
PT-112 Induces Mitochondrial Stress and Immunogenic Cell Death, Targeting Tumor Cells with Mitochondrial Deficiencies

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assays
2.3. Cytotoxicity Assays and Cell Death Quantification
2.4. ROS Production and Mitochondrial Membrane Potential Measurement
2.5. Apoptosis and Necroptosis Inhibition Assays
2.6. Analysis of Caspase-3 Activation
2.7. Cyto-ID® Analysis and Autophagosome Formation Measurement
2.8. Calreticulin (CRT) Surface Expression Measurement
2.9. Immunoblot Analysis
2.10. Coenzyme Q Quantification
2.11. Statistical Analysis and Data Processing
3. Results
3.1. Cell Growth Inhibition by PT-112 and Cisplatin in L929, L929dt and Cybrid Cells
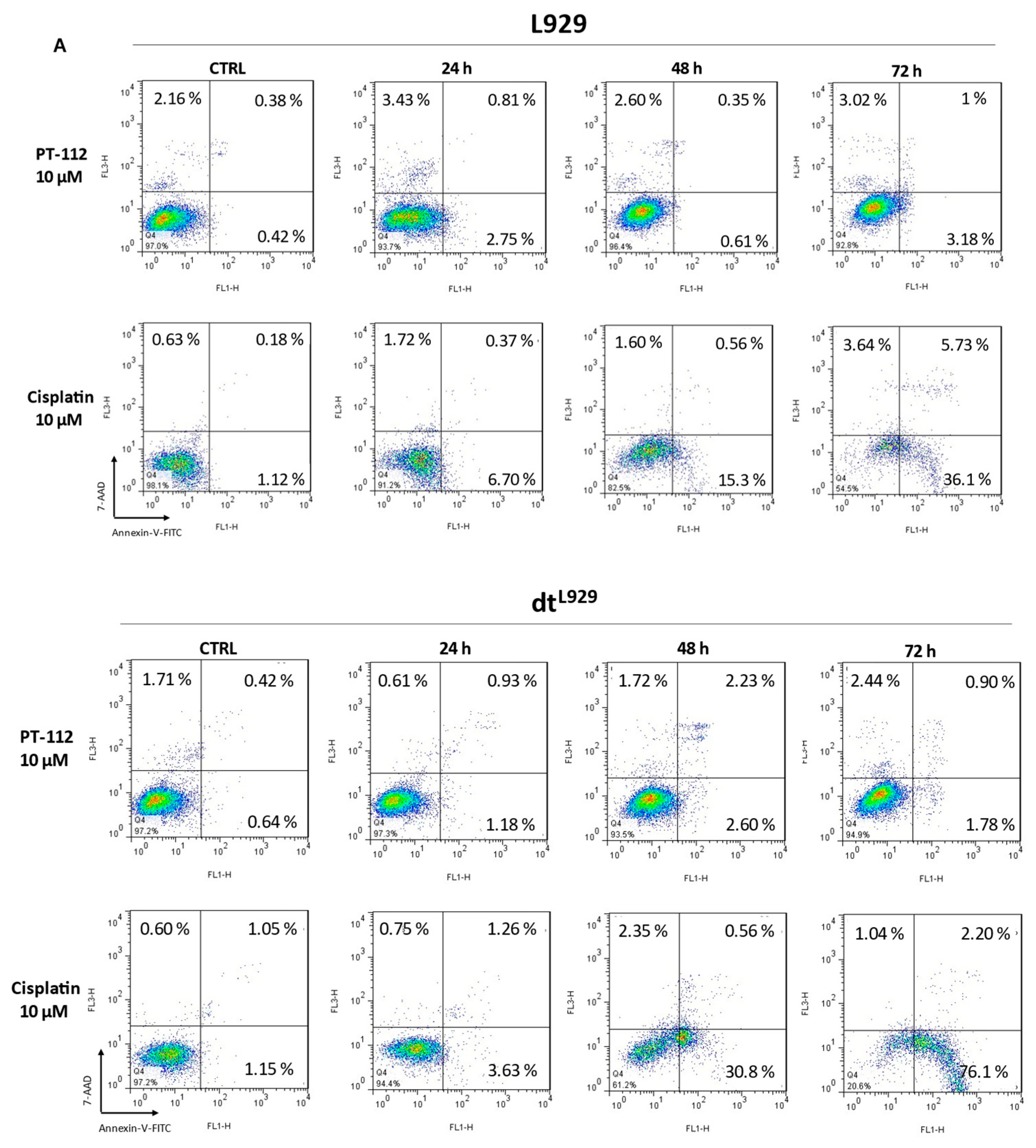
3.2. Cytotoxic Effect of PT-112 and Cisplatin in L929, L929dt and Cybrid Cells and Caspase-3 Activation by PT-112 in Sensitive Cells
3.3. PT-112 Induces Autophagosome Formation
3.4. PT-112 Affects Mitochondrial Membrane Potential and Induces Massive Mitochondrial Reactive Oxygen Species (ROS) Generation in Sensitive Cells
3.5. Effect of PT-112 on Mitochondrial CoQ10 Levels
3.6. PT-112 Induces CRT Exposure on the Surface of Sensitive Cells
3.7. Cells Sensitive to PT-112 Express High Levels of HIF-1α
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bose, R.; Maurmann, L.; Mishur, R.; Yasui, L.; Gupta, S.; Grayburn, W.; Hofstetter, H.; Salley, T. Non-DNA-binding platinum anticancer agents: Cytotoxic activities of platinum-phosphato complexes towards human ovarian cancer cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18314–18319. [Google Scholar] [CrossRef]
- Ames, T.; Slusher, B.; Wozniak, K.; Takase, Y.; Shimizu, H.; Kanada-Sonobe, R.; Kerns, W.; Fong, K.; Pourquier, P.; Nishibata-Kobayashi, K.; et al. Findings across pre-clinical models in the development of PT-112, a novel investigational platinum-pyrophosphate anti-cancer agent. Eur. J. Cancer 2016, 69, S153. [Google Scholar] [CrossRef]
- Bose, R.; Moghaddas, S.; Belkacemi, L.; Tripathi, S.; Adams, N.; Majmudar, P.; McCall, K.; Dezvareh, H.; Nislow, C. Absence of activation of DNA repair genes and excellent efficacy of phosphaplatins against human ovarian cancers: Implications to treat resistant cancers. J. Med. Chem. 2015, 58, 8387–8401. [Google Scholar] [CrossRef]
- Corte-Rodriguez, M.; Espina, M.; Sierra, L.; Blanco, E.; Ames, T.; Montes-Bayon, M.; Sanz-Medel, A. Quantitative evaluation of cellular uptake, DNA incorporation and adduct formation in cisplatin sensitive and resistant cell lines: Comparison of different Pt-containing drugs. Biochem. Pharmacol. 2015, 98, 69–77. [Google Scholar] [CrossRef]
- Ames, T.; Sharik, M.; Rather, G.; Hochart, G.; Bonnel, D.; Linehan, S.; Stauber, J.; Wing, R.; Jimeno, J.; Medina, D.; et al. Translational research of PT-112, a clinical agent in advanced phase i development: Evident bone tropism, synergy in vitro with bortezomib and lenalidomide, and potent efficacy in the Vk*MYC mouse model of multiple myeloma. Blood 2017, 130, 1797. [Google Scholar]
- Moghaddas, S.; Majmudar, P.; Marin, R.; Dezvareh, H.; Qi, C.; Soans, E.; Bose, R. Phosphaplatins, next generation platinum antitumor agents: A paradigm shift in designing and defining molecular targets. Inorg. Chim. Acta 2012, 393, 173–181. [Google Scholar] [CrossRef]
- Yamazaki, T.; Buqué, A.; Ames, T.; Galluzzi, L. PT-112 induces immunogenic cell death and synergizes with immune checkpoint blockers in mouse tumor models. OncoImmunology 2020, 9, e1721810. [Google Scholar] [CrossRef]
- Russell, R.; Watts, N.; Ebetino, F.; Rogers, M. Mechanisms of action of bisphosphonates: Similarities and differences and their potential influence on clinical efficacy. Osteoporos. Int. 2008, 19, 733–759. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kashima, H.; Rahmanto, Y.; Banno, K.; Yu, Y.; Matoba, Y.; Watanabe, K.; Iijima, M.; Takeda, T.; Kunitomi, H.; et al. Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget 2017, 8, 72147–72156. [Google Scholar] [CrossRef]
- Rogers, M.; Crockett, J.; Coxon, F.; Mönkkönen, J. Biochemical and molecular mechanisms of action of bisphosphonates. Bone 2011, 49, 34–41. [Google Scholar] [CrossRef]
- Clézardin, P.; Benzaïd, I.; Croucher, P. Bisphosphonates in preclinical bone oncology. Bone 2011, 49, 66–70. [Google Scholar] [CrossRef] [PubMed]
- van Beek, E.; Pieterman, E.; Cohen, L.; Löwik, C.; Papapoulos, S. Farnesyl Pyrophosphate Synthase Is the Molecular Target of Nitrogen-Containing Bisphosphonates. Biochem. Biophys. Res. Commun. 1999, 264, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Karp, D.; Camidge, D.; Infante, J.; Ames, T.; Jimeno, J.; Bryce, A. A well-tolerated novel immunogenic cell death (ICD) inducer with activity in advanced solid tumors. Ann. Oncol. 2018, 29, viii143. [Google Scholar] [CrossRef]
- Bryce, A.; Dronca, R.; Costello, B.; Infante, J.; Ames, T.; Jimeno, J.; Karp, D. PT-112 in advanced metastatic castrate-resistant prostate cancer (mCRPC), as monotherapy or in combination with PD-L1 inhibitor avelumab: Findings from two phase I studies. J. Clin. Oncol. 2020, 2020, 38. [Google Scholar] [CrossRef]
- Karp, D.; Dronca, R.; Camidge, R.; Costello, B.; Mansfield, A.; Ames, T.; Jimeno, J.; Bryce, A. Phase Ib dose escalation study of novel immunogenic cell death (ICD) inducer PT-112 plus PD-L1 inhibitor avelumab in solid tumours. Ann. Oncol. 2020, 31, S708. [Google Scholar] [CrossRef]
- Kourelis, T.; Ailawadhi, S.; Vogl, D.; Cooper, D.; Ames, T.; Yim, C.; Price, M.; Jimeno, J.; Bergsagel, P. A Phase I Dose Escalation Study of PT-112 in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2020, 136 (Suppl. 1), 9–10. [Google Scholar] [CrossRef]
- Catalán, E.; Charni, S.; Jaime, P.; Aguiló, J.; Enríquez, J.; Naval, J.; Pardo, J.; Villalba, M.; Anel, A. MHC-I modulation due to changes in tumor cell metabolism regulates tumor sensitivity to CTL and NK cells. OncoImmunology 2015, 4, e985924. [Google Scholar] [CrossRef]
- Marco-Brualla, J.; Al-Wasaby, S.; Soler, R.; Romanos, E.; Conde, B.; Justo-Méndez, R.; Enríquez, J.; Fernández-Silva, P.; Martínez-Lostao, L.; Villalba, M.; et al. Mutations in the ND2 subunit of mitochondrial complex I are sufficient to confer increased tumorigenic and metastatic potential to cancer cells. Cancers 2019, 11, 1027. [Google Scholar] [CrossRef]
- Gamen, S.; Anel, A.; Montoya, J.; Marzo, I.; Piñeiro, A.; Naval, J. mtDNA depleted U937 cells are sensitive to TNF- and Fas-induced cytotoxicity. FEBS Lett. 1995, 376, 15–18. [Google Scholar] [CrossRef]
- Loveland, B.E.; Johns, T.G.; Mackay, I.R.; Vaillant, F.; Wang, Z.X.; Hertzog, P.J. Validation of the MTT dye assay for enumeration of cells in proliferative and antiproliferative assays. Biochem. Int. 1992, 27, 501–510. [Google Scholar]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. Embo J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.; Pérez-Martos, A.; Enriquez, J. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Brea-Calvo, G.; Rodríguez-Hernández, A.; Fernández-Ayala, D.; Navas, P.; Sánchez-Alcázar, J. Chemotherapy induces an increase in coenzyme Q10 levels in cancer cell lines. Free Radic. Biol. Med. 2006, 40, 1293–1302. [Google Scholar] [CrossRef]
- Barry, M.; Behnke, C.; Eastman, A. Activation of Programmed Cell Death (Apoptosis) by Cisplatin, Other Anticancer Drugs, Toxins and Hyperthermia. Biochem. Pharmacol. 1990, 40, 2353–2362. [Google Scholar] [CrossRef]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef]
- Chan, L.; Shen, D.; Wilkinson, A.; Patton, W.; Lai, N.; Chan, E.; Kuksin, D.; Lin, B.; Qiu, J. A novel image-based cytometry method for autophagy detection in living cells. Autophagy 2012, 8, 1371–1382. [Google Scholar] [CrossRef]
- Gamen, S.; Anel, A.; Pérez-Galán, P.; Lasierra, P.; Johnson, D.; Piñeiro, A.; Naval, J. Doxorubicin treatment activates a Z-VAD-sensitive caspase, which causes ∆Ψm loss, caspase-9 activity, and apoptosis in Jurkat cells. Exp. Cell Res. 2000, 258, 223–235. [Google Scholar] [CrossRef]
- Aguiló, J.I.; Anel, A.; Catalán, E.; Sebastián, A.; Acín-Pérez, R.; Naval, J.; Wallich, R.; Simon, M.M.; Pardo, J. Granzyme B of cytotoxic T cells induces extramitochondrial reactive oxygen species production via caspase-dependent NADPH oxidase activation. Immunol. Cell Biol. 2010, 88, 545–554. [Google Scholar] [CrossRef]
- Gruenbacher, G.; Thurnher, M. Mevalonate metabolism governs cancer immune surveillance. OncoImmunology 2017, 6, e1342917. [Google Scholar] [CrossRef]
- Tricarico, P.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067–16084. [Google Scholar] [CrossRef]
- Semenza, G. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Patra, K.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef]
- Bathaie, S.; Ashrafi, M.; Azizian, M.; Tamanoi, F. Mevalonate Pathway and Human Cancers. Curr. Mol. Pharmacol. 2017, 10, 77–85. [Google Scholar]
- Antoku, K.; Liu, Z.; Johnson, D. Inhibition of caspase proteases by CrmA enhances the resistance of human leukemic cells to multiple chemotherapeutic agents. Leukemia 1997, 11, 1665–1672. [Google Scholar] [CrossRef]
- Farrell, K.; Karpeisky, A.; Thamm, D.; Zinnen, S. Bisphosphonate conjugation for bone specific drug targeting. Bone Rep. 2017, 9, 47–60. [Google Scholar] [CrossRef]
- Qiu, L.; Yang, H.; Lv, G.; Li, K.; Liu, G.; Wang, W.; Wang, S.; Zhao, X.; Xie, M.; Lin, J. Insights into the mevalonate pathway in the anticancer effect of a platinum complex on human gastric cancer cells. Eur. J. Pharmacol. 2017, 810, 120–127. [Google Scholar] [CrossRef]
- Todenhöfer, T.; Hennenlotter, J.; Kühs, U.; Gerber, V.; Gakis, G.; Vogel, U.; Aufderklamm, S.; Merseburger, A.; Knapp, J.; Stenzl, A.; et al. Altered expression of farnesyl pyrophosphate synthase in prostate cancer: Evidence for a role of the mevalonate pathway in disease progression? World J. Urol. 2013, 31, 345–350. [Google Scholar] [CrossRef]
- Kepp, O.; Kroemer, G. Autophagy induction by thiostrepton for the improvement of anticancer therapy. Autophagy 2020, 16, 1166–1167. [Google Scholar] [CrossRef]
- Liparulo, I.; Bergamini, C.; Bortolus, M.; Calonghi, N.; Gasparre, G.; Kurelac, I.; Masin, L.; Rizzardi, N.; Rugolo, M.; Wang, W.; et al. Coenzyme Q biosynthesis inhibition induces HIF-1α stabilization and metabolic switch toward glycolysis. FEBS J. 2021, 288, 1956–1974. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soler-Agesta, R.; Marco-Brualla, J.; Minjárez-Sáenz, M.; Yim, C.Y.; Martínez-Júlvez, M.; Price, M.R.; Moreno-Loshuertos, R.; Ames, T.D.; Jimeno, J.; Anel, A. PT-112 Induces Mitochondrial Stress and Immunogenic Cell Death, Targeting Tumor Cells with Mitochondrial Deficiencies. Cancers 2022, 14, 3851. https://doi.org/10.3390/cancers14163851
Soler-Agesta R, Marco-Brualla J, Minjárez-Sáenz M, Yim CY, Martínez-Júlvez M, Price MR, Moreno-Loshuertos R, Ames TD, Jimeno J, Anel A. PT-112 Induces Mitochondrial Stress and Immunogenic Cell Death, Targeting Tumor Cells with Mitochondrial Deficiencies. Cancers. 2022; 14(16):3851. https://doi.org/10.3390/cancers14163851
Chicago/Turabian StyleSoler-Agesta, Ruth, Joaquín Marco-Brualla, Martha Minjárez-Sáenz, Christina Y. Yim, Marta Martínez-Júlvez, Matthew R. Price, Raquel Moreno-Loshuertos, Tyler D. Ames, José Jimeno, and Alberto Anel. 2022. "PT-112 Induces Mitochondrial Stress and Immunogenic Cell Death, Targeting Tumor Cells with Mitochondrial Deficiencies" Cancers 14, no. 16: 3851. https://doi.org/10.3390/cancers14163851
APA StyleSoler-Agesta, R., Marco-Brualla, J., Minjárez-Sáenz, M., Yim, C. Y., Martínez-Júlvez, M., Price, M. R., Moreno-Loshuertos, R., Ames, T. D., Jimeno, J., & Anel, A. (2022). PT-112 Induces Mitochondrial Stress and Immunogenic Cell Death, Targeting Tumor Cells with Mitochondrial Deficiencies. Cancers, 14(16), 3851. https://doi.org/10.3390/cancers14163851