
Heterogeneity in Melanoma


Abstract
:Simple Summary
Abstract
1. Introduction
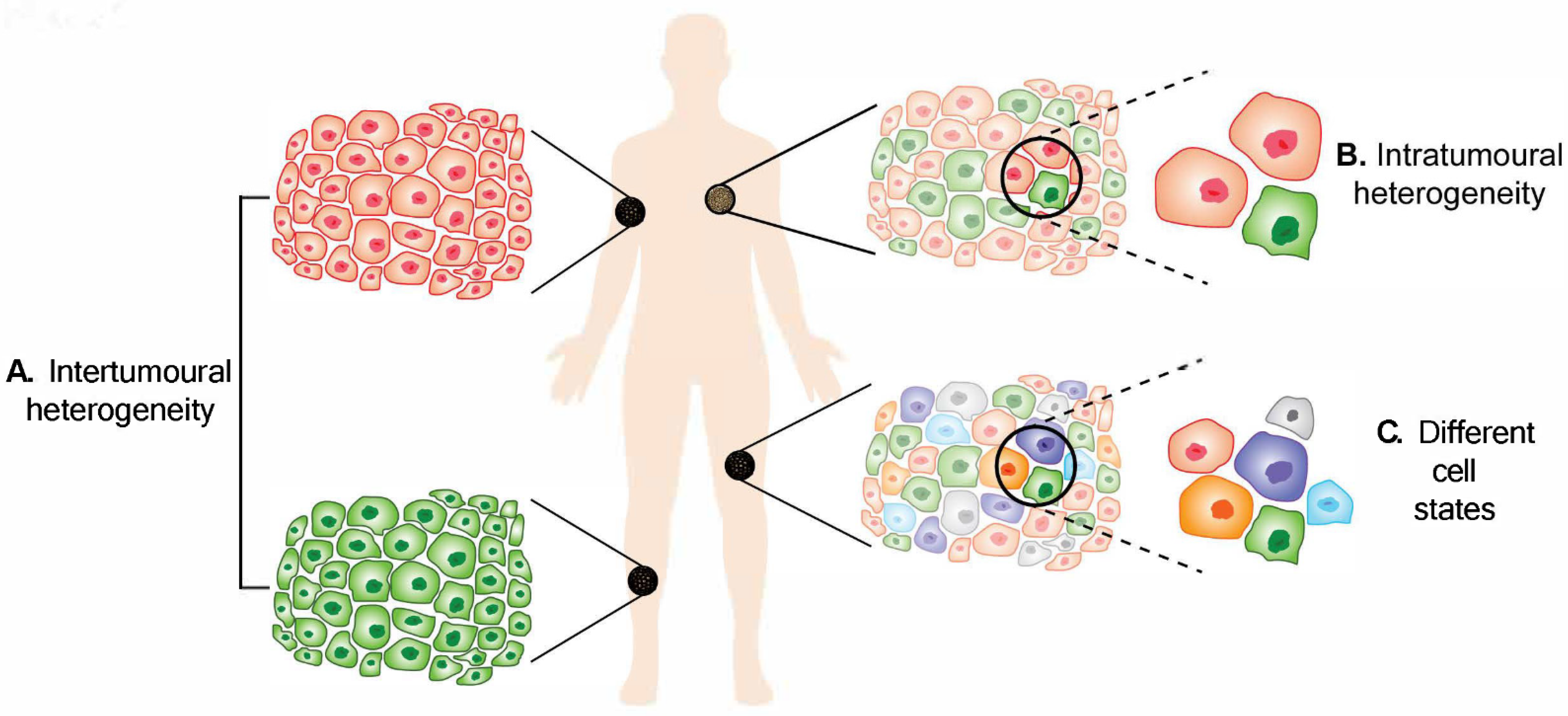
2. Intertumoural Heterogeneity
3. Intratumoural Heterogeneity
3.1. Genetic Contribution to Intratumoural Heterogeneity

{kind=link}
| Heterogeneity | Subpopulations | References |
|---|---|---|
| Genetic | BRAF wild type and BRAF mutants | [25,39,40] |
| Heterogenous expression of BRAFV600E | [41] | |
| KITWT and KITL576P | [42] | |
| BRAFV600E/NRASWT and BRAFWT/NRASQ61R | [43] | |
| NRASwild type and NRASG13R | [44] | |
| Epigenetic | RASSF1A, CDKN2A, DAPK, MGMT and RB1 | [45] |
| Differential methylation leads to heterogeneous expressions of MAGE-A3 | [46] | |
| H3K27 hypermethylation | [47] | |
| JARID1B+ and JARID1B− | [48,49] | |
| Phenotypic | MITFhigh and MITFlow | [50,51,52,53,54,55,56,57,58,59] |
| MITF and BRN2 | [60,61,62,63,64,65,66,67] | |
| MITF and PAX3 | [66] | |
| Transition from MITFhigh/NF-κBlow to MITFlow/NF-κBhigh/AXLhigh during acquisition of resistance | [68,69] | |
| Transition from ZEB2high/SNAIL2high/ZEB1low/TWIST1low to ZEB2low/SNAIL2low/ZEB1high/TWIST1high in primary melanoma to metastatic melanoma | [70] | |
| ABCB5+ and ABCB5- | [71,72,73,74] | |
| CD133+ and CD133- | [74,75,76,77,78] | |
| NGFR+ and NGFR- | [79,80] | |
| Transition from MART-1neg/NGFRhigh to MART-1neg/NGFRneg upon BRAFi treatment | [81] | |
| ALDH+ and ALDH- | [82] | |
| NME1high and NME1low | [83,84] | |
| PGC1αhigh and PGC1αlow | [85,86] | |
| MCT1high and MCT1low | [87] |
3.2. Intratumoural Heterogeneity from Non-Genetic Sources
3.2.1. Epigenetic Heterogeneity
3.2.2. Phenotypic Heterogeneity
3.2.3. Intratumoral Heterogeneity from Other Perspectives
Tumour Microenvironment
Immune Heterogeneity
4. Discoveries from Single-Cell Sequencing
5. Heterogeneity in Melanoma Progression
6. Impact of Heterogeneity on Treatment Responses
7. Conclusions and Implications
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M.; Jimbow, K.; Boissy, R.E.; Slominski, A.; Plonka, P.M.; Slawinski, J.; Wortsman, J.; Tosk, J. What’s the use of generating melanin? Exp. Dermatol. 1999, 8, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Weatherhead, S.C.; Haniffa, M.; Lawrence, C.M. Melanomas arising from naevi and de novo melanomas—Does origin matter? Br. J. Dermatol. 2007, 156, 72–76. [Google Scholar] [CrossRef]
- Kiuru, M.; Tartar, D.M.; Qi, L.; Chen, D.; Yu, L.; Konia, T.; McPherson, J.D.; Murphy, W.J.; Fung, M.A. Improving classification of melanocytic nevi: Association of BRAF V600E expression with distinct histomorphologic features. J. Am. Acad. Dermatol. 2018, 79, 221–229. [Google Scholar] [CrossRef]
- Bauer, J.; Curtin, J.; Pinkel, D.; Bastian, B.C. Congenital Melanocytic Nevi Frequently Harbor NRAS Mutations but no BRAF Mutations. J. Investig. Dermatol. 2007, 127, 179–182. [Google Scholar] [CrossRef]
- Tschandl, P.; Berghoff, A.S.; Preusser, M.; Burgstaller-Muehlbacher, S.; Pehamberger, H.; Okamoto, I.; Kittler, H. NRAS and BRAF Mutations in Melanoma-Associated Nevi and Uninvolved Nevi. PLoS ONE 2013, 8, e69639. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Cadet, J.; Douki, T.; Ravanat, J.-L. Oxidatively Generated Damage to Cellular DNA by UVB and UVA Radiation. Photochem. Photobiol. 2015, 91, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; Soberino-García, J.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. OncoTargets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osella-Abate, S.; Bertero, L.; Senetta, R.; Mariani, S.; Lisa, F.; Coppola, V.; Metovic, J.; Pasini, B.; Puig, S.S.; Fierro, M.T.; et al. TERT Promoter Mutations are Associated with Visceral Spreading in Melanoma of the Trunk. Cancers 2019, 11, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.; Pincus, L.; Ruben, B.; et al. The Genetic Evolution of Melanoma from Precursor Lesions. New Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A Landscape of Driver Mutations in Melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef]
- Ding, L.; Kim, M.; Kanchi, K.L.; Dees, N.D.; Lu, C.; Griffith, M.; Fenstermacher, D.; Sung, H.; Miller, C.A.; Goetz, B.; et al. Clonal Architectures and Driver Mutations in Metastatic Melanomas. PLoS ONE 2014, 9, e111153. [Google Scholar] [CrossRef]
- Janku, F. Tumor heterogeneity in the clinic: Is it a real problem? Ther. Adv. Med. Oncol. 2014, 6, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Tanaka, Y.; Murata, M.; Kaku-Ito, Y.; Furue, K.; Furue, M. BRAF Heterogeneity in Melanoma. Curr. Treat. Options Oncol. 2021, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Sakaizawa, K.; Goto, Y.; Kiniwa, Y.; Uchiyama, A.; Harada, K.; Shimada, S.; Saida, T.; Ferrone, S.; Takata, M.; Uhara, H.; et al. Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br. J. Cancer 2012, 106, 939–946. [Google Scholar] [CrossRef]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS Mutation Frequencies Among Primary Tumors and Metastases in Patients with Melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Yancovitz, M.; Litterman, A.; Yoon, J.; Ng, E.; Shapiro, R.L.; Berman, R.S.; Pavlick, A.C.; Darvishian, F.; Christos, P.; Mazumdar, M.; et al. Intra- and inter-tumor heterogeneity of BRAF(V600E)) mutations in primary and metastatic melanoma. PLoS ONE 2012, 7, e29336. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, L.; Baiter, M.; Kühnapfel, S.; Schuler, G.; Keikavoussi, P.; Agaimy, A.; Kiesewetter, F.; Hartmann, A.; Schneider-Stock, R. Mutation landscape in melanoma patients clinical implications of heterogeneity of BRAF mutations. Br. J. Cancer 2013, 109, 2833–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Jean, M.; Quéreux, G.; Nguyen, J.M.; Peuvrel, L.; Brocard, A.; Vallée, A.; Knol, A.C.; Khammari, A.; Denis, M.G.; Dréno, B. Is a single BRAF wild-type test sufficient to exclude melanoma patients from vemurafenib therapy? J. Investig. Dermatol. 2014, 134, 1468–1470. [Google Scholar] [CrossRef] [Green Version]
- Bradish, J.R.; Richey, J.D.; Post, K.M.; Meehan, K.; Sen, J.D.; Malek, A.J.; Katona, T.M.; Warren, S.; Logan, T.F.; Fecher, L.A.; et al. Discordancy in BRAF mutations among primary and metastatic melanoma lesions: Clinical implications for targeted therapy. Mod. Pathol. 2015, 28, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Yaman, B.; Kandiloglu, G.; Akalin, T. BRAF-V600 Mutation Heterogeneity in Primary and Metastatic Melanoma: A Study with Pyrosequencing and Immunohistochemistry. Am. J. Dermatopathol. 2016, 38, 113–120. [Google Scholar] [CrossRef]
- Eriksson, H.; Zebary, A.; Vassilaki, I.; Omholt, K.; Ghaderi, M.; Hansson, J. BRAFV600E protein expression in primary cutaneous malignant melanomas and paired metastases. JAMA Dermatol. 2015, 151, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Stockwell, P.A.; Ahn, A.; Rodger, E.J.; Leichter, A.L.; Eccles, M.R. Genome-wide methylation sequencing of paired primary and metastatic cell lines identifies common DNA methylation changes and a role for EBF3 as a candidate epigenetic driver of melanoma metastasis. Oncotarget 2016, 8, 6085–6101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campagna, R.; Pozzi, V.; Sartini, D.; Salvolini, E.; Brisigotti, V.; Molinelli, E.; Campanati, A.; Offidani, A.; Emanuelli, M. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef] [PubMed]
- Sanborn, J.Z.; Chung, J.; Purdom, E.; Wang, N.J.; Kakavand, H.; Wilmott, J.S.; Butler, T.; Thompson, J.F.; Mann, G.J.; Haydu, L.E.; et al. Phylogenetic analyses of melanoma reveal complex patterns of metastatic dissemination. Proc. Natl. Acad. Sci. USA 2015, 112, 10995–11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbie, R.; Ansari-Pour, N.; Cast, O.; Lau, D.; Scott, F.; Welsh, S.J.; Parkinson, C.; Khoja, L.; Moore, L.; Tullett, M.; et al. Multi-site clonality analysis uncovers pervasive heterogeneity across melanoma metastases. Nat. Commun. 2020, 11, 4306. [Google Scholar] [CrossRef]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 10. [Google Scholar] [CrossRef]
- Mejbel, H.A.; Arudra, S.K.C.; Pradhan, D.; Torres-Cabala, C.A.; Nagarajan, P.; Tetzlaff, M.T.; Curry, J.L.; Ivan, D.; Duose, D.Y.; Luthra, R.; et al. Immunohistochemical and Molecular Features of Melanomas Exhibiting Intratumor and Intertumor Histomorphologic Heterogeneity. Cancers 2019, 11, 1714. [Google Scholar] [CrossRef] [Green Version]
- Vergara, I.A.; Mintoff, C.P.; Sandhu, S.; McIntosh, L.; Young, R.J.; Wong, S.Q.; Colebatch, A.; Cameron, D.L.; Kwon, J.L.; Wolfe, R.; et al. Evolution of late-stage metastatic melanoma is dominated by aneuploidy and whole genome doubling. Nat. Commun. 2021, 12, 1434. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef]
- Lin, J.; Goto, Y.; Murata, H.; Sakaizawa, K.; Uchiyama, A.; Saida, T.; Takata, M. Polyclonality of BRAF mutations in primary melanoma and the selection of mutant alleles during progression. Br. J. Cancer 2011, 104, 464–468. [Google Scholar] [CrossRef]
- Manfredi, L.; Meyer, N.; Tournier, E.; Grand, D.; Uro-Coste, E.; Rochaix, P.; Brousset, P.; Lamant, L. Highly Concordant Results Between Immunohistochemistry and Molecular Testing of Mutated V600E BRAF in Primary and Metastatic Melanoma. Acta Derm. Venereol. 2016, 96, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Busam, K.J.; Hedvat, C.; Pulitzer, M.; von Deimling, A.; Jungbluth, A.A. Immunohistochemical analysis of BRAF(V600E) expression of primary and metastatic melanoma and comparison with mutation status and melanocyte differentiation antigens of metastatic lesions. Am. J. Surg. Pathol. 2013, 37, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Gremel, G.; Lee, R.J.; Girotti, M.R.; Mandal, A.K.; Valpione, S.; Garner, G.; Ayub, M.; Wood, S.; Rothwell, D.; Fusi, A.; et al. Distinct subclonal tumour responses to therapy revealed by circulating cell-free DNA. Ann. Oncol. 2016, 27, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Sensi, M.; Nicolini, G.; Petti, C.; Bersani, I.; Lozupone, F.; Molla, A.; Vegetti, C.; Nonaka, D.; Mortarini, R.; Parmiani, G.; et al. Mutually exclusive NRASQ61R and BRAFV600E mutations at the single-cell level in the same human melanoma. Oncogene 2006, 25, 3357–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilmott, J.S.; Tembe, V.; Howle, J.R.; Sharma, R.; Thompson, J.F.; Rizos, H.; Lo, R.S.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Intratumoral Molecular Heterogeneity in a BRAF-Mutant, BRAF Inhibitor-Resistant Melanoma: A Case Illustrating the Challenges for Personalized Medicine. Mol. Cancer Ther. 2012, 11, 2704–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastetter, M.; Schagdarsurengin, U.; Lahtz, C.; Fiedler, E.; Marsch, W.C.; Dammann, R.; Helmbold, P. Frequent intra-tumoural heterogeneity of promoter hypermethylation in malignant melanoma. Histol. Histopathol. 2007, 22, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Sigalotti, L.; Fratta, E.; Coral, S.; Tanzarella, S.; Danielli, R.; Colizzi, F.; Fonsatti, E.; Traversari, C.; Altomonte, M.; Maio, M. Intratumor Heterogeneity of Cancer/Testis Antigens Expression in Human Cutaneous Melanoma Is Methylation-Regulated and Functionally Reverted by 5-Aza-2′-deoxycytidine. Cancer Res. 2004, 64, 9167–9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, M.; Reid, M.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Körbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Eichhoff, O.M.; Zipser, M.C.; Xu, M.; Weeraratna, A.T.; Mihic, D.; Dummer, R.; Hoek, K.S. The immunohistochemistry of invasive and proliferative phenotype switching in melanoma: A case report. Melanoma Res. 2010, 20, 349–955. [Google Scholar] [CrossRef] [Green Version]
- Chapman, A.; del Ama, L.F.; Ferguson, J.; Kamarashev, J.; Wellbrock, C.; Hurlstone, A. Heterogeneous Tumor Subpopulations Cooperate to Drive Invasion. Cell Rep. 2014, 8, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Salti, G.I.; Manougian, T.; Farolan, M.; Shilkaitis, A.; Majumdar, D.; Das Gupta, T.K. Micropthalmia transcription factor: A new prognostic marker in intermediate-thickness cutaneous malignant melanoma. Cancer Res. 2000, 60, 5012–5016. [Google Scholar] [PubMed]
- Ennen, M.; Keime, C.; Gambi, G.; Kieny, A.; Coassolo, S.; Thibault-Carpentier, C.; Margerin-Schaller, F.; Davidson, G.; Vagne, C.; Lipsker, D.; et al. MITF-High and MITF-Low Cells and a Novel Subpopulation Expressing Genes of Both Cell States Contribute to Intra- and Intertumoral Heterogeneity of Primary Melanoma. Clin. Cancer Res. 2017, 23, 7097–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., II; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Carreira, S.; Goodall, J.; Denat, L.; Rodriguez, M.; Nuciforo, P.; Hoek, K.S.; Testori, A.; LaRue, L.; Goding, C.R. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006, 20, 3426–3439. [Google Scholar] [CrossRef] [Green Version]
- Goding, C.R. A picture of Mitf in melanoma immortality. Oncogene 2011, 30, 2304–2306. [Google Scholar] [CrossRef]
- Rowling, E.J.; Miskolczi, Z.; Nagaraju, R.; Wilcock, D.; Wang, P.; Telfer, B.; Li, Y.; Lasheras-Otero, I.; Redondo-Muñoz, M.; Sharrocks, A.D.; et al. Cooperative behaviour and phenotype plasticity evolve during melanoma progression. Pigment Cell Melanoma Res. 2020, 33, 695–708. [Google Scholar] [CrossRef]
- Lister, J.A.; Capper, A.; Zeng, Z.; Mathers, M.E.; Richardson, J.; Paranthaman, K.; Jackson, I.J.; Patton, E.E. A Conditional Zebrafish MITF Mutation Reveals MITF Levels Are Critical for Melanoma Promotion vs. Regression In Vivo. J. Investig. Dermatol. 2014, 134, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Travnickova, J.; Wojciechowska, S.; Khamseh, A.; Gautier, P.; Brown, D.V.; Lefevre, T.; Brombin, A.; Ewing, A.; Capper, A.; Spitzer, M.; et al. Zebrafish MITF-Low Melanoma Subtype Models Reveal Transcriptional Subclusters and MITF-Independent Residual Disease. Cancer Res. 2019, 79, 5769–5784. [Google Scholar] [CrossRef] [Green Version]
- Pinner, S.; Jordan, P.; Sharrock, K.; Bazley, L.; Collinson, L.; Marais, R.; Bonvin, E.; Goding, C.; Sahai, E. Intravital Imaging Reveals Transient Changes in Pigment Production and Brn2 Expression during Metastatic Melanoma Dissemination. Cancer Res. 2009, 69, 7969–7977. [Google Scholar] [CrossRef] [Green Version]
- Goodall, J.; Carreira, S.; Denat, L.; Kobi, D.; Davidson, I.; Nuciforo, P.; Sturm, R.A.; Larue, L.; Goding, C.R. Brn-2 Represses Microphthalmia-Associated Transcription Factor Expression and Marks a Distinct Subpopulation of Microphthalmia-Associated Transcription Factor–Negative Melanoma Cells. Cancer Res. 2008, 68, 7788–7794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurber, A.E.; Douglas, G.; Sturm, E.C.; Zabierowski, S.E.; Smit, D.J.; Ramakrishnan, S.N.; Hacker, E.E.; Leonard, J.H.; Herlyn, M.; Sturm, R.A. Inverse expression states of the BRN2 and MITF transcription factors in melanoma spheres and tumour xenografts regulate the NOTCH pathway. Oncogene 2011, 30, 3036–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fane, M.; Chhabra, Y.; Hollingsworth, D.E.; Simmons, J.; Spoerri, L.; Oh, T.G.; Chauhan, J.; Chin, T.; Harris, L.; Harvey, T.J.; et al. NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF. eBioMedicine 2017, 16, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, G.M.; Woods, S.L.; Bonazzi, V.F.; Stark, M.S.; Hacker, E.; Aoude, L.G.; Dutton-Regester, K.; Cook, A.L.; Sturm, R.A.; Hayward, N.K. Melanoma cell invasiveness is regulated by miR-211 suppression of the BRN2 transcription factor. Pigment Cell Melanoma Res. 2011, 24, 525–537. [Google Scholar] [CrossRef]
- Golan, T.; Messer, A.R.; Amitai-Lange, A.; Melamed, Z.; Ohana, R.; Bell, R.E.; Kapitansky, O.; Lerman, G.; Greenberger, S.; Khaled, M.; et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis via Notch Signaling Inhibition of MITF. Mol. Cell 2015, 59, 664–676. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Rana, S.; Ferguson, J.; Rowling, E.J.; Flaherty, K.T.; Wargo, J.A.; Marais, R.; Wellbrock, C. A PAX3/BRN2 rheostat controls the dynamics of BRAF mediated MITF regulation in MITF(high)/AXL(low) melanoma. Pigment Cell Melanoma Res. 2019, 32, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.; Pierce, C.J.; Al-Ejeh, F.; Boyle, G.M. MITF and BRN2 contribute to metastatic growth after dissemination of melanoma. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A Melanoma Cell State Distinction Influences Sensitivity to MAPK Pathway Inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Tang, L.; Lin, J.; Shen, Z.; Yao, Y.; Wang, W.; Tao, S.; Gu, C.; Ma, J.; Xie, Y.; et al. ABCB5 promotes melanoma metastasis through enhancing NF-kappaB p65 protein stability. Biochem. Biophys. Res. Commun. 2017, 492. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.J.; Saab, K.R.; Ma, J.; Schatton, T.; Pütz, P.; Zhan, Q.; Murphy, G.F.; Gasser, M.; Waaga-Gasser, A.M.; Frank, N.Y.; et al. ABCB5 Maintains Melanoma-Initiating Cells through a Proinflammatory Cytokine Signaling Circuit. Cancer Res. 2014, 74, 4196–4207. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.-Y.; Schwartz, B.E.; Hsu, M.-Y. CD133+ Melanoma Subpopulations Contribute to Perivascular Niche Morphogenesis and Tumorigenicity through Vasculogenic Mimicry. Cancer Res. 2012, 72, 5111–5118. [Google Scholar] [CrossRef] [Green Version]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef]
- Jamal, S.M.E.; Alamodi, A.; Wahl, R.U.; Grada, Z.; Shareef, M.A.; Hassan, S.Y.; Murad, F.; Hassan, S.L.; Santourlidis, S.; Gomez, C.R.; et al. Melanoma stem cell maintenance and chemo-resistance are mediated by CD133 signal to PI3K-dependent pathways. Oncogene 2020, 39, 5468–5478. [Google Scholar] [CrossRef] [PubMed]
- Zimmerer, R.M.; Korn, P.; Demougin, P.; Kampmann, A.; Kokemüller, H.; Eckardt, A.M.; Gellrich, N.-C.; Tavassol, F. Functional features of cancer stem cells in melanoma cell lines. Cancer Cell Int. 2013, 13, 78. [Google Scholar] [CrossRef] [Green Version]
- Simbulan-Rosenthal, C.M.; Dougherty, R.; Vakili, S.; Ferraro, A.M.; Kuo, L.-W.; Alobaidi, R.; Aljehane, L.; Gaur, A.; Sykora, P.; Glasgow, E.; et al. CRISPR-Cas9 Knockdown and Induced Expression of CD133 Reveal Essential Roles in Melanoma Invasion and Metastasis. Cancers 2019, 11, 1490. [Google Scholar] [CrossRef] [Green Version]
- Boiko, A.D.; Razorenova, O.V.; van de Rijn, M.; Swetter, S.M.; Johnson, D.L.; Ly, D.P.; Butler, P.D.; Yang, G.P.; Joshua, B.; Kaplan, M.J.; et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nat. Cell Biol. 2010, 466, 133–137. [Google Scholar] [CrossRef]
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Bintz, M.; Yang, Y.; Robert, L.; Ng, A.H.C.; Liu, V.; Ribas, A.; Heath, J.R.; Wei, W. Phenotypic heterogeneity and evolution of melanoma cells associated with targeted therapy resistance. PLoS Comput. Biol. 2019, 15, e1007034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A Isozymes are Markers of Human Melanoma Stem Cells and Potential Therapeutic Targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Leonard, M.K.; Snyder, D.E.; Fisher, M.L.; Eckert, R.L.; Kaetzel, D.M. NME1 Drives Expansion of Melanoma Cells with Enhanced Tumor Growth and Metastatic Properties. Mol. Cancer Res. 2019, 17, 1665–1674. [Google Scholar] [CrossRef]
- Snyder, D.; Wang, Y.; Kaetzel, D.M. A rare subpopulation of melanoma cells with low expression of metastasis suppressor NME1 is highly metastatic in vivo. Sci. Rep. 2020, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Lim, J.-H.; Lee, Y.; Granter, S.R.; Thomas, A.; Vazquez, F.; Widlund, H.; Puigserver, P. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 2016, 537, 422–426. [Google Scholar] [CrossRef] [Green Version]
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115–120. [Google Scholar] [CrossRef]
- Bogdan, I.; Xin, H.; Burg, G.; Böni, R. Heterogeneity of allelic deletions within melanoma metastases. Melanoma Res. 2001, 11, 349–354. [Google Scholar] [CrossRef]
- Harbst, K.; Lauss, M.; Cirenajwis, H.; Winter, C.; Howlin, J.; Törngren, T.; Kvist, A.; Nodin, B.; Olsson, E.; Häkkinen, J.; et al. Molecular and genetic diversity in the metastatic process of melanoma. J. Pathol. 2014, 233, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Mitra, A.; Andrews, M.C.; Roh, W.; De Macedo, M.P.; Hudgens, C.W.; Carapeto, F.; Singh, S.; Reuben, A.; Wang, F.; Mao, X.; et al. Spatially resolved analyses link genomic and immune diversity and reveal unfavorable neutrophil activation in melanoma. Nat. Commun. 2020, 11, 1839. [Google Scholar] [CrossRef] [Green Version]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic Heterogeneity among Tumorigenic Melanoma Cells from Patients that Is Reversible and Not Hierarchically Organized. Cancer Cell 2010, 18, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmer, D.S.; Cheng, P.F.; Eichhoff, O.M.; Belloni, B.C.; Zipser, M.C.; Schlegel, N.C.; Javelaud, D.; Mauviel, A.; Dummer, R.; Hoek, K.S. Systematic classification of melanoma cells by phenotype-specific gene expression mapping. Pigment. Cell Melanoma Res. 2012, 25, 343–353. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [Green Version]
- Campbell, N.R.; Rao, A.; Hunter, M.V.; Sznurkowska, M.K.; Briker, L.; Zhang, M.; Baron, M.; Heilmann, S.; Deforet, M.; Kenny, C.; et al. Cooperation between melanoma cell states promotes metastasis through heterotypic cluster formation. Dev. Cell 2021, 56, 2808–2825.e10. [Google Scholar] [CrossRef]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.-Y.; et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef]
- Perego, M.; Maurer, M.; Wang, J.X.; Shaffer, S.; Müller, A.C.; Parapatics, K.; Li, L.; Hristova, D.; Shin, S.; Keeney, F.; et al. A slow-cycling subpopulation of melanoma cells with highly invasive properties. Oncogene 2018, 37, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernsen, M.R.; Diepstra, J.H.S.; van Mil, P.; Punt, C.J.; Figdor, C.G.; van Muijen, G.N.; Adema, G.J.; Ruiter, D.J. Presence and localization of T-cell subsets in relation to melanocyte differentiation antigen expression and tumour regression as assessed by immunohistochemistry and molecular analysis of microdissected T cells. J. Pathol. 2003, 202, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Bosisio, F.M.; Antoranz, A.; Van Herck, Y.; Bolognesi, M.M.; Marcelis, L.; Chinello, C.; Wouters, J.; Magni, F.; Alexopoulos, L.G.; Stas, M.; et al. Functional heterogeneity of lymphocytic patterns in primary melanoma dissected through single-cell multiplexing. eLife 2020, 9, e53008. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Meng, X.; Wen, J.; Corral, J.M.; Andreev, D.; Kachler, K.; Schett, G.; Chen, X.; Bozec, A. Intratumor Heterogeneity Correlates with Reduced Immune Activity and Worse Survival in Melanoma Patients. Front. Oncol. 2020, 10, 596493. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jiménez-Sánchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235.e21. [Google Scholar] [CrossRef] [Green Version]
- Gerber, T.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Schadendorf, D.; Schartl, M.; Anderegg, U.; Camp, G.; Treutlein, B.; Binder, H.; et al. Mapping heterogeneity in patient-derived melanoma cultures by single-cell RNA-seq. Oncotarget 2016, 8, 846–862. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambow, F.; Marine, J.-C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [Green Version]
- Smalley, I.; Kim, E.; Li, J.; Spence, P.; Wyatt, C.J.; Eroglu, Z.; Sondak, V.K.; Messina, J.L.; Babacan, N.A.; Maria-Engler, S.; et al. Leveraging transcriptional dynamics to improve BRAF inhibitor responses in melanoma. eBioMedicine 2019, 48, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Wouters, J.; Kalender-Atak, Z.; Minnoye, L.; Spanier, K.I.; De Waegeneer, M.; González-Blas, C.B.; Mauduit, D.; Davie, K.; Hulselmans, G.; Najem, A.; et al. Robust gene expression programs underlie recurrent cell states and phenotype switching in melanoma. Nat. Cell Biol. 2020, 22, 986–998. [Google Scholar] [CrossRef]
- Schmidt, M.; Mortensen, L.S.; Loeffler-Wirth, H.; Kosnopfel, C.; Krohn, K.; Binder, H.; Kunz, M. Single-cell trajectories of melanoma cell resistance to targeted treatment. Cancer Biol. Med. 2021, 19, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Tagore, M.; Hunter, M.V.; Kim, I.S.; Moncada, R.; Yan, Y.; Campbell, N.R.; White, R.M.; Yanai, I. The Stress-Like Cancer Cell State Is a Consistent Component of Tumorigenesis. Cell Syst. 2020, 11, 536–546.e7. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature 2021, 596, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468.e4. [Google Scholar] [CrossRef] [Green Version]
- Nirschl, C.J.; Suárez-Fariñas, M.; Izar, B.; Prakadan, S.; Dannenfelser, R.; Tirosh, I.; Liu, Y.; Zhu, Q.; Devi, K.S.P.; Carroll, S.L.; et al. IFNgamma-Dependent Tissue-Immune Homeostasis Is Co-opted in the Tumor Microenvironment. Cell 2017, 170, 127–141.e15. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Ko, M.E.; Cheng, H.; Zhu, R.; Xue, M.; Wang, J.; Lee, J.W.; Frankiw, L.; Xu, A.; Wong, S.; et al. Multi-omic single-cell snapshots reveal multiple independent trajectories to drug tolerance in a melanoma cell line. Nat. Commun. 2020, 11, 2345. [Google Scholar] [CrossRef]
- Hanniford, D.; Ulloa-Morales, A.; Karz, A.; Berzoti-Coelho, M.G.; Moubarak, R.; Sánchez-Sendra, B.; Kloetgen, A.; Davalos, V.; Imig, J.; Wu, P.; et al. Epigenetic Silencing of CDR1as Drives IGF2BP3-Mediated Melanoma Invasion and Metastasis. Cancer Cell 2020, 37, 55–70. [Google Scholar] [CrossRef]
- Vivancos, A.; Caratú, G.; Matito, J.; Muñoz, E.; Ferrer, B.; Hernández-Losa, J.; Bodet, D.; Pérez-Alea, M.; Cortés, J.; Garcia-Patos, V.; et al. Genetic evolution of nevus of Ota reveals clonal heterogeneity acquiring BAP1 and TP53 mutations. Pigment Cell Melanoma Res. 2016, 29, 247–253. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekulic, A.; Hingorani, P.; Lenkiewicz, E.; Holley, T.; Barrett, M.; Zismann, V.; Trent, J. Clonal evolution underlying trans-placental transferand vemurafenib resistance in melanoma. Society for Melanoma Research 2012 Congress. Pigment Cell Melanoma Res. 2012, 25, 836–903. [Google Scholar] [CrossRef]
- Khaliq, M.; Manikkam, M.; Martinez, E.D.; Fallahi-Sichani, M. Epigenetic modulation reveals differentiation state specificity of oncogene addiction. Nat. Commun. 2021, 12, 1536. [Google Scholar] [CrossRef]
- Marin-Bejar, O.; Rogiers, A.; Dewaele, M.; Femel, J.; Karras, P.; Pozniak, J.; Bervoets, G.; Van Raemdonck, N.; Pedri, D.; Swings, T.; et al. Evolutionary predictability of genetic versus nongenetic resistance to anticancer drugs in melanoma. Cancer Cell 2021, 39, 1135–1149.e8. [Google Scholar] [CrossRef] [PubMed]
- El-Khattouti, A.; Sheehan, N.T.; Monico, J.; Drummond, H.A.; Haikel, Y.; Brodell, R.T.; Megahed, M.; Hassan, M. CD133 + melanoma subpopulation acquired resistance to caffeic acid phenethyl ester-induced apoptosis is attributed to the elevated expression of ABCB5: Significance for melanoma treatment. Cancer Lett. 2015, 357, 83–104. [Google Scholar] [CrossRef] [PubMed]
- Ravindran Menon, D.; Das, S.; Krepler, C.; Vultur, A.; Rinner, B.; Schauer, S.; Kashofer, K.; Wagner, K.; Zhang, G.; Bonyadi Rad, E.; et al. A stress-induced early innate response causes multidrug tolerance in melanoma. Oncogene 2015, 34, 4448–4459. [Google Scholar] [CrossRef] [Green Version]
- Romano, G.; Chen, P.L.; Song, P.; McQuade, J.L.; Liang, R.J.; Liu, M.; Roh, W.; Duose, D.Y.; Carapeto, F.C.L.; Li, J.; et al. A Preexisting Rare PIK3CA(E545K) Subpopulation Confers Clinical Resistance to MEK plus CDK4/6 Inhibition in NRAS Melanoma and Is Dependent on S6K1 Signaling. Cancer Discov. 2018, 8, 556–567. [Google Scholar] [CrossRef] [Green Version]
- Campagna, R.; Salvolini, E.; Pompei, V.; Pozzi, V.; Salvucci, A.; Molinelli, E.; Brisigotti, V.; Sartini, D.; Campanati, A.; Offidani, A.; et al. Nicotinamide N-methyltransferase gene silencing enhances chemosensitivity of melanoma cell lines. Pigment Cell Melanoma Res. 2021, 34, 1039–1048. [Google Scholar] [CrossRef]
- Boshuizen, J.; Vredevoogd, D.W.; Krijgsman, O.; Ligtenberg, M.A.; Blankenstein, S.; De Bruijn, B.; Frederick, D.T.; Kenski, J.C.N.; Parren, M.; Brüggemann, M.; et al. Reversal of pre-existing NGFR-driven tumor and immune therapy resistance. Nat. Commun. 2020, 11, 3946. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Asher, N.; Ben-Betzalel, G.; Lev-Ari, S.; Shapira-Frommer, R.; Steinberg-Silman, Y.; Gochman, N.; Schachter, J.; Meirson, T.; Markel, G. Real World Outcomes of Ipilimumab and Nivolumab in Patients with Metastatic Melanoma. Cancers 2020, 12, 2329. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Sondak, V.K.; Smalley, K.S. A brief history of melanoma: From mummies to mutations. Melanoma Res. 2012, 22, 114–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Ref. | Cell States | Samples | ||||||
|---|---|---|---|---|---|---|---|---|
| Cycling/Proliferative | Intermediate | Non-Cycling | ||||||
| [54] | MITFhigh | AXLhigh | Patient tumour samples (advanced stage) | |||||
| [106] | Proliferation | Pigmentation | Stromal | Patient-derived cultures (advanced stage) | ||||
| [107] | C4 melanocytic | C3 transitory | C2 neural crest-like | C1 undifferentiated | Human melanoma cell lines (53) | |||
| [108] | Hyper-differentiated | Melanocytic | Intermediate | Starved | Neural crest stem-cell-like | Undifferentiated | PDXs from advanced-stage patients | |
| [109] | State #1 (high cyclin D1, ERBB3, STAT3/5, ATF1, ATF4, MITF & β-catenin; low c-JUN, Axl & EGFR) | State #2 (high ERBB3 Axl & c-JUN; low MET, RELB, E2F1, BIM, ULK1, SMAD1/9 & XIAP) | State #3 (high Axl, c-JUN, E2F1, WEE1, c-MET & EGFR; low MITF, ERBB3 and SMAD9) | State #4 (low MITF/RTK expression & suppressed cell-death-related gene expression) | Human melanoma cell lines (1205Lu, 1205LuR, WM164 & WM164R) | |||
| [110] | Melanocytic | Intermediate | Mesenchymal-like | Patient-derived cultures (9) and human melanoma cell line (A375) | ||||
| [111] | C2 high cycling (G1/S) | C4 high cycling (G2/M) | C5 translation | C7 reactivation of MAPK | C6 pluripotent | C1 neural crest-like | C3 slow cycling, stroma-like | Human melanoma cell lines (A375) |
| [112] | Mature melanocytic | Stress-like | Neural crest | Human melanoma-like tumour from transgenic zebrafish | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, M.F.; Simmons, J.L.; Boyle, G.M. Heterogeneity in Melanoma. Cancers 2022, 14, 3030. https://doi.org/10.3390/cancers14123030
Ng MF, Simmons JL, Boyle GM. Heterogeneity in Melanoma. Cancers. 2022; 14(12):3030. https://doi.org/10.3390/cancers14123030
Chicago/Turabian StyleNg, Mei Fong, Jacinta L. Simmons, and Glen M. Boyle. 2022. "Heterogeneity in Melanoma" Cancers 14, no. 12: 3030. https://doi.org/10.3390/cancers14123030
APA StyleNg, M. F., Simmons, J. L., & Boyle, G. M. (2022). Heterogeneity in Melanoma. Cancers, 14(12), 3030. https://doi.org/10.3390/cancers14123030