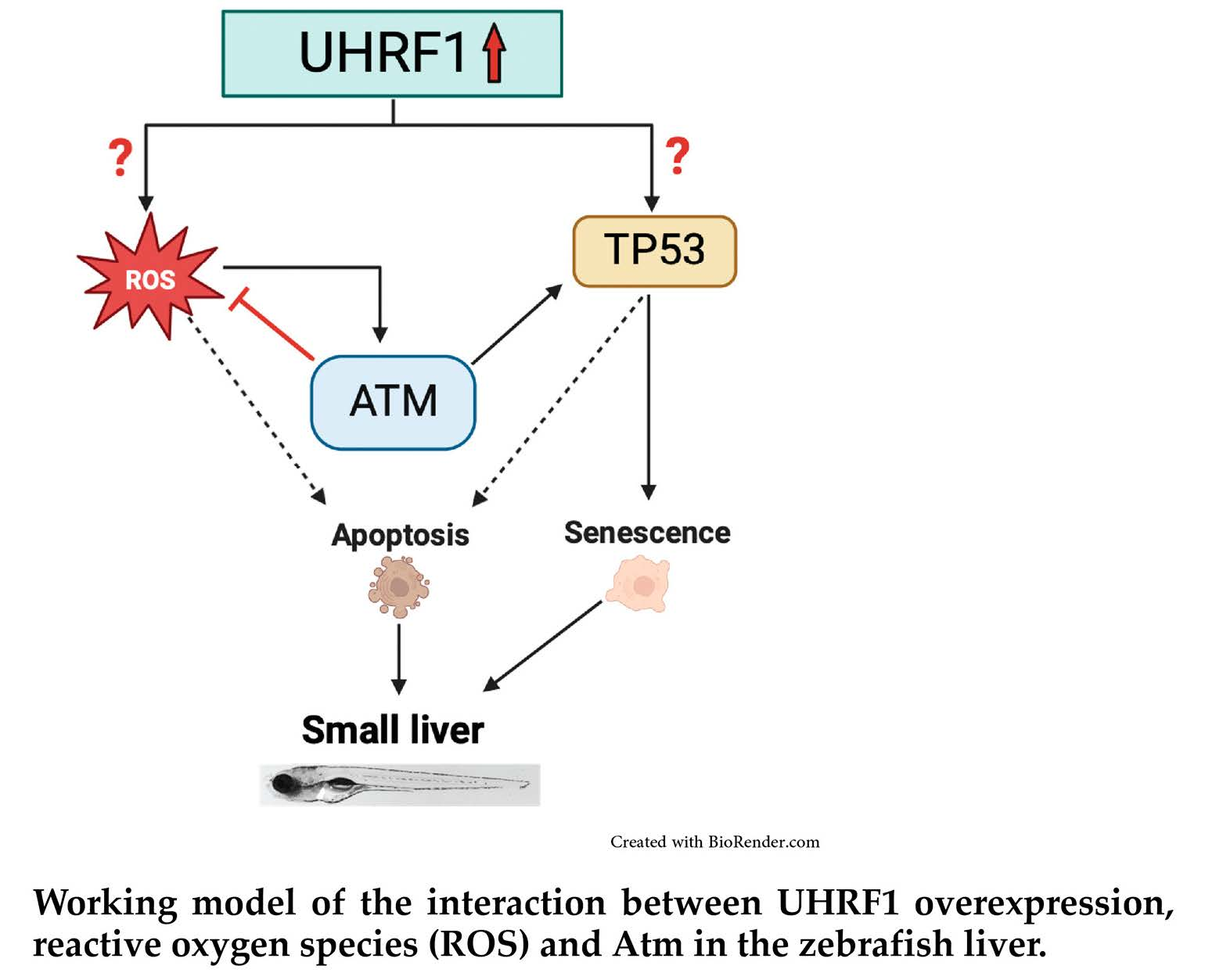
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell. Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoare, M.; Narita, M. The Power Behind the Throne: Senescence and the Hallmarks of Cancer. Annu. Rev. Cancer Biol. 2018, 2, 175–194. [Google Scholar] [CrossRef] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, Q.; Sack, L.; Kang, C.; Elledge, S.J. A gain-of-function senescence bypass screen identifies the homeobox transcription factor DLX2 as a regulator of ATM-p53 signaling. Genes. Dev. 2016, 30, 293–306. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell. Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell. Biol. 2011, 13, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Rezaeian, A.H. ATM: Main Features, Signaling Pathways, and Its Diverse Roles in DNA Damage Response, Tumor Suppression and Cancer Development. Genes 2021, 12, 845. [Google Scholar] [CrossRef]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.S.; Park, S.C. Chemical screening identifies ATM as a target for alleviating senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef]
- Aird, K.M.; Zhang, R. ATM in senescence. Oncotarget 2015, 6, 14729–14730. [Google Scholar] [CrossRef]
- Putti, S.; Giovinazzo, A.; Merolle, M.; Falchetti, M.L.; Pellegrini, M. ATM Kinase Dead: From Ataxia Telangiectasia Syndrome to Cancer. Cancers 2021, 13, 5498. [Google Scholar] [CrossRef]
- Berger, N.D.; Stanley, F.K.T.; Moore, S.; Goodarzi, A.A. ATM-dependent pathways of chromatin remodelling and oxidative DNA damage responses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160283. [Google Scholar] [CrossRef] [Green Version]
- Elson, A.; Wang, Y.; Daugherty, C.J.; Morton, C.C.; Zhou, F.; Campos-Torres, J.; Leder, P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 13084–13089. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ashley, T.; Brainerd, E.E.; Bronson, R.T.; Meyn, M.S.; Baltimore, D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes. Dev. 1996, 10, 2411–2422. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Wang, P.; Chen, J.; Ying, Y.; Chen, Y.; Gilson, E.; Lu, Y.; Ye, J. Loss of atm in Zebrafish as a Model of Ataxia-Telangiectasia Syndrome. Biomedicines 2022, 10, 392. [Google Scholar] [CrossRef]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, L.; Lu, A.; Han, Y.; Colangelo, D.; Bukata, C.; Scibetta, A.; Yousefzadeh, M.J.; Li, X.; Gurkar, A.U.; et al. ATM is a key driver of NF-kappaB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging 2020, 12, 4688–4710. [Google Scholar] [CrossRef]
- Guo, Q.Q.; Wang, S.S.; Zhang, S.S.; Xu, H.D.; Li, X.M.; Guan, Y.; Yi, F.; Zhou, T.T.; Jiang, B.; Bai, N.; et al. ATM-CHK2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- Okuno, Y.; Nakamura-Ishizu, A.; Otsu, K.; Suda, T.; Kubota, Y. Pathological neoangiogenesis depends on oxidative stress regulation by ATM. Nat. Med. 2012, 18, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, J.H.; Paull, T.T.; Gehrke, S.; D’Alessandro, A.; Dou, Q.; Gladyshev, V.N.; Schroeder, E.A.; Steyl, S.K.; Christian, B.E.; et al. Mitochondrial redox sensing by the kinase ATM maintains cellular antioxidant capacity. Sci. Signal. 2018, 11, eaaq0702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Mand, M.R.; Kao, C.H.; Zhou, Y.; Ryu, S.W.; Richards, A.L.; Coon, J.J.; Paull, T.T. ATM directs DNA damage responses and proteostasis via genetically separable pathways. Sci. Signal. 2018, 11, eaan5598. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Paull, T.T. Mitochondria at the crossroads of ATM-mediated stress signaling and regulation of reactive oxygen species. Redox Biol. 2020, 32, 101511. [Google Scholar] [CrossRef]
- Huiting, W.; Dekker, S.L.; van der Lienden, J.C.J.; Mergener, R.; Musskopf, M.K.; Furtado, G.V.; Gerrits, E.; Coit, D.; Oghbaie, M.; Di Stefano, L.H.; et al. Targeting DNA topoisomerases or checkpoint kinases results in an overload of chaperone systems, triggering aggregation of a metastable subproteome. Elife 2022, 11, e70726. [Google Scholar] [CrossRef]
- Fu, X.; Wan, S.; Lyu, Y.L.; Liu, L.F.; Qi, H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE 2008, 3, e2009. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, Y.; Wang, Z.; Wang, S.; Jiang, X.; Cui, H.; Zhou, T.; He, Z.; Feng, H.; Guo, Q.; et al. ATM at the crossroads of reactive oxygen species and autophagy. Int. J. Biol. Sci. 2021, 17, 3080–3090. [Google Scholar] [CrossRef]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.R.; Bagheri, M.; Saini, D.K. Temporally distinct roles of ATM and ROS in genotoxic-stress-dependent induction and maintenance of cellular senescence. J. Cell. Sci. 2015, 128, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Polderman, P.E.; Pagès-Gallego, M.; van Es, R.M.; Vos, H.R.; Burgering, B.M.T.; Dansen, T.B. p53 Forms Redox-Dependent Protein-Protein Interactions through Cysteine 277. Antioxidants 2021, 10, 1578. [Google Scholar] [CrossRef]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; van Soest, D.M.K.; Polderman, P.E.; Burgering, B.M.T.; Dansen, T.B. DNA damage and oxidant stress activate p53 through differential upstream signaling pathways. Free Radic. Biol. Med. 2021, 172, 298–311. [Google Scholar] [CrossRef]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell. Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kowalski, M.L.; Carson, R.P.; Bridges, L.R.; Ess, K.C. Heterozygous inactivation of tsc2 enhances tumorigenesis in p53 mutant zebrafish. Dis. Model. Mech. 2013, 6, 925–933. [Google Scholar] [CrossRef] [Green Version]
- Berghmans, S.; Murphey, R.D.; Wienholds, E.; Neuberg, D.; Kutok, J.L.; Fletcher, C.D.; Morris, J.P.; Liu, T.X.; Schulte-Merker, S.; Kanki, J.P.; et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Dovey, M.; White, R.M.; Zon, L.I. Oncogenic NRAS cooperates with p53 loss to generate melanoma in zebrafish. Zebrafish 2009, 6, 397–404. [Google Scholar] [CrossRef]
- Mudbhary, R.; Hoshida, Y.; Chernyavskaya, Y.; Jacob, V.; Villanueva, A.; Fiel, M.I.; Chen, X.; Kojima, K.; Thung, S.; Bronson, R.T.; et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014, 25, 196–209. [Google Scholar] [CrossRef] [Green Version]
- Sidi, S.; Sanda, T.; Kennedy, R.D.; Hagen, A.T.; Jette, C.A.; Hoffmans, R.; Pascual, J.; Imamura, S.; Kishi, S.; Amatruda, J.F.; et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008, 133, 864–877. [Google Scholar] [CrossRef] [Green Version]
- Imamura, S.; Kishi, S. Molecular cloning and functional characterization of zebrafish ATM. Int. J. Biochem. Cell. Biol. 2005, 37, 1105–1116. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, E.M.; Brugarolas, J.; Jacks, T.; Baltimore, D. Involvement of p53 and p21 in cellular defects and tumorigenesis in Atm-/- mice. Mol. Cell. Biol. 1998, 18, 4385–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, Y.; Hu, X.; Han, P.; Mendez-Bermudez, A.; Bauwens, S.; Eid, R.; Tan, L.; Pousse, M.; Giraud-Panis, M.J.; Lu, Y.; et al. The non-telomeric evolutionary trajectory of TRF2 in zebrafish reveals its specific roles in neurodevelopment and aging. Nucleic Acids Res. 2022, 50, 2081–2095. [Google Scholar] [CrossRef] [PubMed]
- So, J.; Kim, M.; Lee, S.H.; Ko, S.; Lee, D.A.; Park, H.; Azuma, M.; Parsons, M.J.; Prober, D.; Shin, D. Attenuating the Epidermal Growth Factor Receptor-Extracellular Signal-Regulated Kinase-Sex-Determining Region Y-Box 9 Axis Promotes Liver Progenitor Cell-Mediated Liver Regeneration in Zebrafish. Hepatology 2021, 73, 1494–1508. [Google Scholar] [CrossRef] [PubMed]
- Shamma, A.; Suzuki, M.; Hayashi, N.; Kobayashi, M.; Sasaki, N.; Nishiuchi, T.; Doki, Y.; Okamoto, T.; Kohno, S.; Muranaka, H.; et al. ATM mediates pRB function to control DNMT1 protein stability and DNA methylation. Mol. Cell. Biol. 2013, 33, 3113–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramdas Nair, A.; Delaney, P.; Koomson, A.A.; Ranjan, S.; Sadler, K.C. Systematic Evaluation of the Effects of Toxicant Exposure on Survival in Zebrafish Embryos and Larvae. Curr. Protoc. 2021, 1, e231. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Magnani, E.; Macchi, F.; Madakashira, B.P.; Zhang, C.; Alaydaroos, F.; Sadler, K.C. uhrf1 and dnmt1 Loss Induces an Immune Response in Zebrafish Livers Due to Viral Mimicry by Transposable Elements. Front. Immunol. 2021, 12, 627926. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Sadler, K.C.; Amsterdam, A.; Soroka, C.; Boyer, J.; Hopkins, N. A genetic screen in zebrafish identifies the mutants vps18, nf2 and foie gras as models of liver disease. Development 2005, 132, 3561–3572. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair. 2015, 36, 8–12. [Google Scholar] [CrossRef] [Green Version]
- Malaquin, N.; Olivier, M.A.; Martinez, A.; Nadeau, S.; Sawchyn, C.; Coppe, J.P.; Cardin, G.; Mallette, F.A.; Campisi, J.; Rodier, F. Non-canonical ATM/MRN activities temporally define the senescence secretory program. EMBO Rep. 2020, 21, e50718. [Google Scholar] [CrossRef]
- Danilova, N.; Bibikova, E.; Covey, T.M.; Nathanson, D.; Dimitrova, E.; Konto, Y.; Lindgren, A.; Glader, B.; Radu, C.G.; Sakamoto, K.M.; et al. The role of the DNA damage response in zebrafish and cellular models of Diamond Blackfan anemia. Dis. Model. Mech. 2014, 7, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Karapetian, M.; Tsikarishvili, S.; Kulikova, N.; Kurdadze, A.; Zaalishvili, G. Genotoxic effects of topoisomerase poisoning and PARP inhibition on zebrafish embryos. DNA Repair. 2020, 87, 102772. [Google Scholar] [CrossRef]
- Tsedensodnom, O.; Vacaru, A.M.; Howarth, D.L.; Yin, C.; Sadler, K.C. Ethanol metabolism and oxidative stress are required for unfolded protein response activation and steatosis in alcoholic liver disease. Dis. Model. Mech. 2013, 6, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Huff, L.A.; Yan, S.; Clemens, M.G. Mechanisms of Ataxia Telangiectasia Mutated (ATM) Control in the DNA Damage Response to Oxidative Stress, Epigenetic Regulation, and Persistent Innate Immune Suppression Following Sepsis. Antioxidants 2021, 10, 1146. [Google Scholar] [CrossRef]
- Reichenbach, J.; Schubert, R.; Schwan, C.; Müller, K.; Böhles, H.J.; Zielen, S. Anti-oxidative capacity in patients with ataxia telangiectasia. Clin. Exp. Immunol. 1999, 117, 535–539. [Google Scholar] [CrossRef]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Breau, M.; Houssaini, A.; Lipskaia, L.; Abid, S.; Born, E.; Marcos, E.; Czibik, G.; Attwe, A.; Beaulieu, D.; Palazzo, A.; et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight 2019, 4, e127647. [Google Scholar] [CrossRef]
- Rakshit, S.; Bagchi, J.; Mandal, L.; Paul, K.; Ganguly, D.; Bhattacharjee, S.; Ghosh, M.; Biswas, N.; Chaudhuri, U.; Bandyopadhyay, S. N-acetyl cysteine enhances imatinib-induced apoptosis of Bcr-Abl+ cells by endothelial nitric oxide synthase-mediated production of nitric oxide. Apoptosis 2009, 14, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, V.X.; Sze, K.M.-F.; Chan, L.-K.; Ho, D.W.-H.; Tsui, Y.-M.; Chiu, Y.-T.; Lee, E.; Husain, A.; Huang, H.; Tian, L.; et al. Antioxidant supplements promote tumor formation and growth and confer drug resistance in hepatocellular carcinoma by reducing intracellular ROS and induction of TMBIM1. Cell. Biosci. 2021, 11, 217. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Liu, X.-B.; Yu, J.-J.; Hua, F.; Hu, Z.-W. Antioxidant N-Acetylcysteine Attenuates Hepatocarcinogenesis by Inhibiting ROS/ER Stress in TLR2 Deficient Mouse. PLoS ONE 2013, 8, e74130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.K.; Kan, G.; Mao, Y.; Wu, Z.; Tan, X.; He, H.; Lee, C. UHRF1 downmodulation enhances antitumor effects of histone deacetylase inhibitors in retinoblastoma by augmenting oxidative stress-mediated apoptosis. Mol. Oncol. 2020, 14, 329–346. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Wan, X.; Luo, Y.; Liang, F.; Jiang, S.; Yuan, X.; Mo, Z. The vicious circle of UHRF1 down-regulation and KEAP1/NRF2/HO-1 pathway impairment promotes oxidative stress-induced endothelial cell apoptosis in diabetes. Diabet. Med. 2022, 40, e15026. [Google Scholar] [CrossRef]
- Mancini, M.; Magnani, E.; Macchi, F.; Bonapace, I.M. The multi-functionality of UHRF1: Epigenome maintenance and preservation of genome integrity. Nucleic Acids Res. 2021, 49, 6053–6068. [Google Scholar] [CrossRef]
- Ashraf, W.; Ibrahim, A.; Alhosin, M.; Zaayter, L.; Ouararhni, K.; Papin, C.; Ahmad, T.; Hamiche, A.; Mely, Y.; Bronner, C.; et al. The epigenetic integrator UHRF1: On the road to become a universal biomarker for cancer. Oncotarget 2017, 8, 51946–51962. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| hUHRF1 vs. WT at 5 dpf | n. Genes |
|---|---|
| Total identified genes (base mean > 0) | 21,642 |
| Significant differentially expressed genes (DEGs; padj < 0.05) | 5327 |
| Significant upregulated genes (padj < 0.05 & Log2 fold change > 1.5) | 1254 |
| Significant downregulated genes (padj < 0.05 & Log2 fold change < −1.5) | 1069 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ajouaou, Y.; Magnani, E.; Madakashira, B.; Jenkins, E.; Sadler, K.C. atm Mutation and Oxidative Stress Enhance the Pre-Cancerous Effects of UHRF1 Overexpression in Zebrafish Livers. Cancers 2023, 15, 2302. https://doi.org/10.3390/cancers15082302
Ajouaou Y, Magnani E, Madakashira B, Jenkins E, Sadler KC. atm Mutation and Oxidative Stress Enhance the Pre-Cancerous Effects of UHRF1 Overexpression in Zebrafish Livers. Cancers. 2023; 15(8):2302. https://doi.org/10.3390/cancers15082302
Chicago/Turabian StyleAjouaou, Yousra, Elena Magnani, Bhavani Madakashira, Eleanor Jenkins, and Kirsten C. Sadler. 2023. "atm Mutation and Oxidative Stress Enhance the Pre-Cancerous Effects of UHRF1 Overexpression in Zebrafish Livers" Cancers 15, no. 8: 2302. https://doi.org/10.3390/cancers15082302
APA StyleAjouaou, Y., Magnani, E., Madakashira, B., Jenkins, E., & Sadler, K. C. (2023). atm Mutation and Oxidative Stress Enhance the Pre-Cancerous Effects of UHRF1 Overexpression in Zebrafish Livers. Cancers, 15(8), 2302. https://doi.org/10.3390/cancers15082302