
In Vivo Quantitative Imaging of Glioma Heterogeneity Employing Positron Emission Tomography

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
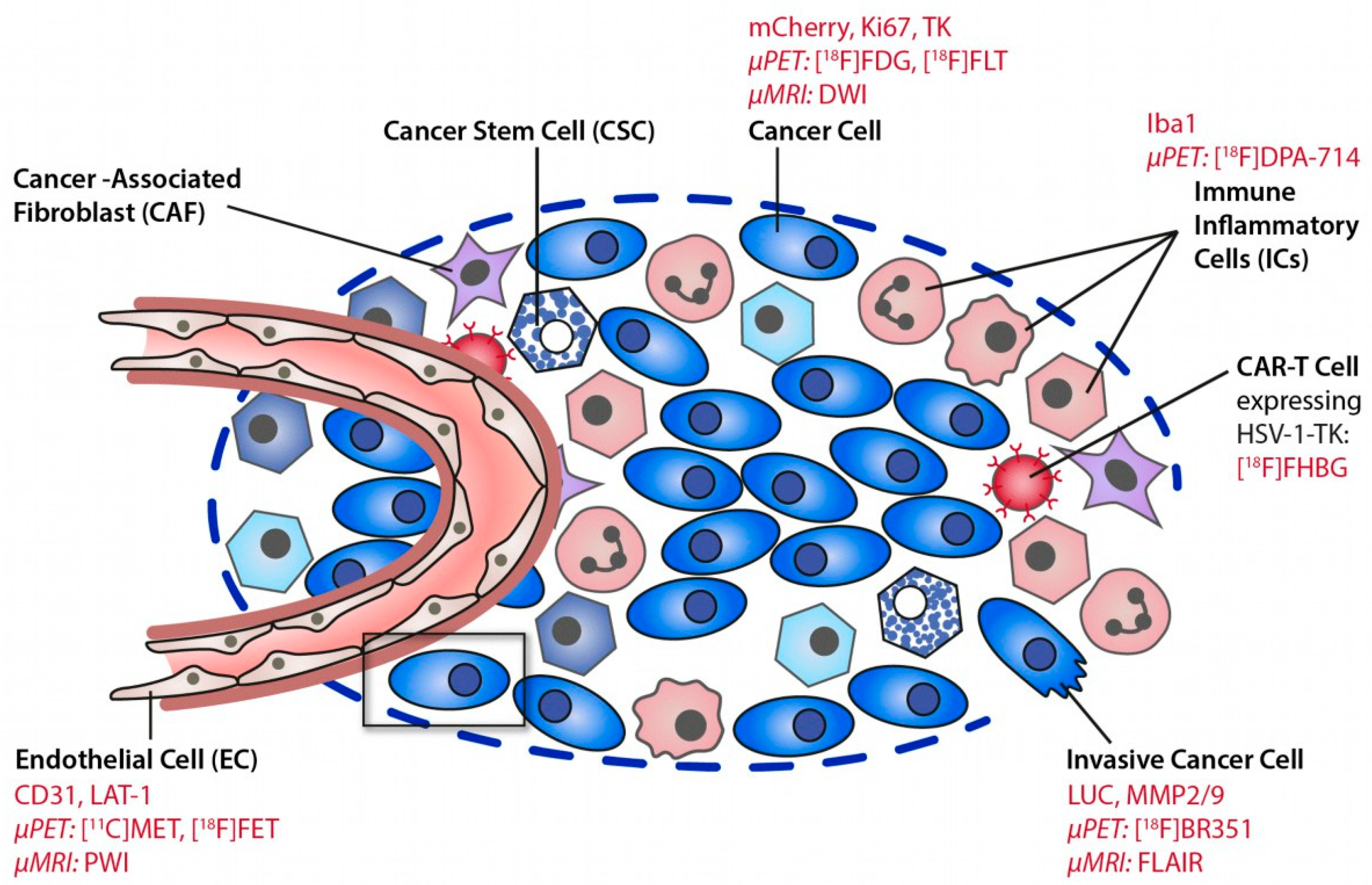
2. The Tumor Microenvironment (TME)
3. Quantitative Imaging of Glioblastoma
3.1. Magnetic Resonance Imaging
3.2. [18F]FET and Other Amino Acid PET Ligands
3.3. Translocator Protein (TSPO) PET Imaging
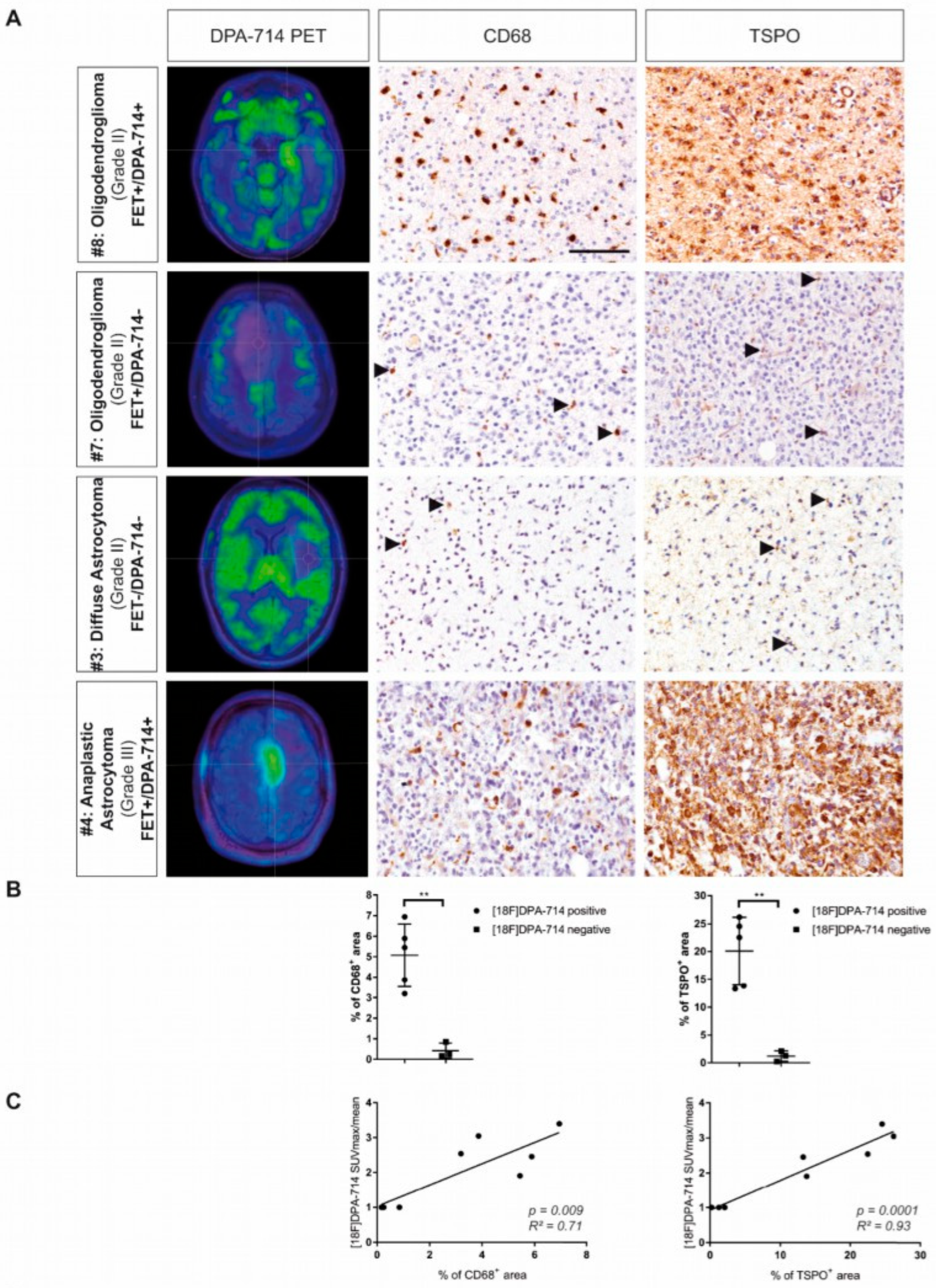
3.3.1. Detecting Areas beyond FET Imaging and MRI
3.3.2. TSPO Imaging of Tumor Progression
3.3.3. Tumor Classification
3.3.4. Detecting Early Infiltration
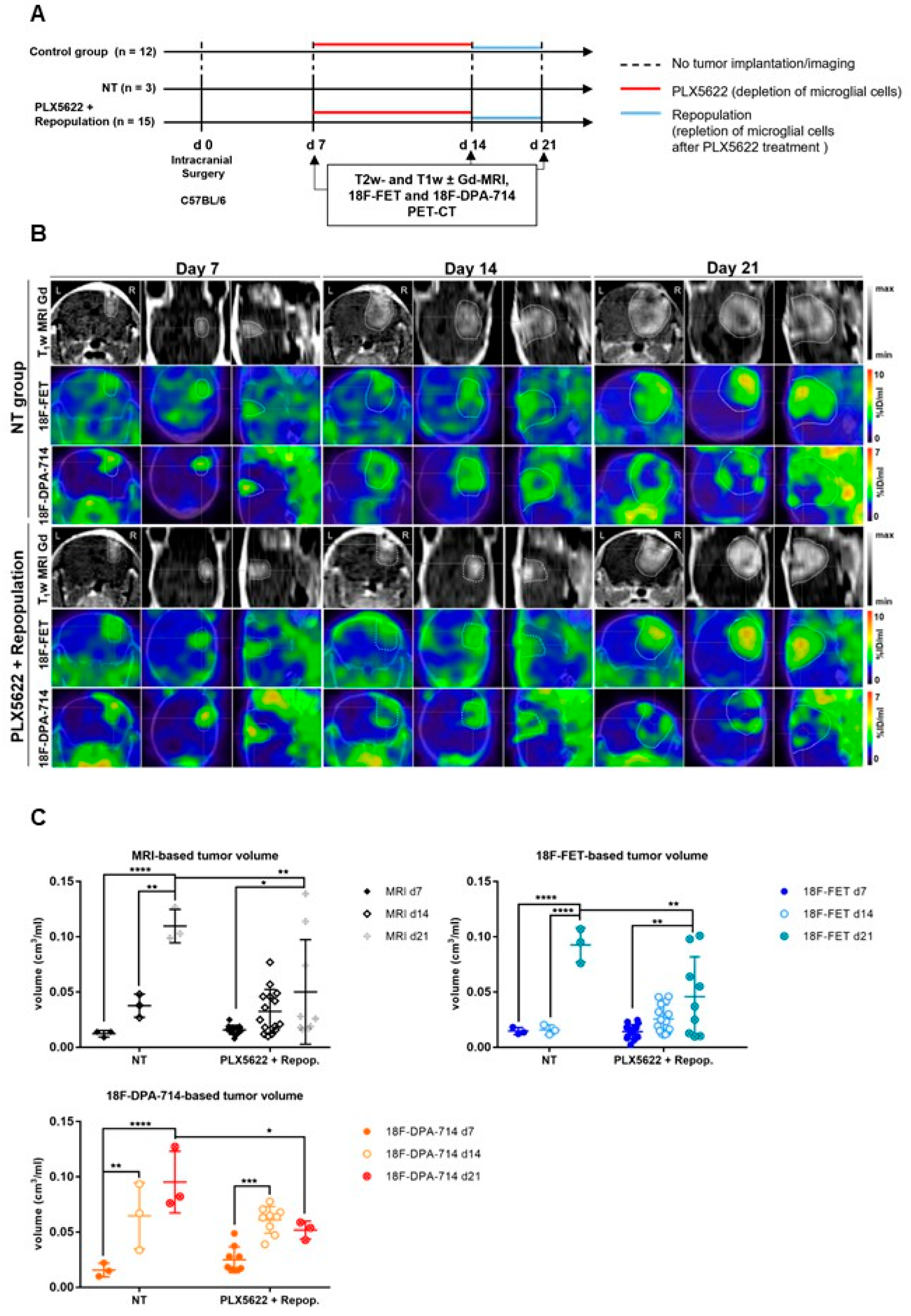
3.3.5. Glioma-Associated Inflammation
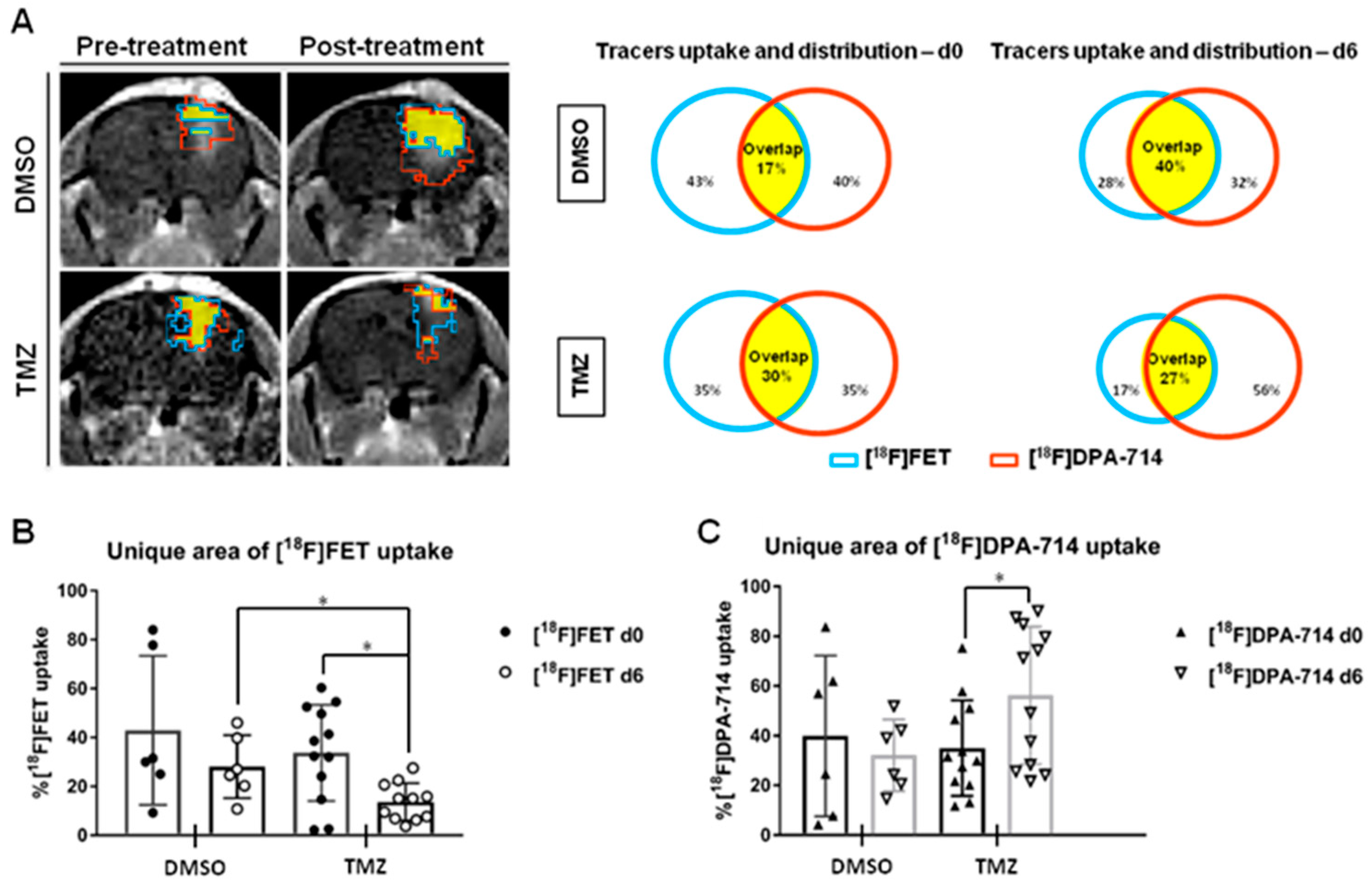
3.3.6. Therapy Readout
3.3.7. Other Features
3.3.8. TSPO and Matrix Metalloproteinases
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verduin, M.; Compter, I.; Steijvers, D.; Postma, A.A.; Eekers, D.B.P.; Anten, M.M.; Ackermans, L.; Ter Laan, M.; Leijenaar, R.T.H.; Van de Weijer, T.; et al. Noninvasive glioblastoma testing: Multimodal approach to monitoring and predicting treatment response. Dis. Markers 2018, 2018, 2908609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priceman, S.J.; Sung, J.L.; Shaposhnik, Z.; Burton, J.B.; Torres-Collado, A.X.; Moughon, D.L.; Johnson, M.; Lusis, A.J.; Cohen, D.A.; Iruela-Arispe, M.L.; et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: Combating tumor evasion of antiangiogenic therapy. Blood 2010, 115, 1461–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R Signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [Green Version]
- Kamran, N.; Alghamri, M.S.; Nunez, F.J.; Shah, D.; Asad, A.S.; Candolfi, M.; Altshuler, D.; Lowenstein, P.R.; Castro, M.G. Current state and future prospects of immunotherapy for glioma. Immunotherapy 2018, 10, 317–339. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.B.; Barros, L.R.C.; Bracco, P.A.; Vigo, A.; Boroni, M.; Bonamino, M.H.; Lenz, G. Transcriptional characterization of immunological infiltrates and their relation with glioblastoma patients overall survival. Oncoimmunology 2018, 7, e1431083. [Google Scholar] [CrossRef] [Green Version]
- Nigam, S.; McCarl, L.; Kumar, R.; Edinger, R.S.; Kurland, B.F.; Anderson, C.J.; Panigrahy, A.; Kohanbash, G.; Edwards, W.B. Preclinical immunoPET imaging of glioblastoma-infiltrating myeloid cells using zirconium-89 labeled anti-CD11b antibody. Mol. Imaging Biol. 2020, 22, 685–694. [Google Scholar] [CrossRef]
- Jacobs, A.H.; Schelhaas, S.; Viel, T.; Waerzeggers, Y.; Winkeler, A.; Zinnhardt, B.; Gelovani, J. Imaging of gene and cell-based therapies: Basis and clinical trials. In Molecular Imaging; Ross, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1539–1587. [Google Scholar]
- Lundy, P.; Domino, J.; Ryken, T.; Fouke, S.; McCracken, D.J.; Ormond, D.R.; Olson, J.J. The role of imaging for the management of newly diagnosed glioblastoma in adults: A systematic review and evidence-based clinical practice guideline update. J. Neurooncol. 2020, 150, 95–120. [Google Scholar] [CrossRef]
- Huang, R.Y.; Neagu, M.R.; Reardon, D.A.; Wen, P.Y. Pitfalls in the neuroimaging of glioblastoma in the era of antiangiogenic and immuno/targeted therapy—Detecting illusive disease, defining response. Front. Neurol. 2015, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Miyake, M.; Takahashi, M.; Hamamoto, R. Observing deep radiomics for the classification of glioma grades. Sci. Rep. 2021, 11, 10942. [Google Scholar] [CrossRef] [PubMed]
- Chaddad, A.; Kucharczyk, M.J.; Daniel, P.; Sabri, S.; Jean-claude, B.J.; Niazi, T.; Abdulkarim, B. Radiomics in glioblastoma: Current status and challenges facing clinical implementation. Front. Oncol. 2019, 9, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Manjila, S.; Sakla, N.; True, A.; Wardeh, A.H.; Beig, N.; Vaysberg, A.; Matthews, J.; Prasanna, P.; Spektor, V. Radiomics and radiogenomics in gliomas: A contemporary update. Br. J. Cancer 2021, 125, 641–657. [Google Scholar] [CrossRef]
- Ginet, M.; Zaragori, T.; Marie, P.Y.; Roch, V.; Gauchotte, G.; Rech, F.; Blonski, M.; Lamiral, Z.; Taillandier, L.; Imbert, L.; et al. Integration of dynamic parameters in the analysis of 18F-FDopa PET imaging improves the prediction of molecular features of gliomas. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Zaragori, T.; Oster, J.; Roch, V.; Hossu, G.; Chawki, M.B.; Grignon, R.; Pouget, C.; Gauchotte, G.; Rech, F.; Blonski, M.; et al. 18F-FDOPA PET for the noninvasive prediction of glioma molecular parameters: A radiomics study. J. Nucl. Med. 2022, 63, 147–157. [Google Scholar] [CrossRef]
- Hutterer, M.; Nowosielski, M.; Putzer, D.; Jansen, N.L.; Seiz, M.; Schocke, M.; McCoy, M.; Göbel, G.; la Fougère, C.; Virgolini, I.J.; et al. [18F]-fluoro-ethyl-l-tyrosine PET: A valuable diagnostic tool in neuro-oncology, but not all that glitters is glioma. Neuro. Oncol. 2013, 15, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Nowosielski, M.; DiFranco, M.D.; Putzer, D.; Seiz, M.; Recheis, W.; Jacobs, A.H.; Stockhammer, G.; Hutterer, M. An Intra-Individual Comparison of MRI, [18F]-FET and [18F]-FLT PET in Patients with High-Grade Gliomas. PLoS ONE 2014, 9, e95830. [Google Scholar] [CrossRef]
- Galldiks, N.; Langen, K.J.; Holy, R.; Pinkawa, M.; Stoffels, G.; Nolte, K.W.; Kaiser, H.J.; Filss, C.P.; Fink, G.R.; Coenen, H.H.; et al. Assessment of treatment response in patients with glioblastoma using O-(2-18F-fluoroethyl)-L-tyrosine PET in comparison to MRI. J. Nucl. Med. 2012, 53, 1048–1057. [Google Scholar] [CrossRef] [Green Version]
- Laukamp, K.R.; Lindemann, F.; Weckesser, M.; Hesselmann, V.; Ligges, S.; Wölfer, J.; Jeibmann, A.; Zinnhardt, B.; Viel, T.; Schäfers, M.; et al. Multimodal imaging of patients with gliomas confirms 11 C-MET PET as a complementary marker to MRI for noninvasive tumor grading and intraindividual follow-up after therapy. Mol. Imaging 2017, 16, 153601211668765. [Google Scholar] [CrossRef] [Green Version]
- Hutterer, M.; Hattingen, E.; Palm, C.; Proescholdt, M.A.; Hau, P. Current standards and new concepts in MRI and PET response assessment of antiangiogenic therapies in high-grade glioma patients. Neuro. Oncol. 2015, 17, 784–800. [Google Scholar] [CrossRef]
- Lohmann, P.; Stavrinou, P.; Lipke, K.; Bauer, E.K.; Ceccon, G.; Werner, J.M.; Neumaier, B.; Fink, G.R.; Shah, N.J.; Langen, K.J.; et al. FET PET reveals considerable spatial differences in tumour burden compared to conventional MRI in newly diagnosed glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 591–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celli, M.; Caroli, P.; Amadori, E.; Arpa, D.; Gurrieri, L.; Ghigi, G.; Cenni, P.; Paganelli, G.; Matteucci, F. Diagnostic and prognostic potential of 18F-FET PET in the differential diagnosis of glioma recurrence and treatment-induced changes after chemoradiation therapy. Front. Oncol. 2021, 11, 721821. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Brucker, D.P.; Stoffels, G.; Filipski, K.; Filss, C.P.; Mottaghy, F.M.; Galldiks, N.; Steinbach, J.P.; Hattingen, E.; Langen, K.J. 18F-FET PET imaging in differentiating glioma progression from treatment-related changes: A single-center experience. J. Nucl. Med. 2020, 61, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Foray, C.; Valtorta, S.; Barca, C.; Winkeler, A.; Roll, W.; Müther, M.; Wagner, S.; Gardner, M.L.; Hermann, S.; Schäfers, M.; et al. Imaging temozolomide-induced changes in the myeloid glioma microenvironment. Theranostics 2021, 11, 2020–2033. [Google Scholar] [CrossRef]
- Ceccon, G.; Lohmann, P.; Werner, J.M.; Tscherpel, C.; Dunkl, V.; Stoffels, G.; Rosen, J.; Rapp, M.; Sabel, M.; Herrlinger, U.; et al. Early treatment response assessment using 18F-FET PET compared with contrast-enhanced MRI in Glioma patients after adjuvant temozolomide chemotherapy. J. Nucl. Med. 2021, 62, 918–925. [Google Scholar] [CrossRef]
- Awde, A.R.; Boisgard, R.; Thézé, B.; Dubois, A.; Zheng, J.; Dollé, F.; Jacobs, A.H.; Tavitian, B.; Winkeler, A. The translocator protein radioligand 18 F-DPA-714 monitors antitumor effect of erufosine in a Rat 9L intracranial glioma model. J. Nucl. Med. 2013, 54, 2125–2131. [Google Scholar] [CrossRef] [Green Version]
- Winkeler, A.; Boisgard, R.; Awde, A.R.; Dubois, A.; Thézé, B.; Zheng, J.; Ciobanu, L.; Dollé, F.; Viel, T.; Jacobs, A.H.; et al. The translocator protein ligand [18F]DPA-714 images glioma and activated microglia in vivo. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Wang, D.; Wang, H.; Cai, M.; Li, C.; Zhang, X.; Chen, H.; Hu, Y.; Zhang, X.; Ying, M.; et al. TSPO deficiency induces mitochondrial dysfunction, leading to hypoxia, angiogenesis, and a growth-promoting metabolic shift toward glycolysis in glioblastoma. Neuro. Oncol. 2020, 22, 240–252. [Google Scholar] [CrossRef]
- Miettinen, H.; Kononen, J.; Haapasalo, H.; Helén, P.; Sallinen, P.; Harjuntausta, T.; Helin, H.; Alho, H. Expression of peripheral-type benzodiazepine receptor and diazepam binding inhibitor in human astrocytomas: Relationship to cell proliferation. Cancer Res. 1995, 55, 2691–2695. [Google Scholar]
- Vlodavsky, E.; Soustiel, J.F. Immunohistochemical expression of peripheral benzodiazepine receptors in human astrocytomas and its correlation with grade of malignancy, proliferation, apoptosis and survival. J. Neurooncol. 2006, 81, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Herholz, K.; Gerhard, A.; Roncaroli, F.; Du Plessis, D.; Jackson, A.; Turkheimer, F.; Hinz, R. [11C]-(R)PK11195 tracer kinetics in the brain of glioma patients and a comparison of two referencing approaches. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 1406–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Hu, K.; Shao, T.; Hou, L.; Zhang, S.; Ye, W.; Josephson, L.; Meyer, J.H.; Zhang, M.R.; Vasdev, N.; et al. Recent developments on PET radiotracers for TSPO and their applications in neuroimaging. Acta Pharm. Sin. B 2021, 11, 373–393. [Google Scholar] [CrossRef]
- Zinnhardt, B.; Müther, M.; Roll, W.; Backhaus, P.; Jeibmann, A.; Foray, C.; Barca, C.; Döring, C.; Tavitian, B.; Dollé, F.; et al. TSPO imaging-guided characterization of the immunosuppressive myeloid tumor microenvironment in patients with malignant glioma. Neuro. Oncol. 2020, 22, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Pigeon, H.; Pérès, E.A.; Truillet, C.; Jego, B.; Boumezbeur, F.; Caillé, F.; Zinnhardt, B.; Jacobs, A.H.; Le Bihan, D.; Winkeler, A. TSPO-PET and diffusion-weighted MRI for imaging a mouse model of infiltrative human glioma. Neuro. Oncol. 2019, 21, 755–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foray, C.; Barca, C.; Winkeler, A.; Wagner, S.; Hermann, S.; Schaefers, M.; Grauer, O.M.; Zinnhardt, B.; Jacobs, A.H. Interrogating glioma-associated microglia/macrophage dynamics under CSF-1R therapy with multi-tracer in vivo PET/MR imaging. J. Nucl. Med. 2022; in press. [Google Scholar] [CrossRef]
- Albert, N.L.; Unterrainer, M.; Fleischmann, D.F.; Lindner, S.; Vettermann, F.; Brunegraf, A.; Vomacka, L.; Brendel, M.; Wenter, V.; Wetzel, C.; et al. TSPO PET for glioma imaging using the novel ligand 18F-GE-180: First results in patients with glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2230–2238. [Google Scholar] [CrossRef]
- Kaiser, L.; Holzgreve, A.; Quach, S.; Ingrisch, M.; Unterrainer, M.; Dekorsy, F.J.; Lindner, S.; Ruf, V.; Brosch-Lenz, J.; Delker, A.; et al. Differential spatial distribution of TSPO or amino acid PET signal and MRI contrast enhancement in gliomas. Cancers. 2021, 14, 53. [Google Scholar] [CrossRef]
- Su, Z.; Roncaroli, F.; Durrenberger, P.F.; Coope, D.J.; Karabatsou, K.; Hinz, R.; Thompson, G.; Turkheimer, F.E.; Janczar, K.; Du Plessis, D.; et al. The 18-kDa mitochondrial translocator protein in human gliomas: An 11 C-(R)PK11195 PET imaging and neuropathology study. J. Nucl. Med. 2015, 56, 512–517. [Google Scholar] [CrossRef] [Green Version]
- Jensen, P.; Feng, L.; Law, I.; Svarer, C.; Knudsen, G.M.; Mikkelsen, J.D.; de Nijs, R.; Larsen, V.A.; Dyssegaard, A.; Thomsen, G.; et al. TSPO Imaging in glioblastoma multiforme: A direct comparison between 123 I-CLINDE SPECT, 18 F-FET PET, and gadolinium-enhanced MR imaging. J. Nucl. Med. 2015, 56, 1386–1390. [Google Scholar] [CrossRef] [Green Version]
- Unterrainer, M.; Fleischmann, D.F.; Diekmann, C.; Vomacka, L.; Lindner, S.; Vettermann, F.; Brendel, M.; Wenter, V.; Ertl-Wagner, B.; Herms, J.; et al. Comparison of 18F-GE-180 and dynamic 18F-FET PET in high grade glioma: A double-tracer pilot study. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 580–590. [Google Scholar] [CrossRef]
- Tran, T.T.; Gallezot, J.-D.; Jilaveanu, L.B.; Zito, C.; Turcu, G.; Lim, K.; Nabulsi, N.; Huang, H.; Huttner, A.; Kluger, H.M.; et al. [11C]Methionine and [11C]PBR28 as PET imaging tracers to differentiate metastatic tumor recurrence or radiation necrosis. Mol. Imaging 2020, 19, 153601212096866. [Google Scholar] [CrossRef] [PubMed]
- Ammer, L.M.; Vollmann-Zwerenz, A.; Ruf, V.; Wetzel, C.H.; Riemenschneider, M.J.; Albert, N.L.; Beckhove, P.; Hau, P. The role of translocator protein TSPO in hallmarks of glioblastoma. Cancers 2020, 12, 2973. [Google Scholar] [CrossRef] [PubMed]
- Holzgreve, A.; Pötter, D.; Brendel, M.; Orth, M.; Weidner, L.; Gold, L.; Kirchner, M.A.; Bartos, L.M.; Unterrainer, L.M.; Unterrainer, M.; et al. Longitudinal [18F]GE-180 PET Imaging facilitates in vivo monitoring of TSPO expression in the GL261 glioblastoma mouse model. Biomedicines 2022, 10, 738. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Gondi, C.S.; Rao, J.S. Proteases and glioma angiogenesis. Brain Pathol. 2006, 15, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Angus McQuibban, G.; Gong, J.H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 2002, 100, 1160–1167. [Google Scholar] [CrossRef]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; Van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, P.A.; Wong, H.; Laing, T.D.; Rewcastle, N.B.; Morris, D.G.; Muzik, H.; Leco, K.J.; Johnston, R.N.; Brasher, P.M.A.; Sutherland, G.; et al. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas. Br. J. Cancer 1999, 79, 1828–1835. [Google Scholar] [CrossRef]
- Wagner, S.; Faust, A.; Breyholz, H.J.; Schober, O.; Schäfers, M.; Kopka, K. The MMP inhibitor (R)-2-(N-benzyl-4-(2-[18F]fluoroethoxy)phenylsulphonamido)-N-hydroxy-3-methylbutanamide: Improved precursor synthesis and fully automated radiosynthesis. Appl. Radiat. Isot. 2011, 69, 862–868. [Google Scholar] [CrossRef]
- Zinnhardt, B.; Pigeon, H.; Thézé, B.; Viel, T.; Wachsmuth, L.; Fricke, I.B.; Schelhaas, S.; Honold, L.; Schwegmann, K.; Wagner, S.; et al. Combined PET Imaging of the inflammatory tumor microenvironment identifies margins of unique radiotracer uptake. Cancer Res. 2017, 77, 1831–1841. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modality | Pros | Cons |
|---|---|---|
| MRI | High resolution, mostly non-invasive, clinical availability | Primarily structural information |
| T1w ± CE | Tumor size and location, indicative for disrupted BBB, edema formation, hemorrhage, necrosis | Contrast dependent on a disrupted BBB, pseudoprogression, pseudoresponse |
| T2w | ||
| FLAIR | ||
| Diffusion | Indicative of early changes in tumor density, differentiation between GBM from lymphoma, narrowing the differential diagnosis, treatment planning | High variability |
| Perfusion | Neovascularization, differentiation of pseudoprogression from tumor progression | Signal quantification |
| PET | Functional/metabolic activity, high sensitivity, quantifiable | Low resolution, radiotracer production |
| Amino acid | Indicative of metabolic active tumor tissue, discriminate between glioma recurrence and treatment-induced changes | |
| TSPO | Indicative of neoplastic cells and GAMs, associated with tumor infiltration and an immunosuppressive TME | Not exclusive to neoplastic cells or GAMs |
| Matrix | Indicative of enhanced intracerebral invasion, neovascularization | No clinical use |
| metalloproteinases |
| Tracers | Results | Main Conclusion | Ref. |
|---|---|---|---|
| [11C]PK11195 | n = 23 glioma patients, 10 healthy volunteers Three different regional kinetics were observed in individual tumors TACs: grey matter-like kinetics, white matter-like kinetics and mixed kinetics. Kinetics differed between LG astrocytoma and oligodendroglioma, independent of the tumor grade. The number of TSPO+ tumor cells increased with tumor grade. Only a minority of microglial cells and newly formed vessels showed TSPO expression. | Tracer kinetics in gliomas could potentially discriminate between LG astrocytomas and oligodendrogliomas | [33] |
| [18F]DPA-714 [18F]FET | n = 9, including 4 LGG and 5 HGG. Both [18F]FET and [18F]DPA-714 uptake patterns showed partial overlap with FLAIR hyperintensities. LGG patients were classified into [18F]FET-positive/[18F]DPA-714-positive and [18F]FET-positive/[18F]DPA-714-negative subgroups while all HGG patients were [18F]FET-positive/[18F]DPA-714-positive. In patients with positive uptake for both tracers, the mean percentage of overlap was 24.56%. Positive correlation between [18F]DPA-714 uptake and the number of (CD68+) GAMs (r = 0.84, p = 0.009). TSPO is strongly upregulated in HLA-DR+ GAMs, including HLA-DR+ TAMs and HLA-DR+ MDSCs. | [18F]DPA-714 may detect the glioma-associated immunosuppressive TME | [35] |
| [18F]GE-180 | n = 10 GBM, 1 AA (confirmed IDH-wt glioma) All gliomas showed positive [18F]GE-180 uptake with high T/B contrast (median SUVBG: 0.47 (0.37–0.93), TBRmax: 6.61 (3.88–9.07)). [18F]GE-180 uptake could be found even in areas without contrast enhancement on MR images. | First [18F]GE-180 imaging in patients | [38] |
| [18F]GE180 [18F]FET | n = 34 newly diagnosed glioma, including 30/34 WHO grade IV and no mutational IDH1/2 gene. Poor correlation between rCE and PET signals was observed but a strong correlation between PET signals. More than 50% of the patients showed a large distance between rCE and TBRGE-180 or TBRFET hottest spots. In most patients, a large proportion of voxels without increased rCE (73 ± 17%) was identified, of which a high fraction was positive in [18F]GE-180 (46 ± 27%) and [18F]FET PET (32 ± 18%), showing a large variance in TBR values for both PET signals. | Amino acid and TSPO PET imaging combined with MRI allow the depiction of tumor heterogeneity | [39] |
| [11C]PK11195 | n = 22 (13 astrocytomas, 9 oligogendrogliomas). BPND of [11C]PK11195, corrected for local blood volume, in HG glioma was significantly higher than in LG astrocytoma (p = 0.007) and oligodendroglioma (p = 0.05). TSPO in gliomas was mostly expressed by neoplastic cells, correlating with BPND in tumor. GAMs accounted for 7.5–44.4% of the total cell density, with only 16.9% of GAMs expressing TSPO. TSPO expression in GAMs did not correlate with BPND. | Tracer kinetics predominantly reflect TSPO+ glioma cells | [40] |
| [123I]CLINDE [18F]FET | n = 3 GBM patients (grade IV) The percentage of overlap between [18F]FET and [123I]CLINDE VOIs was variable (12–42%). VOIs of increased gadolinium-enhanced (Gd-CE) at baseline overlapped to a greater extent with baseline [18F]FET while Gd-CE VOIs at follow-up overlapped to a greater extent with baseline [123I]CLINDE VOIs. | TSPO PET at baseline may predict tumor progression at follow-up. | [41] |
| [18F]GE-180 [18F]FET | n = 20 HGG (9 IDH-wt, 11 IDH-mutant), including n = 8 newly diagnosed and n = 12 recurrent gliomas. IDH-wt gliomas showed a higher median TBRmax in [18F]GE-180 PET compared to IDH-mutant gliomas (median: 5.44 vs. 3.97), without reaching significance (p = 0.08). No difference in [18F]GE-180 TBRmax or BTVGE-180 was observed between newly diagnosed and recurrent HGG. The spatial correlation between BTVGE-180 and BTVFET was only moderate, independently of the IDH mutation. | [18F]GE-180 may be susceptible to the IDH-mutational status | [42] |
| [11C]PBR28 [11C]MET | n = 5 patients with intracranial metastatic lesions. [11C]MET was accurate for detecting tumor regrowth in 7/7 brain metastases, whereas [11C]PBR28 was only accurate in 3/7 lesions. | [11C]PBR28 is not reliable to detect radiation necrosis | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barca, C.; Foray, C.; Zinnhardt, B.; Winkeler, A.; Herrlinger, U.; Grauer, O.M.; Jacobs, A.H. In Vivo Quantitative Imaging of Glioma Heterogeneity Employing Positron Emission Tomography. Cancers 2022, 14, 3139. https://doi.org/10.3390/cancers14133139
Barca C, Foray C, Zinnhardt B, Winkeler A, Herrlinger U, Grauer OM, Jacobs AH. In Vivo Quantitative Imaging of Glioma Heterogeneity Employing Positron Emission Tomography. Cancers. 2022; 14(13):3139. https://doi.org/10.3390/cancers14133139
Chicago/Turabian StyleBarca, Cristina, Claudia Foray, Bastian Zinnhardt, Alexandra Winkeler, Ulrich Herrlinger, Oliver M. Grauer, and Andreas H. Jacobs. 2022. "In Vivo Quantitative Imaging of Glioma Heterogeneity Employing Positron Emission Tomography" Cancers 14, no. 13: 3139. https://doi.org/10.3390/cancers14133139
APA StyleBarca, C., Foray, C., Zinnhardt, B., Winkeler, A., Herrlinger, U., Grauer, O. M., & Jacobs, A. H. (2022). In Vivo Quantitative Imaging of Glioma Heterogeneity Employing Positron Emission Tomography. Cancers, 14(13), 3139. https://doi.org/10.3390/cancers14133139