
HSF1 Stimulates Glutamine Transport by Super-Enhancer-Driven lncRNA LINC00857 in Colorectal Cancer


Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Antibodies, Reagents, and Kits
2.3. Tissue Samples
2.4. CCK8 Assays and Colony Formation Assays
2.5. siRNA and shRNA Transfection
2.6. RNA Isolation, Reverse Transcription, and Quantitative PCR
2.7. Western Blot and Immunohistochemistry
2.8. Fluorescence In Situ Hybridization (FISH)
2.9. Luciferase Activity Assays
2.10. Chromatin Immunoprecipitation (ChIP) and Sequencing Analysis (ChIP-Seq)
2.11. lncRNA Microarray Analysis
2.12. RNA Immunoprecipitation Assays (RIP)
2.13. Amino Acid Metabolism Analysis
2.14. Xenograft Model
2.15. AOM/DSS Model
2.16. Statistical Analysis
3. Results
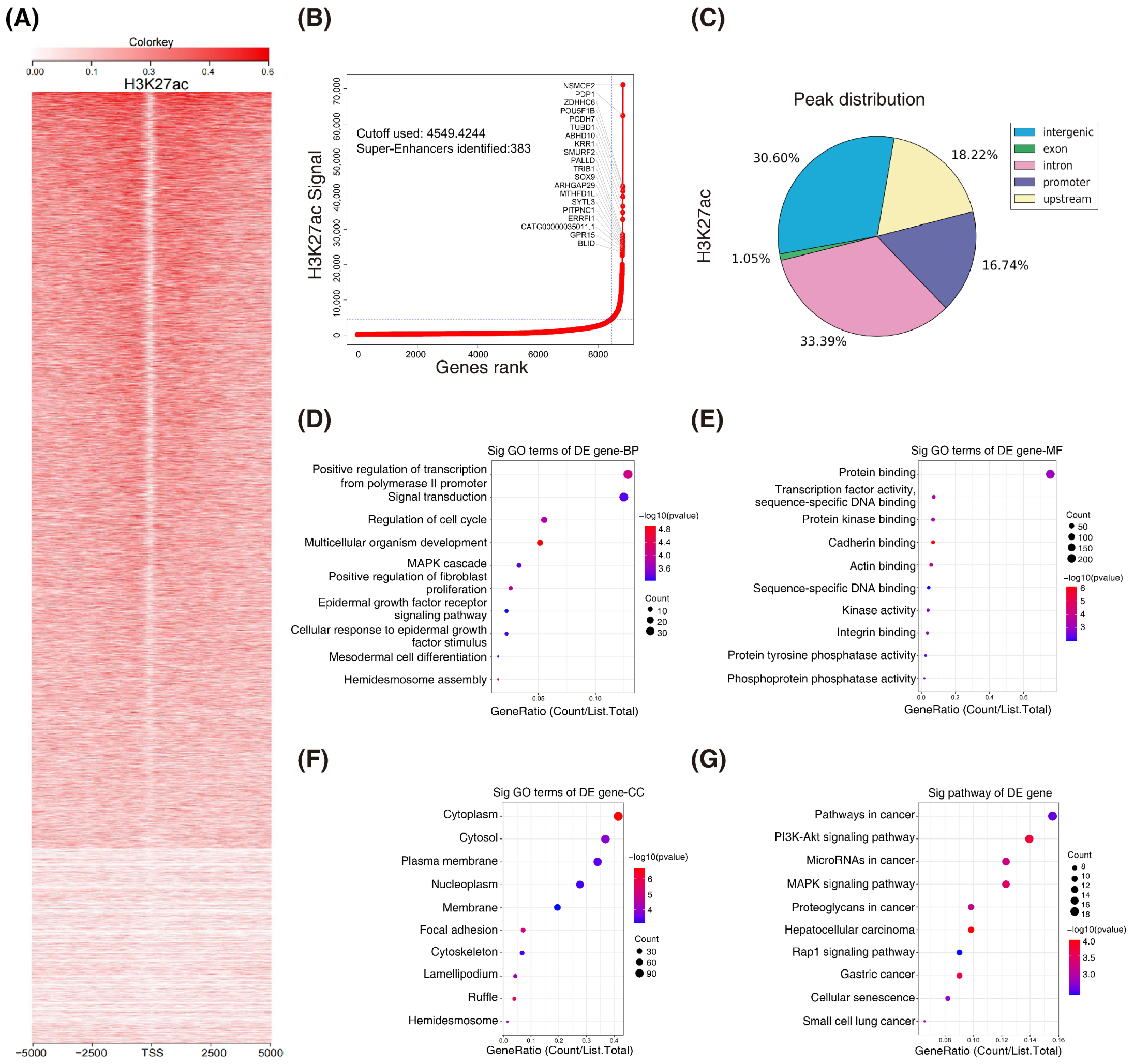
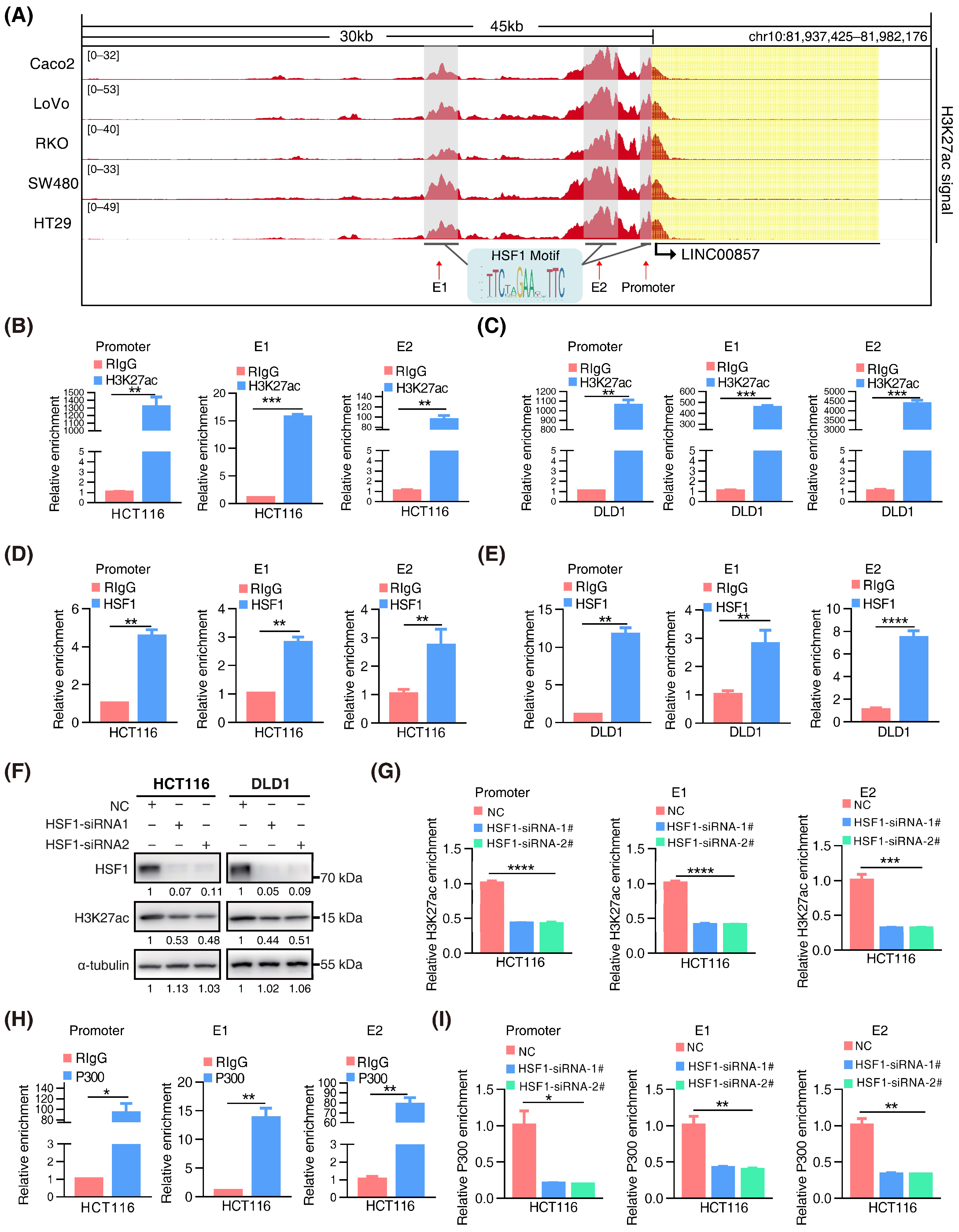
3.1. H3K27ac Landscape in Colorectal Cancer
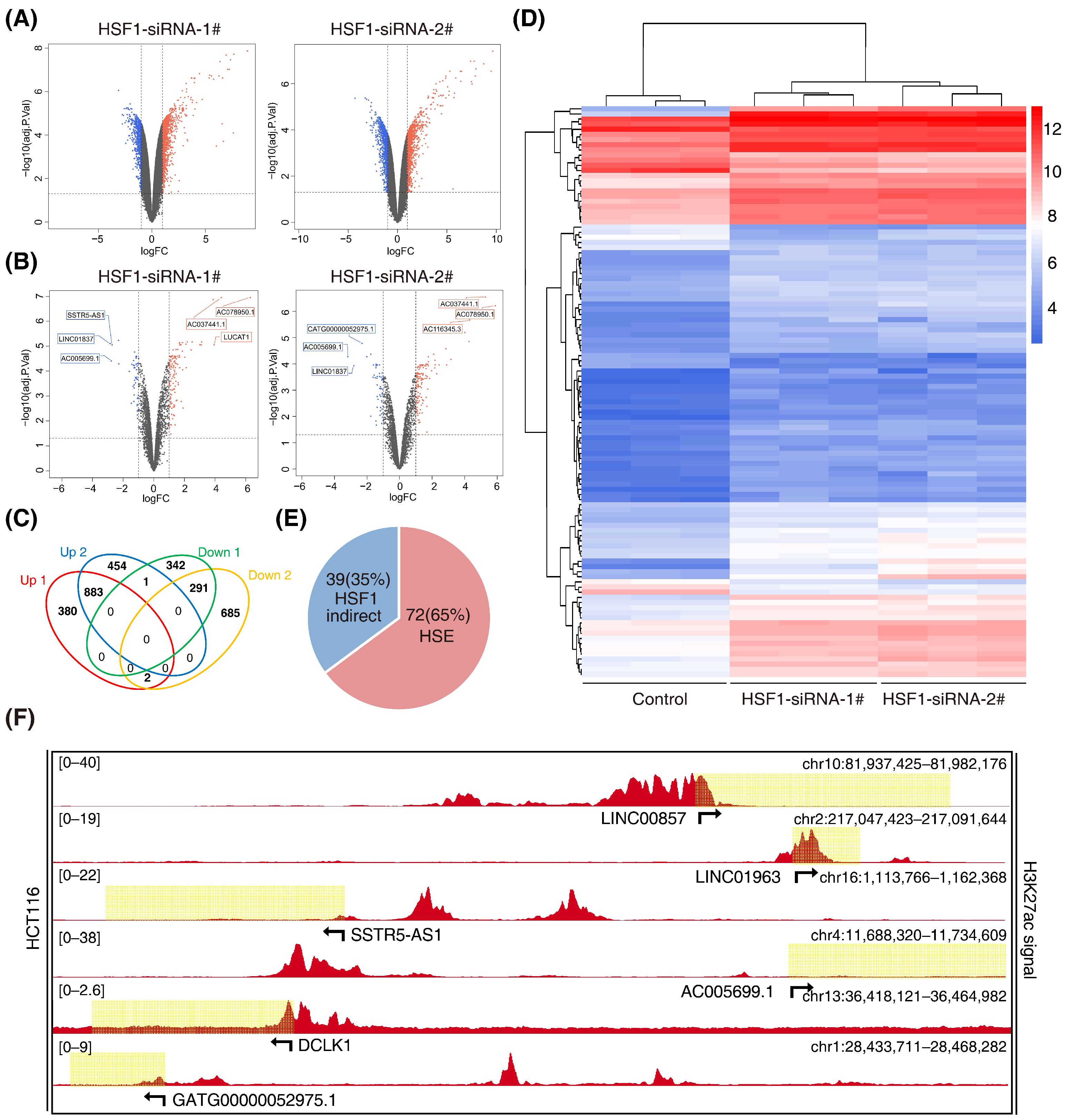
3.2. Screening for HSF1-Associated lncRNAs
3.3. HSF1 Activates the Transcription of LINC00857 via Promoting Its Super-Enhancer Activity
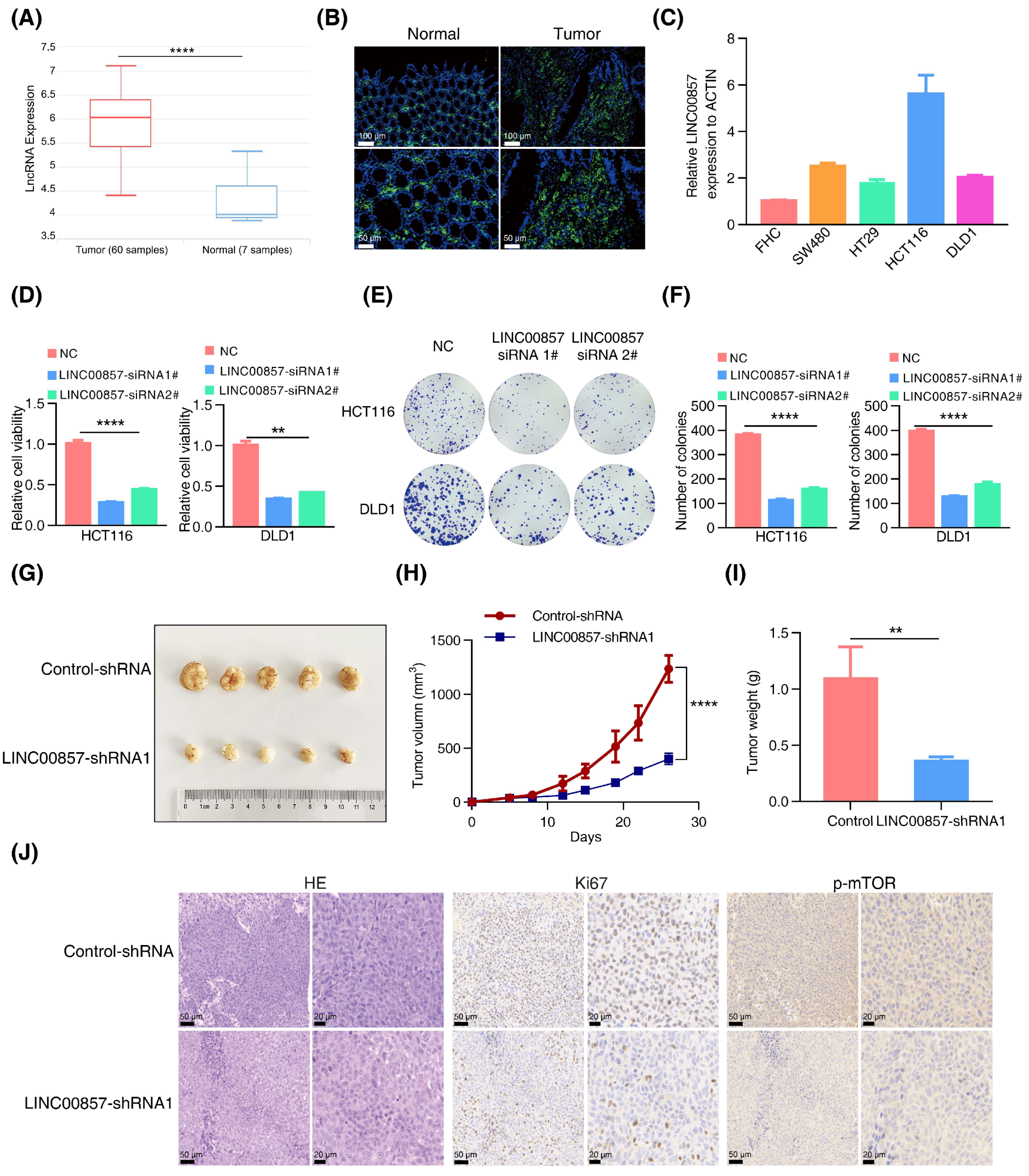
3.4. LINC00857 Is Upregulated and Beneficial for CRC Carcinogenesis
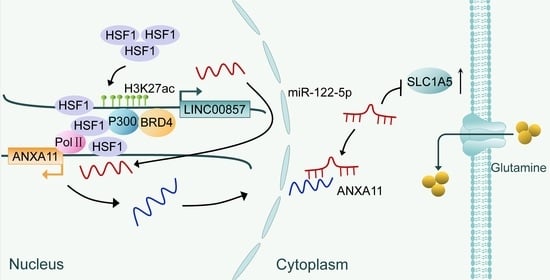
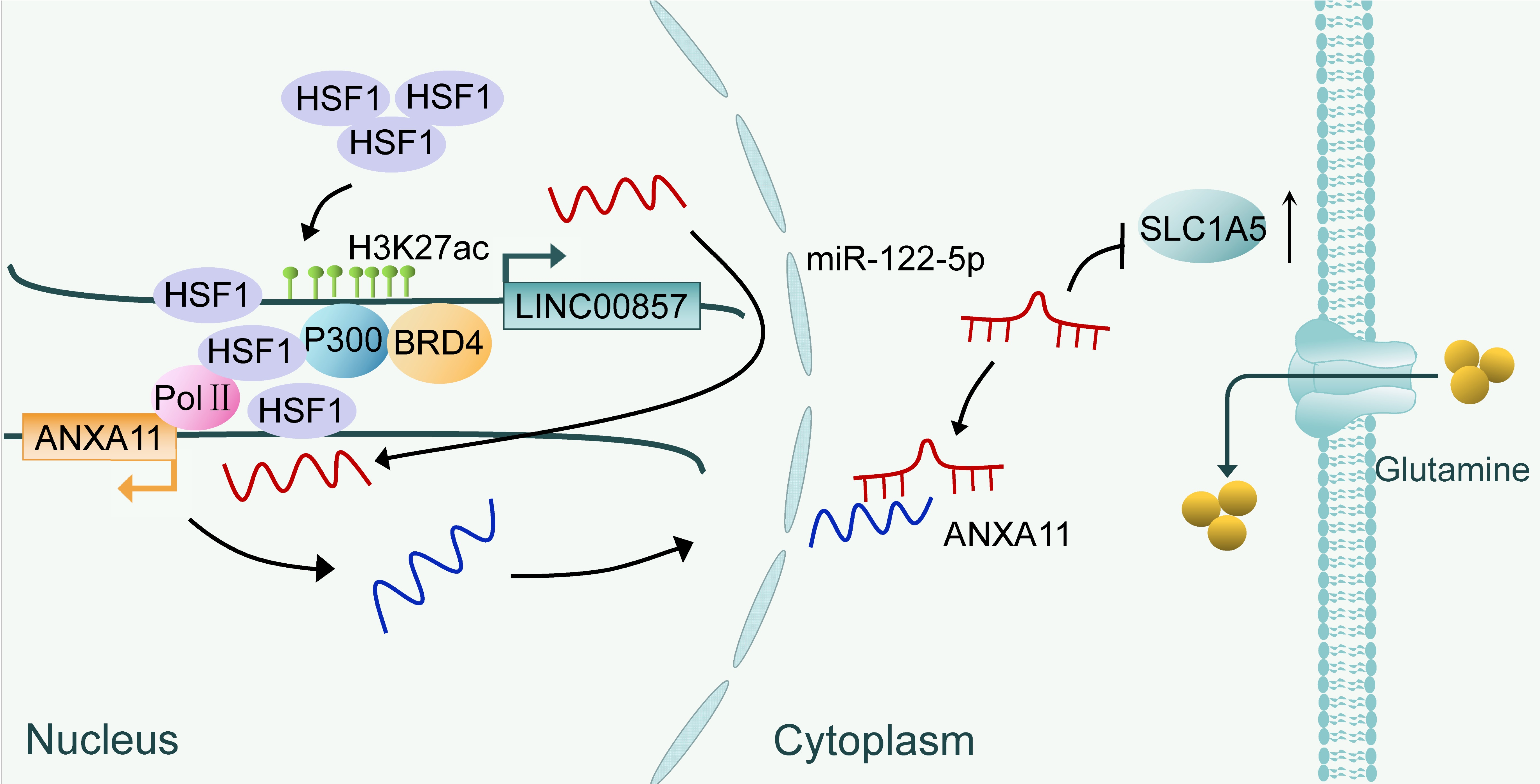
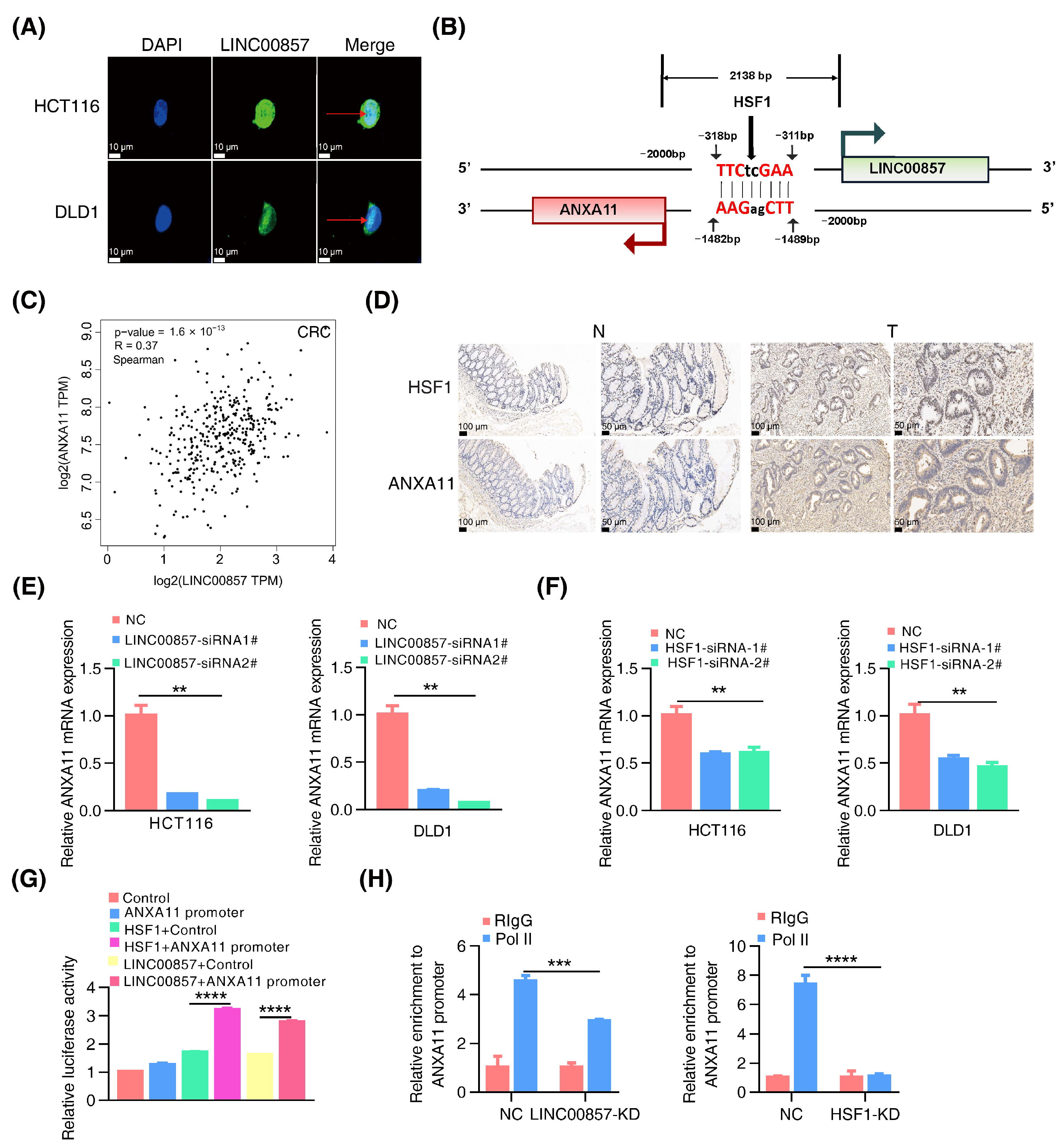
3.5. The HSF1/LINC00857 Axis Promotes the Transcription of ANXA11
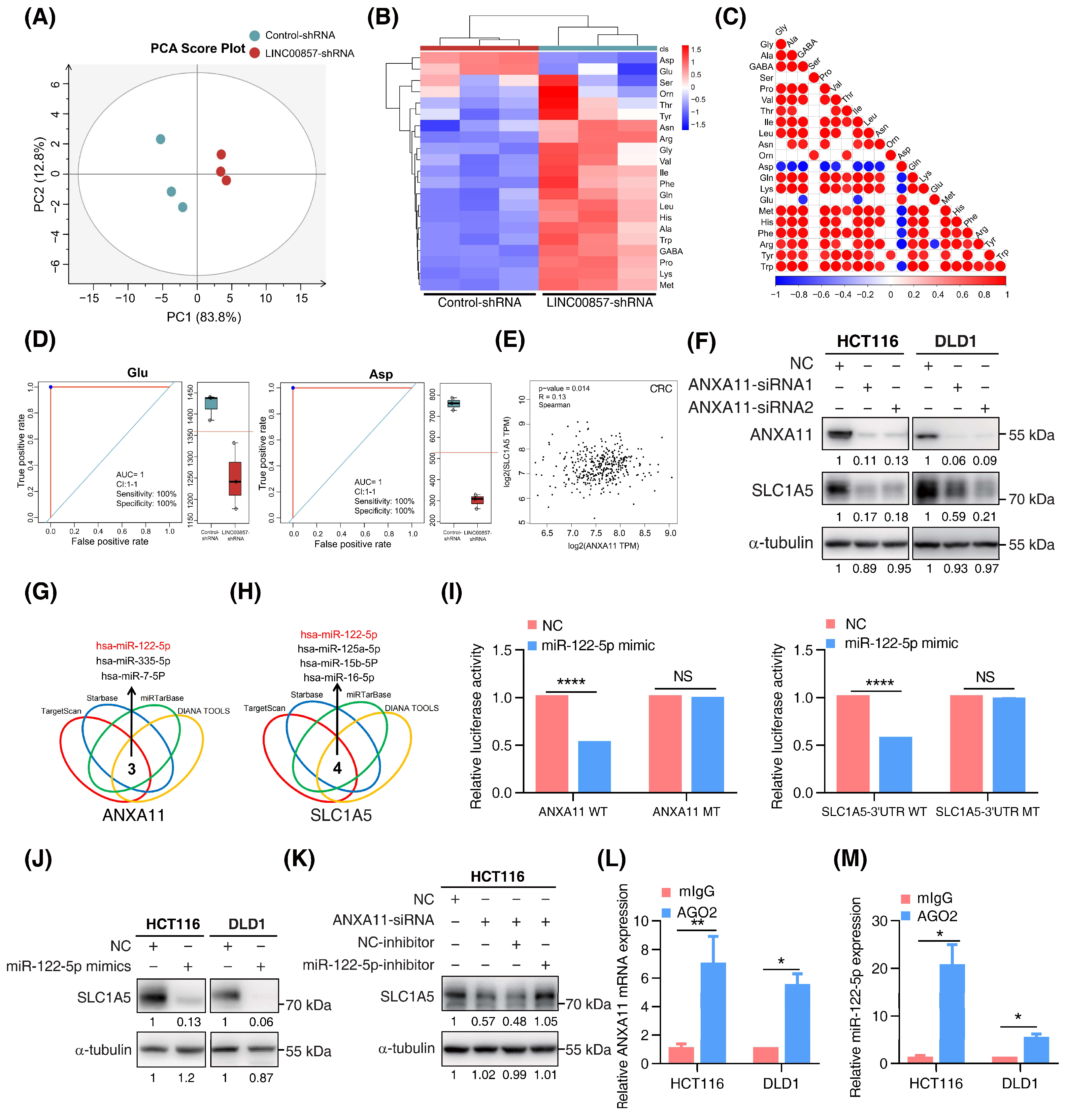
3.6. The LINC00857/ANXA11 Axis Promotes SLC1A5-Mediated Glutamine Transport
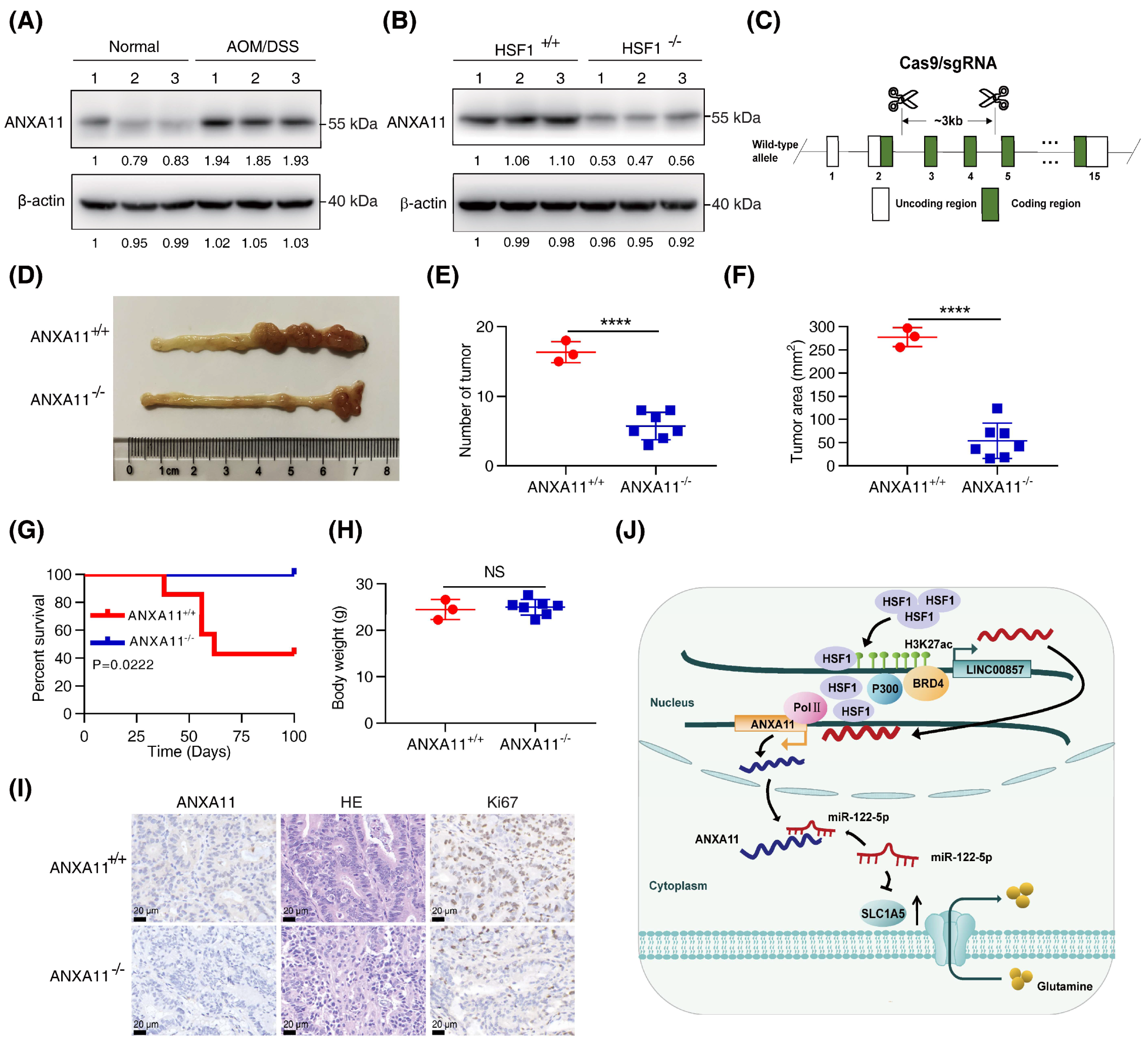
3.7. Knockout of ANXA11 Attenuated CRC Carcinogenesis In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sears, C.L.; Garrett, W.S. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014, 15, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; George, R.E. Super-Enhancer-Driven Transcriptional Dependencies in Cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Bannister, A.J. Re-place your BETs: The dynamics of super enhancers. Mol. Cell 2014, 56, 187–189. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, N.; Banito, A.; Roe, J.S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.H.; Aksoy, O.; Bolden, J.E.; Chen, C.C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Kutschat, A.P.; Yamada, M.; Prokakis, E.; Bottcher, P.; Tanaka, K.; Doki, Y.; Hamdan, F.H.; Johnsen, S.A. Bromodomain protein BRDT directs DeltaNp63 function and super-enhancer activity in a subset of esophageal squamous cell carcinomas. Cell Death Differ. 2021, 28, 2207–2220. [Google Scholar] [CrossRef]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirguet, O.; Gosmini, R.; Toum, J.; Clement, C.A.; Barnathan, M.; Brusq, J.M.; Mordaunt, J.E.; Grimes, R.M.; Crowe, M.; Pineau, O.; et al. Discovery of epigenetic regulator I-BET762: Lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013, 56, 7501–7515. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [Green Version]
- Fernando, T.M.; Marullo, R.; Pera Gresely, B.; Phillip, J.M.; Yang, S.N.; Lundell-Smith, G.; Torregroza, I.; Ahn, H.; Evans, T.; Gyorffy, B.; et al. BCL6 Evolved to Enable Stress Tolerance in Vertebrates and Is Broadly Required by Cancer Cells to Adapt to Stress. Cancer Discov. 2019, 9, 662–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janus, P.; Toma-Jonik, A.; Vydra, N.; Mrowiec, K.; Korfanty, J.; Chadalski, M.; Widlak, P.; Dudek, K.; Paszek, A.; Rusin, M.; et al. Pro-death signaling of cytoprotective heat shock factor 1: Upregulation of NOXA leading to apoptosis in heat-sensitive cells. Cell Death Differ. 2020, 27, 2280–2292. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Kryczek, I.; Nam, J.; Li, X.; Li, S.; Li, J.; Wei, S.; Grove, S.; Vatan, L.; Zhou, J.; et al. LIMIT is an immunogenic lncRNA in cancer immunity and immunotherapy. Nat. Cell Biol. 2021, 23, 526–537. [Google Scholar] [CrossRef]
- Yang, T.; Ren, C.; Lu, C.; Qiao, P.; Han, X.; Wang, L.; Wang, D.; Lv, S.; Sun, Y.; Yu, Z. Phosphorylation of HSF1 by PIM2 Induces PD-L1 Expression and Promotes Tumor Growth in Breast Cancer. Cancer Res. 2019, 79, 5233–5244. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Shi, Q.; Xu, W.; Zhou, Q.; Shi, R.; Ma, Y.; Chen, D.; Zhu, L.; Feng, L.; Cheng, A.S.; et al. Metabolic enzyme PDK3 forms a positive feedback loop with transcription factor HSF1 to drive chemoresistance. Theranostics 2019, 9, 2999–3013. [Google Scholar] [CrossRef]
- Jin, X.; Moskophidis, D.; Mivechi, N.F. Heat shock transcription factor 1 is a key determinant of HCC development by regulating hepatic steatosis and metabolic syndrome. Cell Metab. 2011, 14, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Song, P.; Jiang, T.; Dai, D.; Wang, H.; Sun, J.; Zhu, L.; Xu, W.; Feng, L.; Shin, V.Y.; et al. Heat Shock Factor 1 Epigenetically Stimulates Glutaminase-1-Dependent mTOR Activation to Promote Colorectal Carcinogenesis. Mol. Ther. 2018, 26, 1828–1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, L.; Eeckhoute, J.; Lupien, M. Pioneer factors: Directing transcriptional regulators within the chromatin environment. Trends Genet. 2011, 27, 465–474. [Google Scholar] [CrossRef]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Feng, L.; Li, J.; Dai, D.; Zhu, L.; Wang, C.; Li, J.; Li, L.; Zhou, Q.; Shi, R.; et al. beta-catenin represses miR455-3p to stimulate m6A modification of HSF1 mRNA and promote its translation in colorectal cancer. Mol. Cancer 2020, 19, 129. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [Green Version]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Xu, Q.; Liu, M.; Hu, H.; Xie, Y.; Zuo, Z.; Ren, J. lnCAR: A Comprehensive Resource for lncRNAs from Cancer Arrays. Cancer Res. 2019, 79, 2076–2083. [Google Scholar] [CrossRef] [Green Version]
- Lanczky, A.; Gyorffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J Med Internet Res 2021, 23, e27633. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [Green Version]
- Narita, T.; Ito, S.; Higashijima, Y.; Chu, W.K.; Neumann, K.; Walter, J.; Satpathy, S.; Liebner, T.; Hamilton, W.B.; Maskey, E.; et al. Enhancers are activated by p300/CBP activity-dependent PIC assembly, RNAPII recruitment, and pause release. Mol. Cell 2021, 81, 2166–2182 e6. [Google Scholar] [CrossRef]
- Kahya, U.; Koseer, A.S.; Dubrovska, A. Amino Acid Transporters on the Guard of Cell Genome and Epigenome. Cancers 2021, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, B.; Song, J.; Yu, S.; Li, Y.; Su, H.; He, S. Lnc-PDZD7 contributes to stemness properties and chemosensitivity in hepatocellular carcinoma through EZH2-mediated ATOH8 transcriptional repression. J. Exp. Clin. Cancer Res. 2019, 38, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghini, F.; Rubolino, C.; Climent, M.; Simeone, I.; Marzi, M.J.; Nicassio, F. Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat. Commun. 2018, 9, 3119. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Zhang, L.; Ma, X.; Li, J.; Lu, Z. Integrated bioinformatics and experiments reveal the roles and driving forces for HSF1 in colorectal cancer. Bioengineered 2022, 13, 2536–2552. [Google Scholar] [CrossRef]
- Chen, F.; Fan, Y.; Cao, P.; Liu, B.; Hou, J.; Zhang, B.; Tan, K. Pan-Cancer Analysis of the Prognostic and Immunological Role of HSF1: A Potential Target for Survival and Immunotherapy. Oxid. Med. Cell Longev. 2021, 2021, 5551036. [Google Scholar] [CrossRef]
- Levi-Galibov, O.; Lavon, H.; Wassermann-Dozorets, R.; Pevsner-Fischer, M.; Mayer, S.; Wershof, E.; Stein, Y.; Brown, L.E.; Zhang, W.; Friedman, G.; et al. Heat Shock Factor 1-dependent extracellular matrix remodeling mediates the transition from chronic intestinal inflammation to colon cancer. Nat. Commun. 2020, 11, 6245. [Google Scholar] [CrossRef]
- Reinfeld, B.I.; Madden, M.Z.; Wolf, M.M.; Chytil, A.; Bader, J.E.; Patterson, A.R.; Sugiura, A.; Cohen, A.S.; Ali, A.; Do, B.T.; et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 2021, 593, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Chen, J.; Zhang, X.; Wang, Z.; Chen, J.; Lin, X.; Huang, H.; Fu, W.; Liang, J.; Wu, W.; et al. The HIF-1alpha antisense long non-coding RNA drives a positive feedback loop of HIF-1alpha mediated transactivation and glycolysis. Nat. Commun. 2021, 12, 1341. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.J.; Jiang, Y.Y.; Jiang, Y.; Li, C.Q.; Lim, M.C.; An, O.; Mayakonda, A.; Ding, L.W.; Long, L.; Sun, C.; et al. Super-Enhancer-Driven Long Non-Coding RNA LINC01503, Regulated by TP63, Is Over-Expressed and Oncogenic in Squamous Cell Carcinoma. Gastroenterology 2018, 154, 2137–2151.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, A.; Singhal, U.; Chinnaiyan, A.M. Long noncoding RNAs in cancer: From function to translation. Trends Cancer 2015, 1, 93–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.; Cui, Y.; Liang, X.; Han, D.; Zheng, X.; Wu, A.; Qian, L. Long Noncoding RNA LINC00857 Promotes Proliferation, Migration, and Invasion of Colorectal Cancer Cell through miR-1306/Vimentin Axis. Comput. Math. Methods Med. 2021, 2021, 5525763. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, C.; Liu, S.; Qi, H.; Yin, Y.; Liang, R.; Sun, M.Z.; Greenaway, F.T. Annexin A11 in disease. Clin. Chim. Acta. 2014, 431, 164–168. [Google Scholar] [CrossRef]
- Huang, X.; Luo, Y.; Li, X. Circ_0072995 Promotes Ovarian Cancer Progression Through Regulating miR-122-5p/SLC1A5 Axis. Biochem. Genet. 2022, 60, 153–172. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Q.; Wang, R.; Liu, X.; Song, P.; Zheng, M.; Ren, X.; Ma, J.; Lu, Z.; Li, J. HSF1 Stimulates Glutamine Transport by Super-Enhancer-Driven lncRNA LINC00857 in Colorectal Cancer. Cancers 2022, 14, 3855. https://doi.org/10.3390/cancers14163855
Shen Q, Wang R, Liu X, Song P, Zheng M, Ren X, Ma J, Lu Z, Li J. HSF1 Stimulates Glutamine Transport by Super-Enhancer-Driven lncRNA LINC00857 in Colorectal Cancer. Cancers. 2022; 14(16):3855. https://doi.org/10.3390/cancers14163855
Chicago/Turabian StyleShen, Qi, Rui Wang, Xinling Liu, Ping Song, Mingzhu Zheng, Xiaomin Ren, Jingang Ma, Zhong Lu, and Jiaqiu Li. 2022. "HSF1 Stimulates Glutamine Transport by Super-Enhancer-Driven lncRNA LINC00857 in Colorectal Cancer" Cancers 14, no. 16: 3855. https://doi.org/10.3390/cancers14163855
APA StyleShen, Q., Wang, R., Liu, X., Song, P., Zheng, M., Ren, X., Ma, J., Lu, Z., & Li, J. (2022). HSF1 Stimulates Glutamine Transport by Super-Enhancer-Driven lncRNA LINC00857 in Colorectal Cancer. Cancers, 14(16), 3855. https://doi.org/10.3390/cancers14163855