
The Role of Extracellular Vesicles in Melanoma Progression


Abstract
:Simple Summary
Abstract
1. Introduction
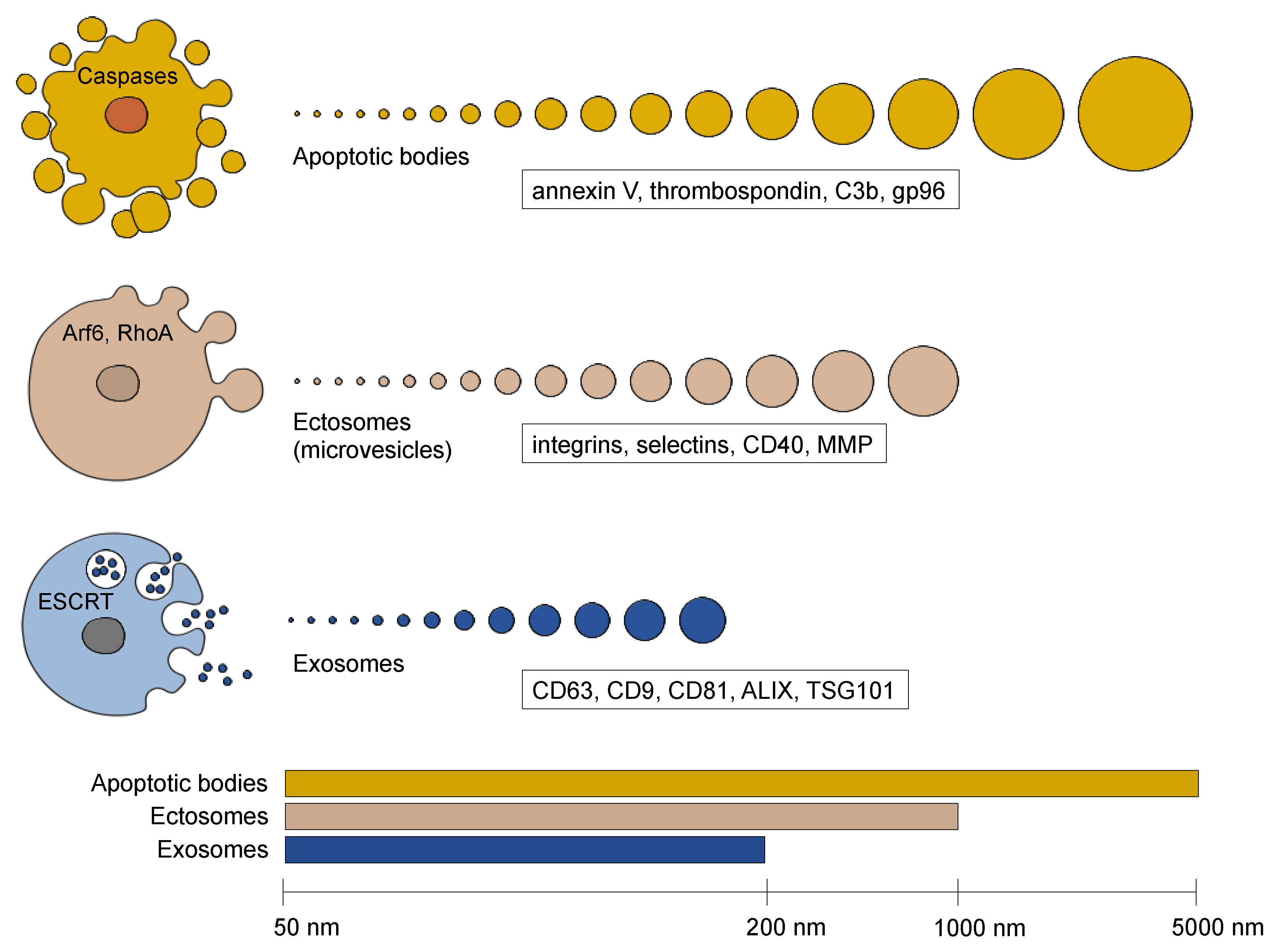
2. Extracellular Vesicle Diversity
2.1. Apoptotic Bodies
2.2. Ectosomes (Microvesicles)
2.3. Exosomes
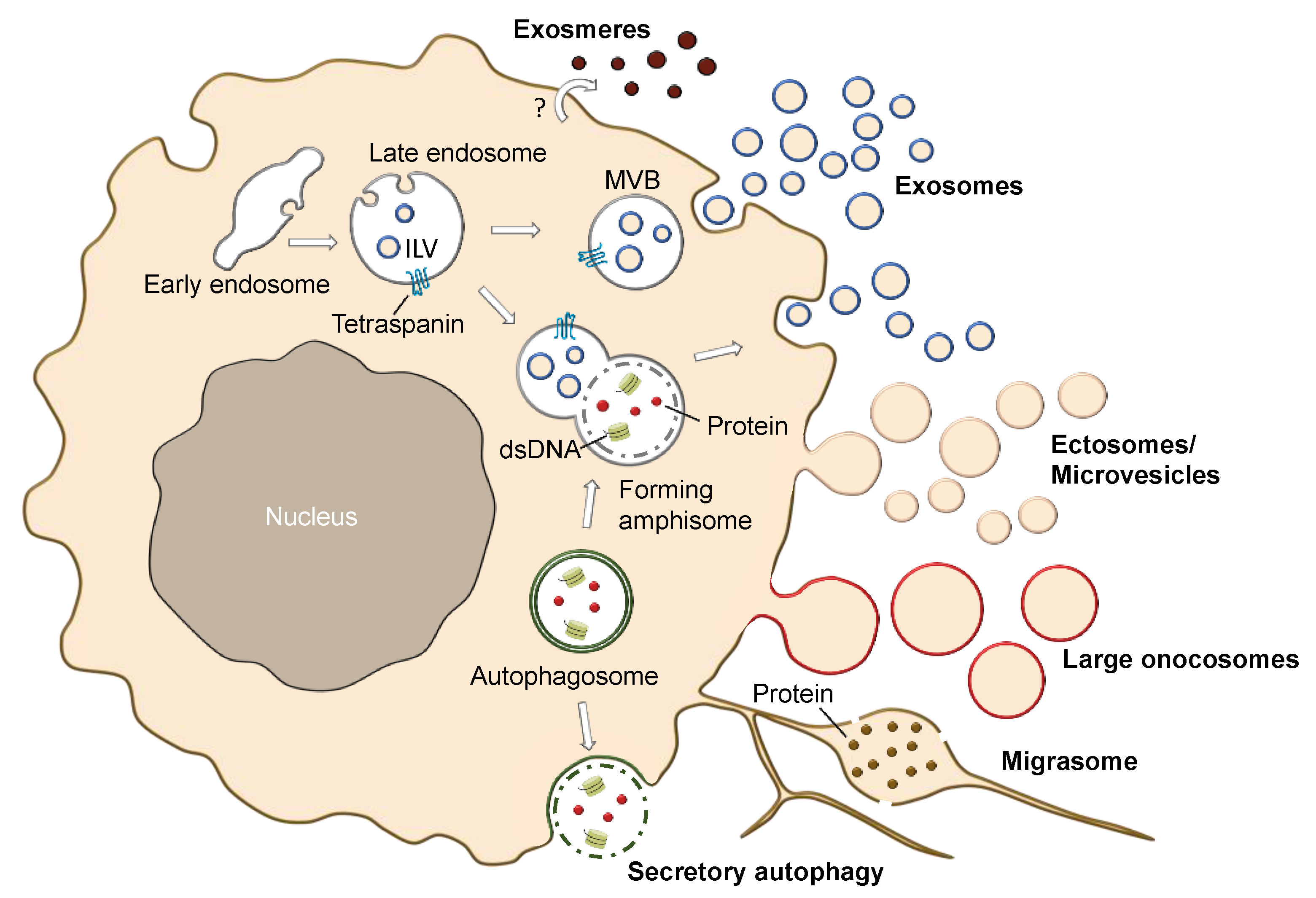
2.4. Different Subtypes of EVs and EV-Like Nanoparticles
3. Melanosomes Are EV-like Organelles in the Skin
3.1. Melanosomes
3.2. The Role of Melanosomes in Melanoma Homeostasis and Inter-Cellular Communication
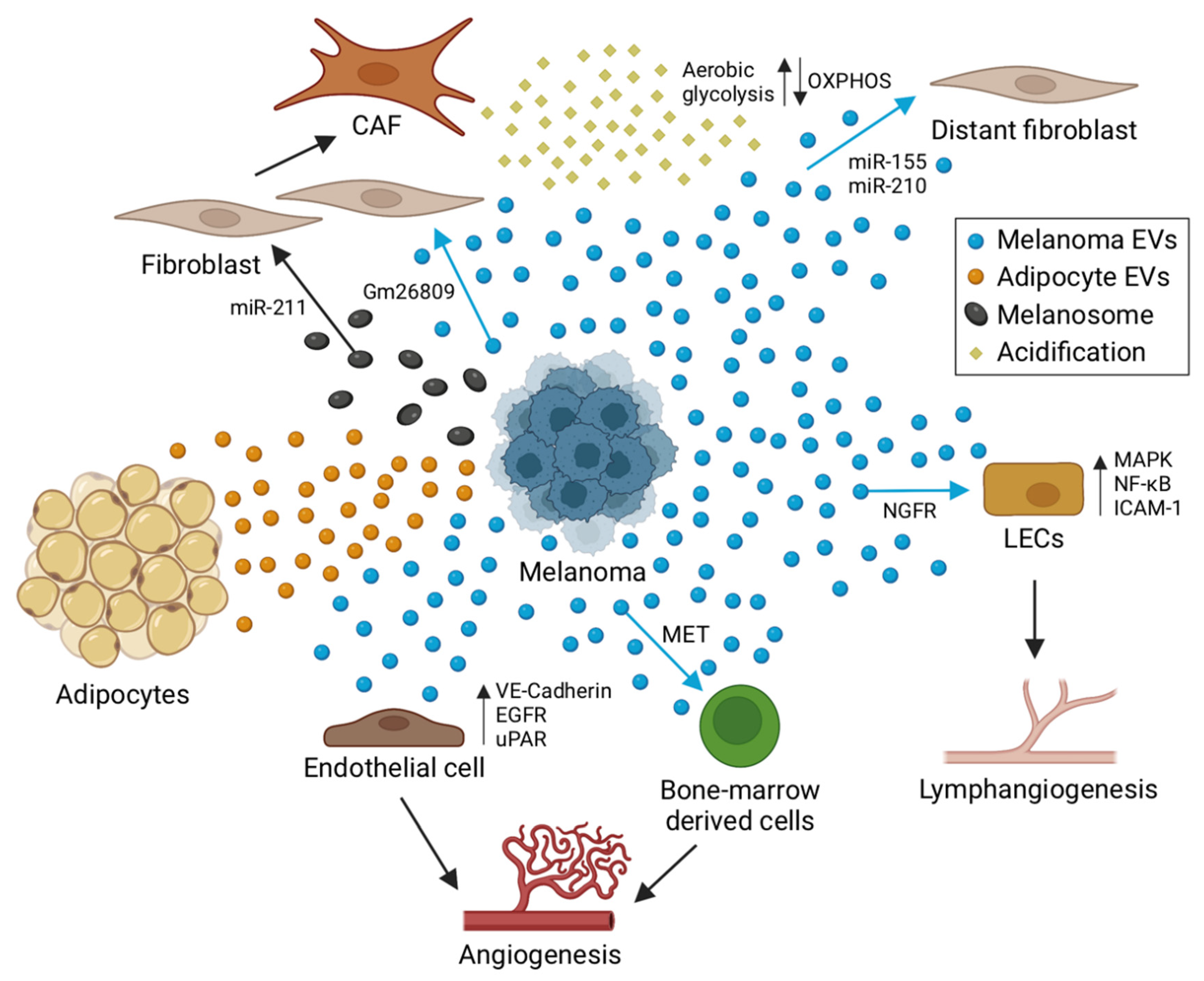
4. The Role of EVs in Melanoma Tumor Niche Formation
4.1. Angiogenesis
4.2. Lymphangiogenesis
4.3. Tumor–Stroma Interactions
5. Profiling of EV Cargo for Potential Biomarker and Drug Target Discovery
5.1. EV-Derived miRNA Profiling in Melanoma Cells
5.2. EV-Derived miRNA Profiling of Melanoma Liquid Biopsies
5.3. EV-Derived Protein Profiling in Melanoma Cells
5.4. EV-Derived Protein Profiling in Liquid Biopsies
5.5. Profiling of Less Studied EV Cargo
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Program—NCI. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 14 June 2022).
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-Genome Landscapes of Major Melanoma Subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef]
- Lovly, C.M.; Dahlman, K.B.; Fohn, L.E.; Su, Z.; Dias-Santagata, D.; Hicks, D.J.; Hucks, D.; Berry, E.; Terry, C.; Duke, M.; et al. Routine Multiplex Mutational Profiling of Melanomas Enables Enrollment in Genotype-Driven Therapeutic Trials. PLoS ONE 2012, 7, e35309. [Google Scholar] [CrossRef] [Green Version]
- Ghafouri-Fard, S.; Gholipour, M.; Taheri, M. MicroRNA Signature in Melanoma: Biomarkers and Therapeutic Targets. Front. Oncol. 2021, 11, 608987. [Google Scholar] [CrossRef]
- Ambros, V. The Functions of Animal MicroRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. Elegans Heterochronic Gene Lin-4 Encodes Small RNAs with Antisense Complementarity to Lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-Nucleotide Let-7 RNA Regulates Developmental Timing in Caenorhabditis Elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of MicroRNAs in Vivo with ‘Antagomirs’. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Lau, N.C.; Garrett-Engele, P.; Grimson, A.; Schelter, J.M.; Castle, J.; Bartel, D.P.; Linsley, P.S.; Johnson, J.M. Microarray Analysis Shows That Some MicroRNAs Downregulate Large Numbers of Target MRNAs. Nature 2005, 433, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Sood, P.; Krek, A.; Zavolan, M.; Macino, G.; Rajewsky, N. Cell-Type-Specific Signatures of MicroRNAs on Target MRNA Expression. Proc. Natl. Acad. Sci. USA 2006, 103, 2746–2751. [Google Scholar] [CrossRef] [Green Version]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of MicroRNA–Target Recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA Expression Profiles Classify Human Cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-Mediated Transfer of MRNAs and MicroRNAs Is a Novel Mechanism of Genetic Exchange between Cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L.; et al. Induction of Lymphocyte Apoptosis by Tumor Cell Secretion of FasL-Bearing Microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 Contributes to Immunosuppression and Is Associated with Anti-PD-1 Response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- García-Silva, S.; Benito-Martín, A.; Nogués, L.; Hernández-Barranco, A.; Mazariegos, M.S.; Santos, V.; Hergueta-Redondo, M.; Ximénez-Embún, P.; Kataru, R.P.; Lopez, A.A.; et al. Melanoma-Derived Small Extracellular Vesicles Induce Lymphangiogenesis and Metastasis through an NGFR-Dependent Mechanism. Nat. Cancer 2021, 2, 1387–1405. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of Extracellular Vesicles (EV): Exosomes, Microvesicles, Retrovirus-like Vesicles, and Apoptotic Bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hauser, P.; Wang, S.; Didenko, V.V. Apoptotic Bodies: Selective Detection in Extracellular Vesicles. In Signal Transduction Immunohistochemistry: Methods and Protocols; Kalyuzhny, A.E., Ed.; Springer: New York, NY, USA, 2017; pp. 193–200. ISBN 978-1-4939-6759-9. [Google Scholar]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic Bodies from Endothelial Cells Enhance the Number and Initiate the Differentiation of Human Endothelial Progenitor Cells in Vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Sirois, I.; Raymond, M.-A.; Brassard, N.; Cailhier, J.-F.; Fedjaev, M.; Hamelin, K.; Londono, I.; Bendayan, M.; Pshezhetsky, A.V.; Hébert, M.-J. Caspase-3-Dependent Export of TCTP: A Novel Pathway for Antiapoptotic Intercellular Communication. Cell Death Differ. 2011, 18, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.-L.; Holmgren, L. Horizontal Transfer of Oncogenes by Uptake of Apoptotic Bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, L.; Bergsmedh, A.; Spetz, A.-L. Horizontal Transfer of DNA by the Uptake of Apoptotic Bodies. Vox Sang. 2002, 83 (Suppl. 1), 305–306. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Wang, H.; Li, F.; Li, C.-Y. Regulation of Mammalian Horizontal Gene Transfer by Apoptotic DNA Fragmentation. Br. J. Cancer 2006, 95, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Vittorelli, M.L. Shed Membrane Vesicles and Clustering of Membrane-Bound Proteolytic Enzymes. In Current Topics in Developmental Biology; Academic Press: Cambridge, MA, USA, 2003; Volume 54, pp. 411–432. [Google Scholar]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding Microvesicles: Artefacts No More. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, C.; Clancy, J.; D’Souza-Schorey, C. Biology and Biogenesis of Shed Microvesicles. Small GTPases 2016, 8, 220–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-Regulated Shedding of Tumor-Cell Derived Plasma Membrane Microvesicles. Curr. Biol. CB 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, A.E.; Clancy, J.W.; Olivia Balmert, M.; D’Souza-Schorey, C. Extracellular Microvesicles and Invadopodia Mediate Non-Overlapping Modes of Tumor Cell Invasion. Sci. Rep. 2015, 5, 14748. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-Inducible Factors and RAB22A Mediate Formation of Microvesicles That Stimulate Breast Cancer Invasion and Metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT Pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Raiborg, C.; Stenmark, H. The ESCRT Machinery in Endosomal Sorting of Ubiquitylated Membrane Proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Hurley, J.H. ESCRTs Are Everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollert, T.; Wunder, C.; Lippincott-Schwartz, J.; Hurley, J.H. Membrane Scission by the ESCRT-III Complex. Nature 2009, 458, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Villarroya-Beltri, C.; Baixauli, F.; Gutiérrez-Vázquez, C.; Sánchez-Madrid, F.; Mittelbrunn, M. Sorting It out: Regulation of Exosome Loading. Semin. Cancer Biol. 2014, 28, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissig, C.; Gruenberg, J. ALIX and the Multivesicular Endosome: ALIX in Wonderland. Trends Cell Biol. 2014, 24, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Odorizzi, G. Get on the Exosome Bus with ALIX. Nat. Cell Biol. 2012, 14, 654–655. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, Biogenesis and Function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Lötvall, J.; Hill, A.F.; Hochberg, F.; Buzás, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal Experimental Requirements for Definition of Extracellular Vesicles and Their Functions: A Position Statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef]
- Liangsupree, T.; Multia, E.; Riekkola, M.-L. Modern Isolation and Separation Techniques for Extracellular Vesicles. J. Chromatogr. A 2021, 1636, 461773. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Wandelmer, J.; Reggiori, F. Amphisomes: Out of the Autophagosome Shadow? EMBO J. 2013, 32, 3116–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory Autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenouchi, T.; Nakai, M.; Iwamaru, Y.; Sugama, S.; Tsukimoto, M.; Fujita, M.; Wei, J.; Sekigawa, A.; Sato, M.; Kojima, S.; et al. The Activation of P2X7 Receptor Impairs Lysosomal Functions and Stimulates the Release of Autophagolysosomes in Microglial Cells. J. Immunol. 2009, 182, 2051–2062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos, S.O.; Leal Santos, N.; Chammas, R.; de Sousa Andrade, L.N. Secretory Autophagy Forges a Therapy Resistant Microenvironment in Melanoma. Cancers 2022, 14, 234. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of Distinct Nanoparticles and Subsets of Extracellular Vesicles by Asymmetric Flow Field-Flow Fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef] [Green Version]
- Di Vizio, D.; Kim, J.; Hager, M.H.; Morello, M.; Yang, W.; Lafargue, C.J.; True, L.; Rubin, M.A.; Adam, R.M.; Beroukhim, R.; et al. Oncosome Formation in Prostate Cancer: Association with a Region of Frequent Chromosomal Deletion in Metastatic Disease. Cancer Res. 2009, 69, 5601–5609. [Google Scholar] [CrossRef] [Green Version]
- Minciacchi, V.R.; You, S.; Spinelli, C.; Morley, S.; Zandian, M.; Aspuria, P.-J.; Cavallini, L.; Ciardiello, C.; Sobreiro, M.R.; Morello, M.; et al. Large Oncosomes Contain Distinct Protein Cargo and Represent a Separate Functional Class of Tumor-Derived Extracellular Vesicles. Oncotarget 2015, 6, 11327–11341. [Google Scholar] [CrossRef] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular Transfer of the Oncogenic Receptor EGFRvIII by Microvesicles Derived from Tumour Cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the Migrasome, an Organelle Mediating Release of Cytoplasmic Contents during Cell Migration. Cell Res. 2015, 25, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Lei, Y.; Zheng, J.; Peng, J.; Li, Y.; Yu, L.; Chen, Y. Identification of Markers for Migrasome Detection. Cell Discov. 2019, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardman-Frank, J.M.; Fisher, D.E. Skin Pigmentation and Its Control: From Ultraviolet Radiation to Stem Cells. Exp. Dermatol. 2021, 30, 560–571. [Google Scholar] [CrossRef] [PubMed]
- Theos, A.C.; Truschel, S.T.; Raposo, G.; Marks, M.S. The Silver Locus Product Pmel17/Gp100/Silv/ME20: Controversial in Name and in Function. Pigment Cell Res. 2005, 18, 322–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delevoye, C.; Marks, M.S.; Raposo, G. Lysosome-Related Organelles as Functional Adaptations of the Endolysosomal System. Curr. Opin. Cell Biol. 2019, 59, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Ohbayashi, N.; Fukuda, M. Recent Advances in Understanding the Molecular Basis of Melanogenesis in Melanocytes. F1000Research 2020, 9, F1000 Faculty Rev-608. [Google Scholar] [CrossRef]
- Fukuda, M. Rab GTPases: Key Players in Melanosome Biogenesis, Transport, and Transfer. Pigment Cell Melanoma Res. 2021, 34, 222–235. [Google Scholar] [CrossRef]
- Van Gele, M.; Dynoodt, P.; Lambert, J. Griscelli Syndrome: A Model System to Study Vesicular Trafficking. Pigment Cell Melanoma Res. 2009, 22, 268–282. [Google Scholar] [CrossRef]
- Moreiras, H.; Seabra, M.C.; Barral, D.C. Melanin Transfer in the Epidermis: The Pursuit of Skin Pigmentation Control Mechanisms. Int. J. Mol. Sci. 2021, 22, 4466. [Google Scholar] [CrossRef]
- Basrur, V.; Yang, F.; Kushimoto, T.; Higashimoto, Y.; Yasumoto, K.; Valencia, J.; Muller, J.; Vieira, W.D.; Watabe, H.; Shabanowitz, J.; et al. Proteomic Analysis of Early Melanosomes: Identification of Novel Melanosomal Proteins. J. Proteome Res. 2003, 2, 69–79. [Google Scholar] [CrossRef]
- Boissy, R.E. Melanosome Transfer to and Translocation in the Keratinocyte. Exp. Dermatol. 2003, 12, 5–12. [Google Scholar] [CrossRef]
- Lazova, R.; Pawelek, J.M. Why Do Melanomas Get so Dark? Exp. Dermatol. 2009, 18, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.G.; Valencia, J.C.; Lai, B.; Zhang, G.; Paterson, J.K.; Rouzaud, F.; Berens, W.; Wincovitch, S.M.; Garfield, S.H.; Leapman, R.D.; et al. Melanosomal Sequestration of Cytotoxic Drugs Contributes to the Intractability of Malignant Melanomas. Proc. Natl. Acad. Sci. USA 2006, 103, 9903–9907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, S.; Sander, L.; Schwartz, H.; Sheinboim, D.; Barzilai, A.; Dishon, Y.; Apcher, S.; Golan, T.; Greenberger, S.; Barshack, I.; et al. Melanoma MiRNA Trafficking Controls Tumour Primary Niche Formation. Nat. Cell Biol. 2016, 18, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- García-Silva, S.; Peinado, H. Melanosomes Foster a Tumour Niche by Activating CAFs. Nat. Cell Biol. 2016, 18, 911–913. [Google Scholar] [CrossRef] [PubMed]
- Lunavat, T.R.; Cheng, L.; Einarsdottir, B.O.; Bagge, R.O.; Muralidharan, S.V.; Sharples, R.A.; Lässer, C.; Gho, Y.S.; Hill, A.F.; Nilsson, J.A.; et al. BRAFV600 Inhibition Alters the MicroRNA Cargo in the Vesicular Secretome of Malignant Melanoma Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E5930–E5939. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-Positive Haematopoietic Bone Marrow Progenitors Initiate the Pre-Metastatic Niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massagué, J. Mediators of Vascular Remodelling Co-Opted for Sequential Steps in Lung Metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal Fibroblasts Present in Invasive Human Breast Carcinomas Promote Tumor Growth and Angiogenesis through Elevated SDF-1/CXCL12 Secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Aguado, B.A.; Caffe, J.R.; Nanavati, D.; Rao, S.S.; Bushnell, G.G.; Azarin, S.M.; Shea, L.D. Extracellular Matrix Mediators of Metastatic Cell Colonization Characterized Using Scaffold Mimics of the Pre-Metastatic Niche. Acta Biomater. 2016, 33, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-Associated Adipocytes Exhibit an Activated Phenotype and Contribute to Breast Cancer Invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-Metastatic Niches: Organ-Specific Homes for Metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Pan, J.; Barsky, L.; Jacob, J.C.; Zheng, Y.; Gao, C.; Wang, S.; Zhu, W.; Sun, H.; Lu, L.; et al. Characteristics of Pre-Metastatic Niche: The Landscape of Molecular and Cellular Pathways. Mol. Biomed. 2021, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, D.; Cerezo-Wallis, D.; Riveiro-Falkenbach, E.; Pennacchi, P.C.; Contreras-Alcalde, M.; Ibarz, N.; Cifdaloz, M.; Catena, X.; Calvo, T.G.; Cañón, E.; et al. Whole-Body Imaging of Lymphovascular Niches Identifies Pre-Metastatic Roles of Midkine. Nature 2017, 546, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Filippou, P.S.; Karagiannis, G.S.; Constantinidou, A. Midkine (MDK) Growth Factor: A Key Player in Cancer Progression and a Promising Therapeutic Target. Oncogene 2020, 39, 2040–2054. [Google Scholar] [CrossRef]
- Dong, Q.; Liu, X.; Cheng, K.; Sheng, J.; Kong, J.; Liu, T. Pre-Metastatic Niche Formation in Different Organs Induced by Tumor Extracellular Vesicles. Front. Cell Dev. Biol. 2021, 9, 733627. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic Cancer Exosomes Initiate Pre-Metastatic Niche Formation in the Liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma Exosomes Educate Bone Marrow Progenitor Cells toward a Pro-Metastatic Phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Afshari, A.; Sengupta, R.; Sebastiano, V.; Gupta, A.; Kim, Y.H. Replication Study: Melanoma Exosomes Educate Bone Marrow Progenitor Cells toward a pro-Metastatic Phenotype through MET. Elife 2018, 7, e39944. [Google Scholar] [CrossRef]
- Biagioni, A.; Laurenzana, A.; Menicacci, B.; Peppicelli, S.; Andreucci, E.; Bianchini, F.; Guasti, D.; Paoli, P.; Serratì, S.; Mocali, A.; et al. UPAR-Expressing Melanoma Exosomes Promote Angiogenesis by VE-Cadherin, EGFR and UPAR Overexpression and Rise of ERK1,2 Signaling in Endothelial Cells. Cell. Mol. Life Sci. 2021, 78, 3057–3072. [Google Scholar] [CrossRef]
- Rinderknecht, M.; Detmar, M. Tumor Lymphangiogenesis and Melanoma Metastasis. J. Cell. Physiol. 2008, 216, 347–354. [Google Scholar] [CrossRef]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes Released by Melanoma Cells Prepare Sentinel Lymph Nodes for Tumor Metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, N.; Walser, S.; He, Y.; Cousin, N.; Pereira, P.; Gallo, A.; Collado-Diaz, V.; Halin, C.; Garcia-Silva, S.; Peinado, H.; et al. Melanoma-derived Extracellular Vesicles Mediate Lymphatic Remodelling and Impair Tumour Immunity in Draining Lymph Nodes. J. Extracell. Vesicles 2022, 11, e12197. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Hu, J. Melanoma-Derived Exosomes Induce Reprogramming Fibroblasts into Cancer-Associated Fibroblasts via Gm26809 Delivery. Cell Cycle 2019, 18, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Strnadová, K.; Pfeiferová, L.; Přikryl, P.; Dvořánková, B.; Vlčák, E.; Frýdlová, J.; Vokurka, M.; Novotný, J.; Šáchová, J.; Hradilová, M.; et al. Exosomes Produced by Melanoma Cells Significantly Influence the Biological Properties of Normal and Cancer-Associated Fibroblasts. Histochem. Cell Biol. 2022, 157, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, S.L.; Yang, Y.; Allen, C.L.; Maguire, O.; Minderman, H.; Sen, A.; Ciesielski, M.J.; Collins, K.A.; Bush, P.J.; Singh, P.; et al. Metabolic Reprogramming of Stromal Fibroblasts by Melanoma Exosome MicroRNA Favours a Pre-Metastatic Microenvironment. Sci. Rep. 2018, 8, 12905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Yan, T.; Huang, C.; Xu, Z.; Wang, L.; Jiang, E.; Wang, H.; Chen, Y.; Liu, K.; Shao, Z.; et al. Melanoma Cell-Secreted Exosomal MiR-155-5p Induce Proangiogenic Switch of Cancer-Associated Fibroblasts via SOCS1/JAK2/STAT3 Signaling Pathway. J. Exp. Clin. Cancer Res. 2018, 37, 242. [Google Scholar] [CrossRef] [Green Version]
- Lazar, I.; Clement, E.; Dauvillier, S.; Milhas, D.; Ducoux-Petit, M.; LeGonidec, S.; Moro, C.; Soldan, V.; Dalle, S.; Balor, S.; et al. Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res. 2016, 76, 4051–4057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, E.; Lazar, I.; Attané, C.; Carrié, L.; Dauvillier, S.; Ducoux-Petit, M.; Esteve, D.; Menneteau, T.; Moutahir, M.; Le Gonidec, S.; et al. Adipocyte Extracellular Vesicles Carry Enzymes and Fatty Acids That Stimulate Mitochondrial Metabolism and Remodeling in Tumor Cells. EMBO J. 2020, 39, e102525. [Google Scholar] [CrossRef]
- Xiao, D.; Ohlendorf, J.; Chen, Y.; Taylor, D.D.; Rai, S.N.; Waigel, S.; Zacharias, W.; Hao, H.; McMasters, K.M. Identifying MRNA, MicroRNA and Protein Profiles of Melanoma Exosomes. PLoS ONE 2012, 7, e46874. [Google Scholar] [CrossRef] [Green Version]
- Asangani, I.A.; Harms, P.W.; Dodson, L.; Pandhi, M.; Kunju, L.P.; Maher, C.A.; Fullen, D.R.; Johnson, T.M.; Giordano, T.J.; Palanisamy, N.; et al. Genetic and Epigenetic Loss of MicroRNA-31 Leads to Feed-Forward Expression of EZH2 in Melanoma. Oncotarget 2012, 3, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, E.; Hershkovitz, L.; Itzhaki, O.; Hajdu, S.; Nemlich, Y.; Ortenberg, R.; Gefen, N.; Edry, L.; Modai, S.; Keisari, Y.; et al. Regulation of Cancer Aggressive Features in Melanoma Cells by MicroRNAs. PLoS ONE 2011, 6, e18936. [Google Scholar] [CrossRef] [PubMed]
- Migliore, C.; Petrelli, A.; Ghiso, E.; Corso, S.; Capparuccia, L.; Eramo, A.; Comoglio, P.M.; Giordano, S. MicroRNAs Impair MET-Mediated Invasive Growth. Cancer Res. 2008, 68, 10128–10136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajos-Michniewicz, A.; Czyz, M. Role of MiRNAs in Melanoma Metastasis. Cancers 2019, 11, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajos-Michniewicz, A.; Duechler, M.; Czyz, M. MiRNA in Melanoma-Derived Exosomes. Cancer Lett. 2014, 347, 29–37. [Google Scholar] [CrossRef]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. MiR221/222 in Cancer: Their Role in Tumor Progression and Response to Therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef]
- Felicetti, F.; Errico, M.C.; Bottero, L.; Segnalini, P.; Stoppacciaro, A.; Biffoni, M.; Felli, N.; Mattia, G.; Petrini, M.; Colombo, M.P.; et al. The Promyelocytic Leukemia Zinc Finger–MicroRNA-221/-222 Pathway Controls Melanoma Progression through Multiple Oncogenic Mechanisms. Cancer Res. 2008, 68, 2745–2754. [Google Scholar] [CrossRef] [Green Version]
- Igoucheva, O.; Alexeev, V. MicroRNA-Dependent Regulation of CKit in Cutaneous Melanoma. Biochem. Biophys. Res. Commun. 2009, 379, 790–794. [Google Scholar] [CrossRef]
- Felicetti, F.; De Feo, A.; Coscia, C.; Puglisi, R.; Pedini, F.; Pasquini, L.; Bellenghi, M.; Errico, M.C.; Pagani, E.; Carè, A. Exosome-Mediated Transfer of MiR-222 Is Sufficient to Increase Tumor Malignancy in Melanoma. J. Transl. Med. 2016, 14, 56. [Google Scholar] [CrossRef] [Green Version]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Pope, R.M.; Lorico, A. Biochemical and Biological Characterization of Exosomes Containing Prominin-1/CD133. Mol. Cancer 2013, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Bland, C.L.; Byrne-Hoffman, C.N.; Fernandez, A.; Rellick, S.L.; Deng, W.; Klinke II, D.J. Exosomes Derived from B16F0 Melanoma Cells Alter the Transcriptome of Cytotoxic T Cells That Impacts Mitochondrial Respiration. FEBS J. 2018, 285, 1033–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wozniak, M.; Peczek, L.; Czernek, L.; Düchler, M. Analysis of the MiRNA Profiles of Melanoma Exosomes Derived Under Normoxic and Hypoxic Culture Conditions. Anticancer Res. 2017, 37, 6779–6789. [Google Scholar] [PubMed] [Green Version]
- Lunavat, T.R.; Cheng, L.; Kim, D.-K.; Bhadury, J.; Jang, S.C.; Lässer, C.; Sharples, R.A.; López, M.D.; Nilsson, J.; Gho, Y.S.; et al. Small RNA Deep Sequencing Discriminates Subsets of Extracellular Vesicles Released by Melanoma Cells--Evidence of Unique MicroRNA Cargos. RNA Biol. 2015, 12, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Gyukity-Sebestyén, E.; Harmati, M.; Dobra, G.; Németh, I.B.; Mihály, J.; Zvara, Á.; Hunyadi-Gulyás, É.; Katona, R.; Nagy, I.; Horváth, P.; et al. Melanoma-Derived Exosomes Induce PD-1 Overexpression and Tumor Progression via Mesenchymal Stem Cell Oncogenic Reprogramming. Front. Immunol. 2019, 10, 2459. [Google Scholar] [CrossRef] [PubMed]
- Gerloff, D.; Lützkendorf, J.; Moritz, R.K.C.; Wersig, T.; Mäder, K.; Müller, L.P.; Sunderkötter, C. Melanoma-Derived Exosomal MiR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers 2020, 12, 464. [Google Scholar] [CrossRef] [Green Version]
- Alegre, E.; Sanmamed, M.F.; Rodriguez, C.; Carranza, O.; Martín-Algarra, S.; González, A. Study of Circulating MicroRNA-125b Levels in Serum Exosomes in Advanced Melanoma. Arch. Pathol. Lab. Med. 2014, 138, 828–832. [Google Scholar] [CrossRef]
- Pfeffer, S.R.; Grossmann, K.F.; Cassidy, P.B.; Yang, C.H.; Fan, M.; Kopelovich, L.; Leachman, S.A.; Pfeffer, L.M. Detection of Exosomal MiRNAs in the Plasma of Melanoma Patients. J. Clin. Med. 2015, 4, 2012–2027. [Google Scholar] [CrossRef]
- Xiao, D.; Barry, S.; Kmetz, D.; Egger, M.; Pan, J.; Rai, S.N.; Qu, J.; McMasters, K.M.; Hao, H. Melanoma Cell-Derived Exosomes Promote Epithelial-Mesenchymal Transition in Primary Melanocytes through Paracrine/Autocrine Signaling in the Tumor Microenvironment. Cancer Lett. 2016, 376, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, X.; Wang, L.; Li, M.; Shen, M.; Zhou, Z.; Zhu, S.; Li, K.; Fang, Z.; Yan, B.; et al. The Plasma Exosomal MiR-1180-3p Serves as a Novel Potential Diagnostic Marker for Cutaneous Melanoma. Cancer Cell Int. 2021, 21, 487. [Google Scholar] [CrossRef]
- Li, J.; Chen, J.; Wang, S.; Li, P.; Zheng, C.; Zhou, X.; Tao, Y.; Chen, X.; Sun, L.; Wang, A.; et al. Blockage of Transferred Exosome-Shuttled MiR-494 Inhibits Melanoma Growth and Metastasis. J. Cell. Physiol. 2019, 234, 15763–15774. [Google Scholar] [CrossRef]
- Tengda, L.; Shuping, L.; Mingli, G.; Jie, G.; Yun, L.; Weiwei, Z.; Anmei, D. Serum Exosomal MicroRNAs as Potent Circulating Biomarkers for Melanoma. Melanoma Res. 2018, 28, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, M.; Barbagallo, C.; Statello, L.; Caltabiano, R.; Russo, A.; Puzzo, L.; Avitabile, T.; Longo, A.; Toro, M.D.; Barbagallo, D.; et al. MiRNA Profiling in Vitreous Humor, Vitreal Exosomes and Serum from Uveal Melanoma Patients: Pathological and Diagnostic Implications. Cancer Biol. Ther. 2015, 16, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Fodstad, O.; Lorico, A. The Stem Cell-Associated Antigen CD133 (Prominin-1) Is a Molecular Therapeutic Target for Metastatic Melanoma. Stem Cells Dayt. Ohio 2008, 26, 3008–3017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehler, D.; Høyland-Kroghsbo, N.M.; Lund, A.H. The MiR-10 MicroRNA Precursor Family. RNA Biol. 2011, 8, 728–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, A.H. MiR-10 in Development and Cancer. Cell Death Differ. 2010, 17, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Peter, M.E. Let-7 and MiR-200 MicroRNAs. Cell Cycle Georget. Tex 2009, 8, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Mears, R.; Craven, R.A.; Hanrahan, S.; Totty, N.; Upton, C.; Young, S.L.; Patel, P.; Selby, P.J.; Banks, R.E. Proteomic Analysis of Melanoma-Derived Exosomes by Two-Dimensional Polyacrylamide Gel Electrophoresis and Mass Spectrometry. Proteomics 2004, 4, 4019–4031. [Google Scholar] [CrossRef]
- Lokman, N.A.; Ween, M.P.; Oehler, M.K.; Ricciardelli, C. The Role of Annexin A2 in Tumorigenesis and Cancer Progression. Cancer Microenviron. 2011, 4, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Prakash, M.; Kale, S.; Ghosh, I.; Kundu, G.C.; Datta, K. Hyaluronan-Binding Protein 1 (HABP1/P32/GC1qR) Induces Melanoma Cell Migration and Tumor Growth by NF-Kappa B Dependent MMP-2 Activation through Integrin Avβ3 Interaction. Cell. Signal. 2011, 23, 1563–1577. [Google Scholar] [CrossRef]
- Rondepierre, F.; Bouchon, B.; Bonnet, M.; Moins, N.; Chezal, J.M.; D’Incan, M.; Degoul, F. B16 Melanoma Secretomes and in Vitro Invasiveness: Syntenin as an Invasion Modulator. Melanoma Res. 2010, 20, 77–84. [Google Scholar] [CrossRef]
- Yi, M.; Schnitzer, J.E. Impaired Tumor Growth, Metastasis, Angiogenesis and Wound Healing in Annexin A1-Null Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 17886–17891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Xie, C. The Role of OXCT1 in the Pathogenesis of Cancer as a Rate-Limiting Enzyme of Ketone Body Metabolism. Life Sci. 2017, 183, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Nakazaki, Y.; Carrasco, D.R.; Draganov, D.; Souders, N.; Johnson, M.; Mihm, M.C.; Dranoff, G. Milk Fat Globule EGF-8 Promotes Melanoma Progression through Coordinated Akt and Twist Signaling in the Tumor Microenvironment. Cancer Res. 2008, 68, 8889–8898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, I.; Clement, E.; Ducoux-Petit, M.; Denat, L.; Soldan, V.; Dauvillier, S.; Balor, S.; Burlet-Schiltz, O.; Larue, L.; Muller, C.; et al. Proteome Characterization of Melanoma Exosomes Reveals a Specific Signature for Metastatic Cell Lines. Pigment Cell Melanoma Res. 2015, 28, 464–475. [Google Scholar] [CrossRef]
- Guerreiro, E.M.; Øvstebø, R.; Thiede, B.; Costea, D.E.; Søland, T.M.; Galtung, H.K. Cancer Cell Line-Specific Protein Profiles in Extracellular Vesicles Identified by Proteomics. PLoS ONE 2020, 15, e0238591. [Google Scholar] [CrossRef]
- Boussadia, Z.; Lamberti, J.; Mattei, F.; Pizzi, E.; Puglisi, R.; Zanetti, C.; Pasquini, L.; Fratini, F.; Fantozzi, L.; Felicetti, F.; et al. Acidic Microenvironment Plays a Key Role in Human Melanoma Progression through a Sustained Exosome Mediated Transfer of Clinically Relevant Metastatic Molecules. J. Exp. Clin. Cancer Res. 2018, 37, 245. [Google Scholar] [CrossRef]
- Surman, M.; Stȩpień, E.; Przybyło, M. Melanoma-Derived Extracellular Vesicles: Focus on Their Proteome. Proteomes 2019, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Alegre, E.; Zubiri, L.; Perez-Gracia, J.L.; González-Cao, M.; Soria, L.; Martín-Algarra, S.; González, A. Circulating Melanoma Exosomes as Diagnostic and Prognosis Biomarkers. Clin. Chim. Acta 2016, 454, 28–32. [Google Scholar] [CrossRef]
- Logozzi, M.; De Milito, A.; Lugini, L.; Borghi, M.; Calabrò, L.; Spada, M.; Perdicchio, M.; Marino, M.L.; Federici, C.; Iessi, E.; et al. High Levels of Exosomes Expressing CD63 and Caveolin-1 in Plasma of Melanoma Patients. PLoS ONE 2009, 4, e5219. [Google Scholar] [CrossRef] [Green Version]
- Pietrowska, M.; Zebrowska, A.; Gawin, M.; Marczak, L.; Sharma, P.; Mondal, S.; Mika, J.; Polańska, J.; Ferrone, S.; Kirkwood, J.M.; et al. Proteomic Profile of Melanoma Cell-Derived Small Extracellular Vesicles in Patients’ Plasma: A Potential Correlate of Melanoma Progression. J. Extracell. Vesicles 2021, 10, e12063. [Google Scholar] [CrossRef]
- Campoli, M.R.; Chang, C.-C.; Kageshita, T.; Wang, X.; McCarthy, J.B.; Ferrone, S. Human High Molecular Weight-Melanoma-Associated Antigen (HMW-MAA): A Melanoma Cell Surface Chondroitin Sulfate Proteoglycan (MSCP) with Biological and Clinical Significance. Crit. Rev. Immunol. 2004, 24. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sabbatino, F.; Wang, X.; Ferrone, S. Detection of Chondroitin Sulfate Proteoglycan 4 (CSPG4) in Melanoma. In Molecular Diagnostics for Melanoma: Methods and Protocols; Methods in Molecular, Biology; Thurin, M., Marincola, F.M., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 523–535. ISBN 978-1-62703-727-3. [Google Scholar]
- Sharma, P.; Ludwig, S.; Muller, L.; Hong, C.S.; Kirkwood, J.M.; Ferrone, S.; Whiteside, T.L. Immunoaffinity-Based Isolation of Melanoma Cell-Derived Exosomes from Plasma of Patients with Melanoma. J. Extracell. Vesicles 2018, 7, 1435138. [Google Scholar] [CrossRef] [PubMed]
- García-Silva, S.; Benito-Martín, A.; Sánchez-Redondo, S.; Hernández-Barranco, A.; Ximénez-Embún, P.; Nogués, L.; Mazariegos, M.S.; Brinkmann, K.; López, A.A.; Meyer, L.; et al. Use of Extracellular Vesicles from Lymphatic Drainage as Surrogate Markers of Melanoma Progression and BRAFV600E Mutation. J. Exp. Med. 2019, 216, 1061–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, A.; Kasumova, G.G.; Michaud, W.A.; Cintolo-Gonzalez, J.; Díaz-Martínez, M.; Ohmura, J.; Mehta, A.; Chien, I.; Frederick, D.T.; Cohen, S.; et al. Plasma-Derived Extracellular Vesicle Analysis and Deconvolution Enable Prediction and Tracking of Melanoma Checkpoint Blockade Outcome. Sci. Adv. 2020, 6, eabb3461. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Vila, M.; Yoshioka, Y.; Ochiya, T. Biological Functions Driven by MRNAs Carried by Extracellular Vesicles in Cancer. Front. Cell Dev. Biol. 2021, 9, 620498. [Google Scholar] [CrossRef]
- Born, L.J.; Harmon, J.W.; Jay, S.M. Therapeutic Potential of Extracellular Vesicle-Associated Long Noncoding RNA. Bioeng. Transl. Med. 2020, 5, e10172. [Google Scholar] [CrossRef]
- Zocco, D.; Bernardi, S.; Novelli, M.; Astrua, C.; Fava, P.; Zarovni, N.; Carpi, F.M.; Bianciardi, L.; Malavenda, O.; Quaglino, P.; et al. Isolation of Extracellular Vesicles Improves the Detection of Mutant DNA from Plasma of Metastatic Melanoma Patients. Sci. Rep. 2020, 10, 15745. [Google Scholar] [CrossRef]
- Lobasso, S.; Tanzarella, P.; Mannavola, F.; Tucci, M.; Silvestris, F.; Felici, C.; Ingrosso, C.; Corcelli, A.; Lopalco, P. A Lipidomic Approach to Identify Potential Biomarkers in Exosomes From Melanoma Cells With Different Metastatic Potential. Front. Physiol. 2021, 12, 748895. [Google Scholar] [CrossRef]
- Palacios-Ferrer, J.L.; García-Ortega, M.B.; Gallardo-Gómez, M.; García, M.Á.; Díaz, C.; Boulaiz, H.; Valdivia, J.; Jurado, J.M.; Almazan-Fernandez, F.M.; Arias-Santiago, S.; et al. Metabolomic Profile of Cancer Stem Cell-Derived Exosomes from Patients with Malignant Melanoma. Mol. Oncol. 2021, 15, 407–428. [Google Scholar] [CrossRef]
- Marar, C.; Starich, B.; Wirtz, D. Extracellular Vesicles in Immunomodulation and Tumor Progression. Nat. Immunol. 2021, 22, 560–570. [Google Scholar] [CrossRef]
- Vergani, E.; Daveri, E.; Vallacchi, V.; Bergamaschi, L.; Lalli, L.; Castelli, C.; Rodolfo, M.; Rivoltini, L.; Huber, V. Extracellular Vesicles in Anti-Tumor Immunity. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNAs | sEV Source | sEV Marker | sEV Isolation | RNA Isolation Kit/Quantitation Method | Study |
|---|---|---|---|---|---|
| miR-31, miR-185, miR-34b | HEMa-LP, NHEM-c, A375, SK-MEL-28 | CD81+, HSC70+ Calnexin-, cytochrome c- | UF 1 + UC 2 | mirVana/Microarray | [103] |
| miR-222 | PD cell lines | HSP90 TSG101 LAMP2 CD63 RAB5B | UC or EQ 3 | NorgenBioteK/qRT-PCR 4 | [112] |
| miR-216b, let-7i, miR-10a | FEMX-I | Alix | ImaSep 5 + UC | Qiazol/Microarray | [113] |
| let-7c, let-7b, let-7d, let-7a | B16F0 | Hsp70+,CD63+ CD9+ CD81- | UC | RNAeasy, Trizol/Microarray | [114] |
| miR-494-5p, miR-4497, miR-513a-5p (high in hypoxic sEVs) vs. miR-125b-5p, miR-21-5p, miR-3934-5p (high in normoxic sEVs) | DMBC9, -10, -11, -12 | CD9+, CD63+ | UC | miRCURY/Microarray | [115] |
| miR-214-3p, miR-199a-3p, miR-155-5p | A375, MML-1, SK-MEL-28 | Exo 6: FLOT1+, TSG101+ MV7: FLOT1+, TSG101 APB 8: FLOT1+, BCL2+, Calnexin+, TSG101 | UC | miRCURY/Ion Torrent | [116] |
| miR-211 | MML-1, A375 | Exo: CD81+, TSG-101+, CD81- MV: absence of TSG-101- APB: Calnexin+ | UC | miRCURY/Ion Torrent | [78] |
| 168 miRNAs | B16F1 | CD9+, CD63+ CD81+, HSP70+ | UC | Zymo Research/SOLiD 5500 | [117] |
| miR-100-5p, miR-99b-5p, miR-221-3p, miR-24-3p, miR-125b-5p, | WM9, WM35, WM902B, NHEM | CD63+, CD81+ Calnexin- | UC | TriFast™/Illumina | [118] |
| miR-125b, miR-16 | Plasma | - | EQ | TRIzol/ qRT PCR | [119] |
| miR-17, miR-19a, miR-21, miR-126, miR-149 | Plasma | - | EQ | Qiazol + Qiagen miRNeasy/ NanoString | [120] |
| miR-191 and let-7a | HEMa-LP, NHEM, A375 SK-MEL-28 Serum | - | UC+EQ | mirVana, SeraMir, qRT-PCR | [121] |
| miR-1180-3p | ME4405, A375, SK-MEL-5, SK-MEL-28, plasma | - | UC | - | [122] |
| miR-494 | A375, serum | - | UC | TRIzol/qRT-PCR | [123] |
| miRNA-532-5p, miRNA-106b | serum | CD63+ | UC | Sangon Biotech/qRT-PCR | [124] |
| miR-146a 11 | VH 9, serum, FFPE 10 | CD9+/−, CD63++, CD81+ | UC | Qiagen miRNeasy/Microarray | [125] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lattmann, E.; Levesque, M.P. The Role of Extracellular Vesicles in Melanoma Progression. Cancers 2022, 14, 3086. https://doi.org/10.3390/cancers14133086
Lattmann E, Levesque MP. The Role of Extracellular Vesicles in Melanoma Progression. Cancers. 2022; 14(13):3086. https://doi.org/10.3390/cancers14133086
Chicago/Turabian StyleLattmann, Evelyn, and Mitchell P. Levesque. 2022. "The Role of Extracellular Vesicles in Melanoma Progression" Cancers 14, no. 13: 3086. https://doi.org/10.3390/cancers14133086
APA StyleLattmann, E., & Levesque, M. P. (2022). The Role of Extracellular Vesicles in Melanoma Progression. Cancers, 14(13), 3086. https://doi.org/10.3390/cancers14133086