1. Background
Bladder cancer (BCa) ranks 12th in cancer incidence globally, with nearly 570,000 new cases each year and 13th in terms of deaths [
1]. A strong male predominance is observed in bladder cancer, where three-fourths of cases occur in men [
2]. The increased risk for bladder cancer correlates to factors including age, smoking, exposure to some industrial chemicals and hormonal differences, particularly androgens [
3]. The treatment of bladder cancer depends on stage and grade to a great extent, which are also strongly associated with the prognosis of patients: the treatment of non-muscle invasive bladder cancer is usually through resection and immunotherapy with intravesical drugs such as Bacillus Calmette-Guerin, whilst more aggressive methods, such as radical cystectomy coupled with chemotherapy, are necessary for muscle invasive bladder cancer [
4]. Unfortunately, little improvement has been achieved in the treatment for bladder cancer with a flat 5-year survival rate until recently [
5]. Therefore, it is urgent to get a deeper understanding of molecular mechanisms underlying bladder cancer, thus exploring novel targets for bladder cancer treatment.
It is well-established that long noncoding RNA small nucleolar RNA host gene 1 (lncRNA SNHG1) is involved in tumor stage, size and overall survival [
6]. The oncogenic role of SNHG1 has been elucidated in various cancers. For instance, a prior study also reported the tumor-promoting potential of SNHG1 in pancreatic cancer with the results that SNHG1 silencing triggered repression of cell proliferative, metastatic, and invasive capacities by inactivating Notch-1 pathway [
7]. In addition, Li et al. observed the suppressive effect of SNHG1 on prostate cancer development by promoting cell proliferation [
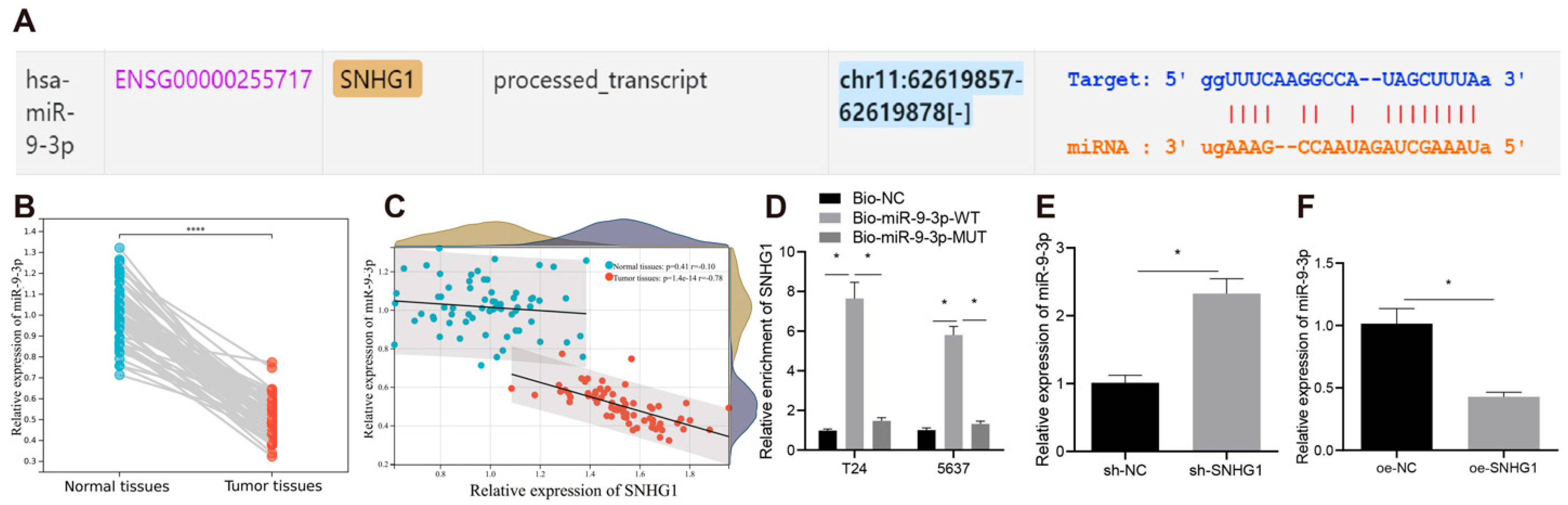
8]. These findings indirectly support the possibility that SNHG1 might promote development of bladder cancer. Moreover, the starBase website used in our study predicted a binding relationship between SNHG1 and microRNA-9-3p (miR-9-3p).
MiR-9-3p (previously known as miR-9) is widely known for its altered expression and function in multiple diseases, such as Huntington’s disease and cancers [
9]. Interestingly, it was detected that ectopically expressed miR-9-3p possessed antitumor potential in bladder cancer by diminishing cell invasion, migration, and proliferation [
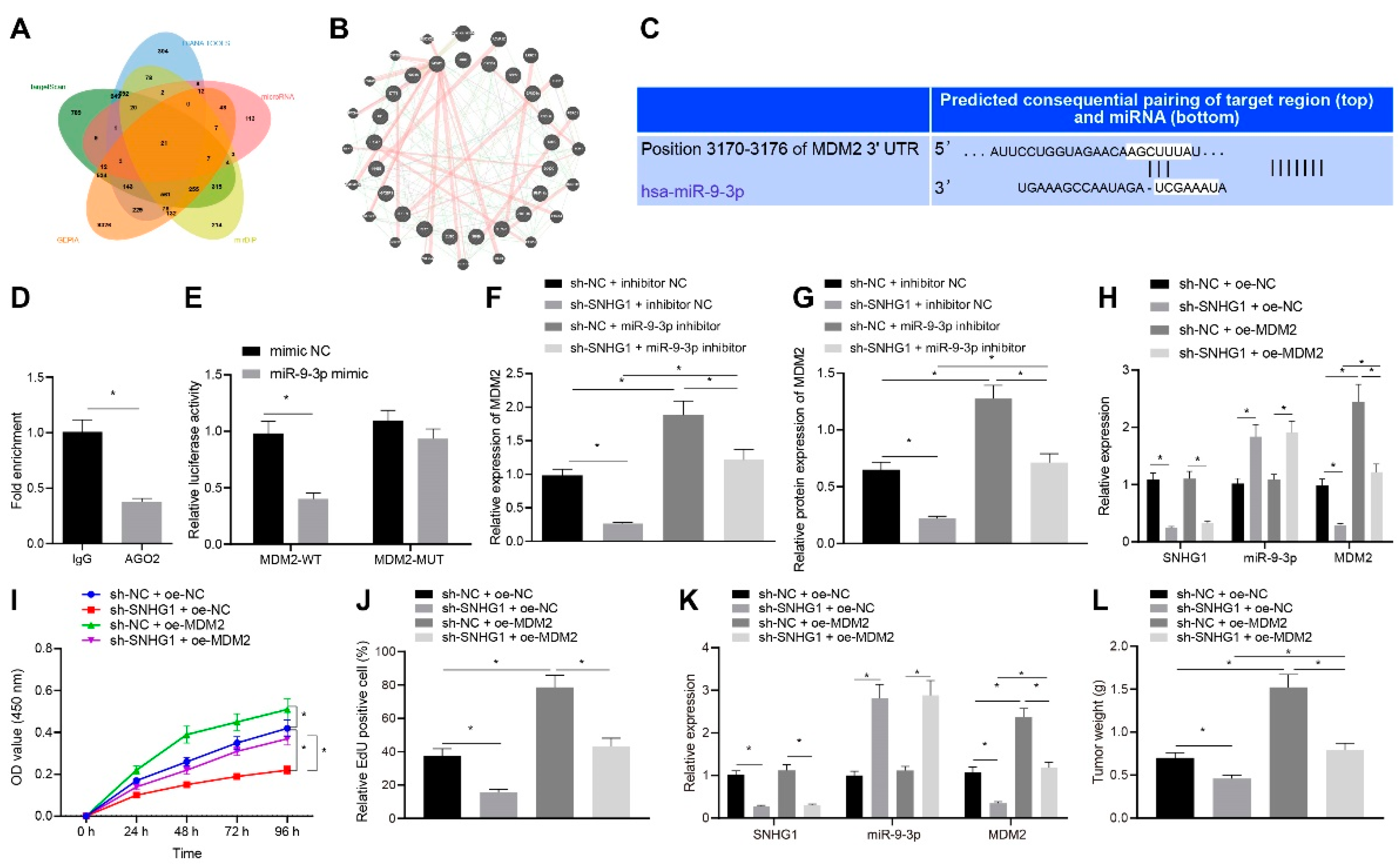
10]. In our study, the binding sites between miR-9-3p and 3′ untranslated region (UTR) of murine double minute 2 (MDM2) were predicted by TargetScan. Overexpression of MDM2 could reportedly neutralize the depressive effect of miR-379-5p on bladder cancer cell proliferative, migratory and invasive capacities [
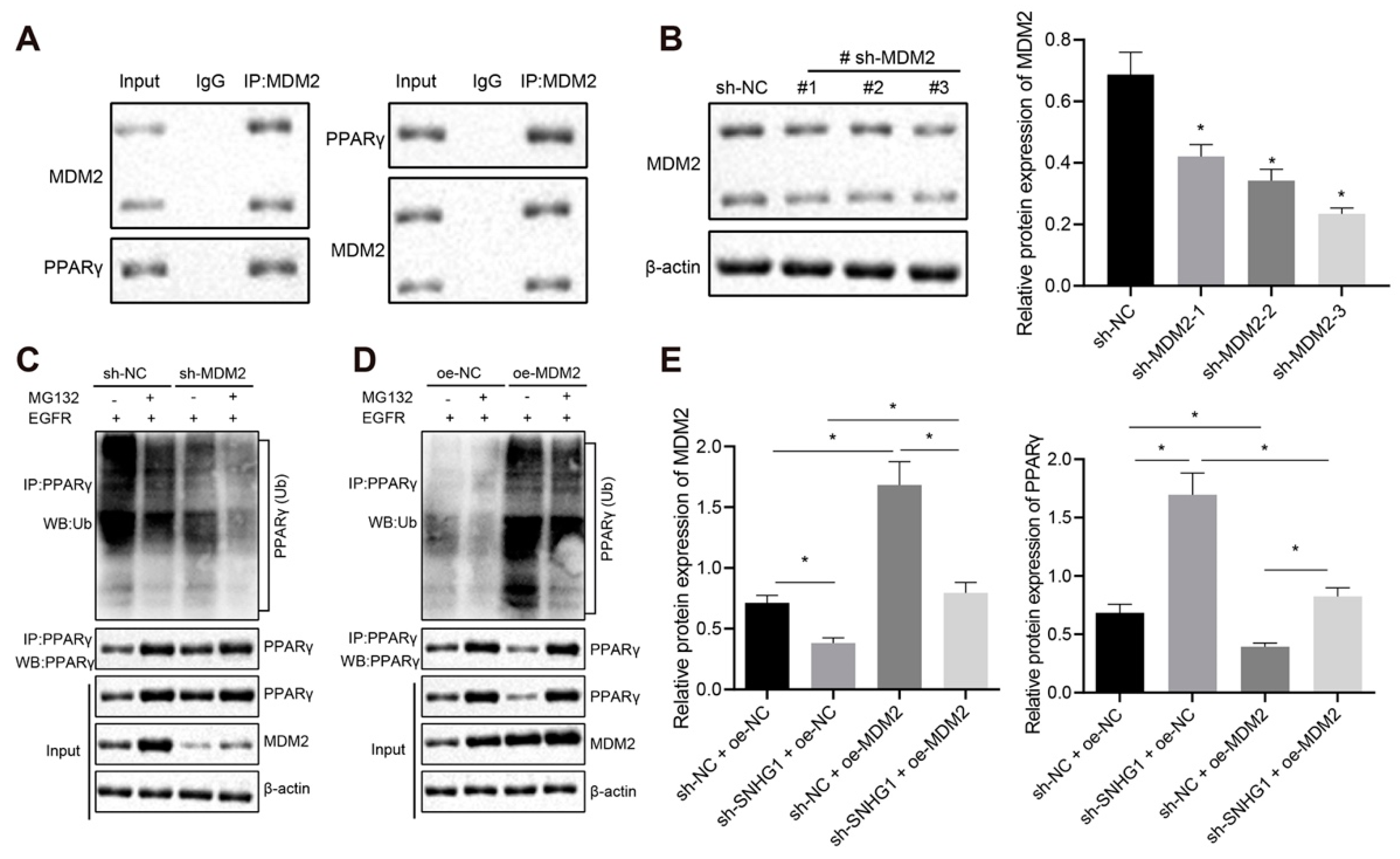
11]. In the presence of EGFR, MDM2 can bind to peroxisome proliferator-activated receptor-gamma (PPARγ) and regulate the ubiquitination of PPARγ protein in colon cancer cells [
12]. More importantly, it was elaborated in a prior study that an antagonist of PPARγ promoted cell cycle entry and decreased cell apoptosis in bladder cancer [
13].
Taken these findings into account, we hypothesized that the SNHG1/miR-9-3p/MDM2/PPARγ axis correlated to the progression of bladder cancer. Therefore, the present study was implemented by focusing on the alteration in SNHG1 expression and function in bladder cancer. Moreover, we investigated the underlying mechanism of SNHG1 in the bladder cancer process via miR-9-3p/MDM2/PPARγ axis.
2. Methods
2.1. Compliance with Ethical Standards
The experiments involving 67 human being samples were approved with ratification of the Ethics Committee of Nanjing Medical University (NO. 402, 2021) by conforming to the principles outlined in the Declaration of Helsinki. Ethical agreements were obtained from the donors or their families through written informed consent. Animal experiments (20 healthy nude mice aged 6–8 weeks) were ratified by the Animal Ethics Committee of Nanjing Medical University (No. 402, 2021) and concurred with the Guidelines for Animal Experiments of Peking University Health Science Center.
2.2. Patients and Tissue Samples
Sixty-seven patients diagnosed with bladder cancer undergoing radical cystectomy in Suqian First Hospital and Jiangsu Cancer Hospital from June 2016 to June 2017 were enrolled. Fresh bladder cancer tissues and corresponding adjacent normal tissues were preserved in liquid nitrogen immediately after resection. None of patients received preoperative radiotherapy, chemotherapy or immunotherapy. Follow-up information was obtained from outpatient clinics and regular telephone interviews.
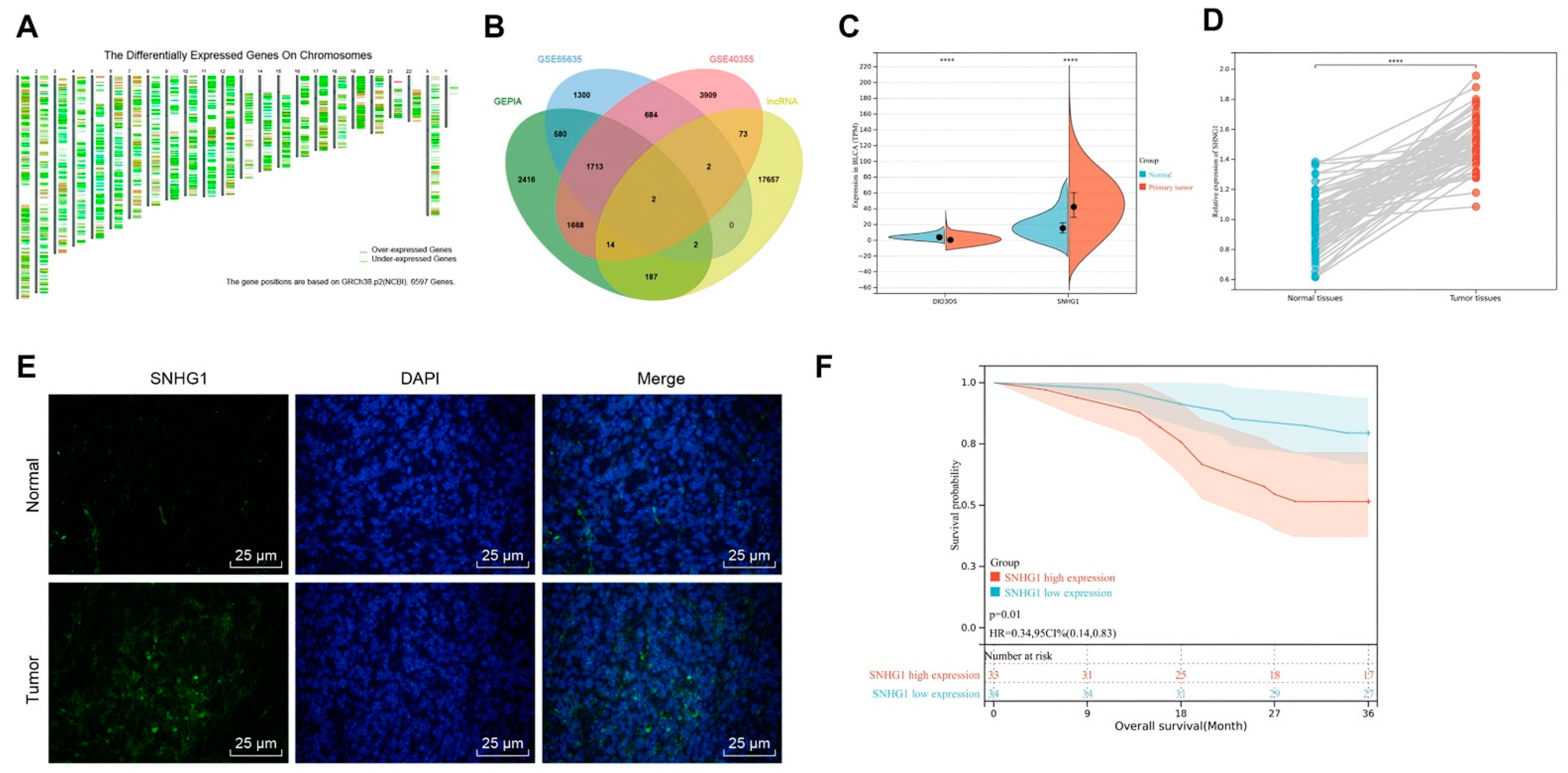
2.3. Bioinformatics Methods
Gene Expression Profiling Interactive Analysis (GEPIA) was adopted to analyze the BLCA dataset of The Cancer Genome Atlas (TCGA) database to obtain the genes with significant differences (
p < 0.05), from which the genes with |logFC| > 0.5 were screened out. The Gene Expression Omnibus (GEO) database:
https://www.ncbi.nlm.nih.gov/gds (accessed on 6 March 2021) was also analyzed by using “limma” package:
http://www.bioconductor.org/packages/release/bioc/html/limma.html (accessed on 6 March 2021) of the R language with |logFC| > 0.5 and
p < 0.05 as thresholds for differential analysis of bladder cancer microarray data GSE65635 and GSE40355. There were 12 samples in microarray data GSE65635, including 4 normal samples and 8 bladder cancer samples. There were 24 samples in microarray data GSE40355, including 8 normal samples and 16 bladder cancer samples. Human lncRNA names were obtained from GENCODE, followed by finding the intersection of significantly differential genes and lncRNA names. A Venn diagram was drawn to screen out the lncRNAs from intersection. LncRNA expression trends were collected in Ualcan, and the key lncRNA was determined by comparing the expression trends and combining with the existing literature. The possible downstream miR of the key lncRNA was discovered by starBase and their binding sites were obtained. The databases TargetScan (cumulative weighted context++ score < 0), DIANA TOOLS (miTG score > 0.6), microRNA (conservation > 0.65, energy < −14, Mirsvr_score < −0.65), and mirDIP (integrated score > 0.1) was applied to predict downstream genes of miR. Intersection of downstream genes with significantly differential genes was taken to find critical downstream genes. The relevant genes of the critical downstream gene were predicted in GeneMANIA:
http://genemania.org/ (3 April 2021), followed by construction of a protein-protein interaction (PPI) network. The most core genes in PPI network (
Table 1) were chosen as the key gene, and the binding sites of the miR to the gene were predicted by TargetScan.
2.4. Fluorescence In Situ Hybridization (FISH)
SNHG1 cDNA fragments were amplified from the SNHG1 plasmid as templates by utilizing high fidelity DNA polymerase (Takara, Kyoto, Japan). Based on this template, fluorescein-labeled lncTCF7 FISH probe DNA was prepared with fluorescein-12-dUTP (Roche, Mannheim, Germany) and Klenow DNA polymerase (Vazyme, Nanjing, China) as per the manufacturer’s protocol. Then, 4-µm frozen sections were made from bladder cancer tissues and adjacent normal tissues. Subsequent to 5-min immersion in proteinase K (MCE, NJ, USA), the slides were washed twice in 2× saline sodium citrate (SSC) (Merck KGaA, Darmstadt, Germany). The FISH hybridization solution encompassing 30 ng/µL lncTCF7 FISH probe DNA (Beijing Dingguo Changsheng Biotechnology Co., Ltd., Beijing, China) was dripped onto the tissue sections before 16-h incubation at 37 °C. The slides were then washed in 0.4 × SSC/0.001% NP-40 (Merck KGaA, Darmstadt, Germany) for 5 min at 56 °C, followed by another 2-min washing in 0.4 × SSC/0.001% NP-40. After being dripped with 4′,6-Diamidino-2-Phenylindole (DAPI)-encompassing sealing agent, the slide was mounted and observed under a fluorescence microscope (Olympus, Tokyo, Japan).
2.5. Cell Incubation
Human normal urothelial cell line SV-HUC-1 (ATCC® CRL-9520), and bladder cancer cell lines [5637 (ATCC® HTB-9), T24 (ATCC® HTB-4™), SW780 (ATCC® CRL-2169™), and UM-UC-3 (ATCC® CRL-1749™) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). The medium used for SV-HUC-1 was ATCC-formulated F-12K (Gibco, Thermo Fisher Scientific, Waltham, MD, USA) Medium (Catalog No. 30-2004) encompassing 10% fetal bovine serum (FBS, ATCC 30-2020). The medium used for 5637 was ATCC-formulated RPMI-1640 Medium (Gibco, Thermo Fisher Scientific, Waltham, MD, USA) (ATCC 30-2001) encompassing 10% FBS. The medium for T24 was ATCC-formulated McCoy’s 5A Medium Modified (Gibco, Thermo Fisher Scientific, Waltham, MD, USA) (Catalog No. 30-2007) with 10% FBS. The medium for SW780 was ATCC-formulated Leibovitz’s L-15 Medium (Gibco, Thermo Fisher Scientific, Waltham, MD, USA) (Catalog No. 30-2008) with 10% FBS. The medium for UM-UC-3 was ATCC-formulated Eagle’s Minimum Essential Medium (Gibco, Thermo Fisher Scientific, Waltham, MD, USA) (Catalog No. 30-2003) with 10% FBS. All media for cell lines encompassed 100 μg/mL streptomycin (MCE, N.J, USA) and 100 U/mL penicillin (MCE, NJ, USA). Cell culture was performed at 37 °C with 5% CO2. The media were positioned in humid air and replaced every 2–3 days according to the growth of cells. Cells were subcultured when 80–90% of the culture plate was covered by cells. Cells were utilized when they reached the logarithmic growth stage.
2.6. Cell Transfection
Lentiviruses expressing specific targeted knockdown SNHG1 [short hairpin-SNHG1 (sh-SNHG1)] and MDM2 (sh-MDM2) sequences and a scramble shRNA (sh-NC; control shRNA) were constructed by GenePharma (Shanghai, China) (
Table 2). Lentiviruses overexpressing SNHG1 (oe-SNHG1), MDM2 (oe-MDM2), and PPARγ (oe-PPARγ), oe-NC, Inhibitor NC, and miR-9-3p inhibitor were obtained from GenePharma. Transfection was implemented using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA).
2.7. Reverse Transcription Quantitative Polymerase Chain Reaction (qRT-PCR)
Subsequent to isolation using RNeasy Mini Kit (Qiagen, Valencia, CA, USA), total RNA underwent reverse transcription to generate cDNA using First Strand cDNA Synthesis Kit (RR047A, Takara). For the detection of miR, the cDNA was obtained by reverse transcription using the miRNA First Strand cDNA Synthesis (Tailing Reaction) kit (B532451-0020, Sangon, Shanghai, China). qRT-PCR reactions were performed using SYBR
® Premix Ex TaqTM II (Perfect Real Time) kit (DRR081, Takara) on real-time fluorescence quantitative PCR instrument (ABI 7500, Applied Biosystems, Foster City, CA, USA). The universal reverse primers for miR and the upstream primers for U6 internal reference were provided in the miRNA First Strand cDNA Synthesis (Tailing Reaction) kit, and the other primers were synthesized by Sangon (
Table 3). After recording the Ct value of each well, the relative expression of mRNAs or miR was calculated using the 2
−ΔΔCt method by normalizing to U6 expression.
2.8. Cell Counting Kit (CCK)-8 Assay
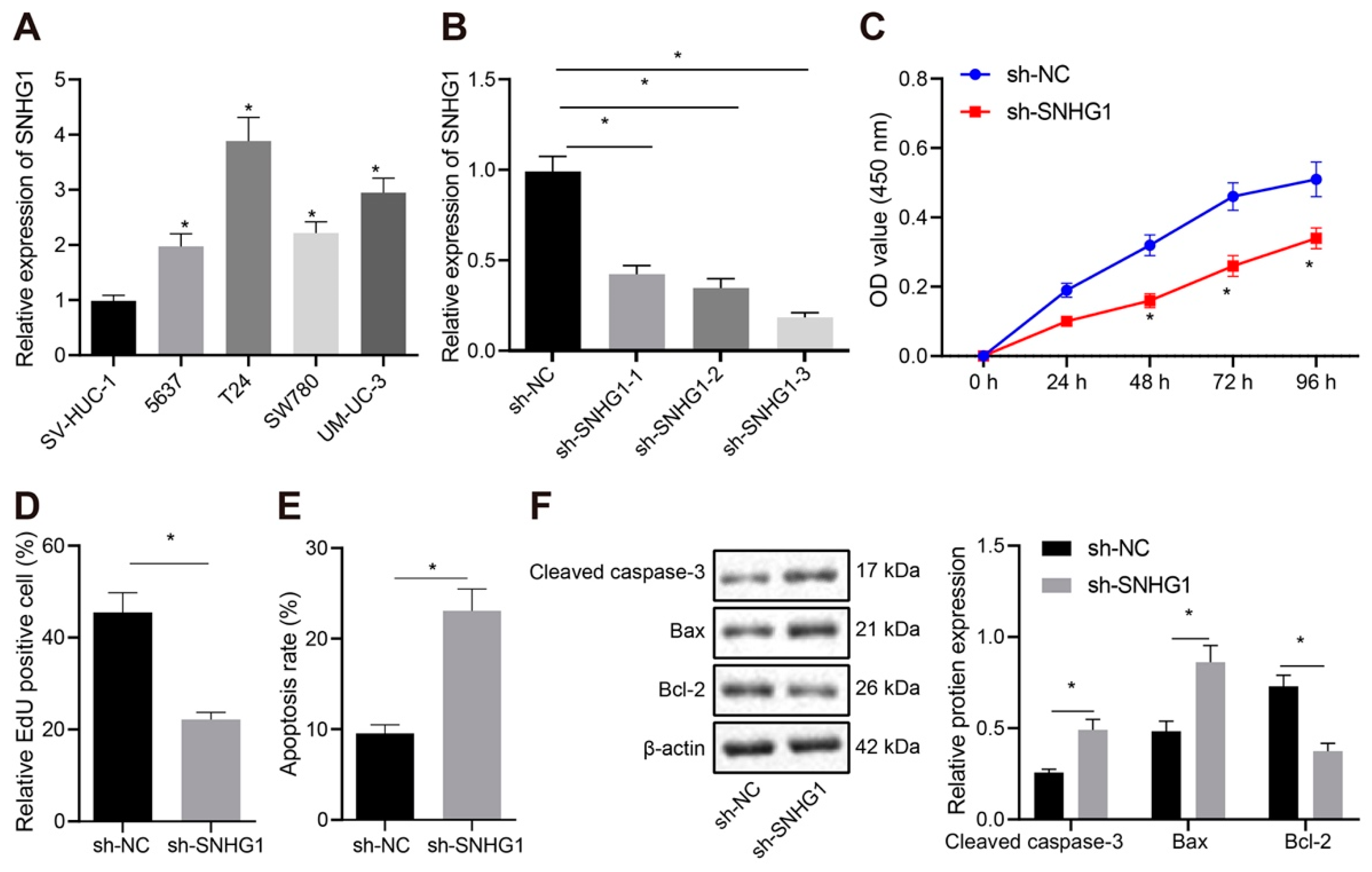
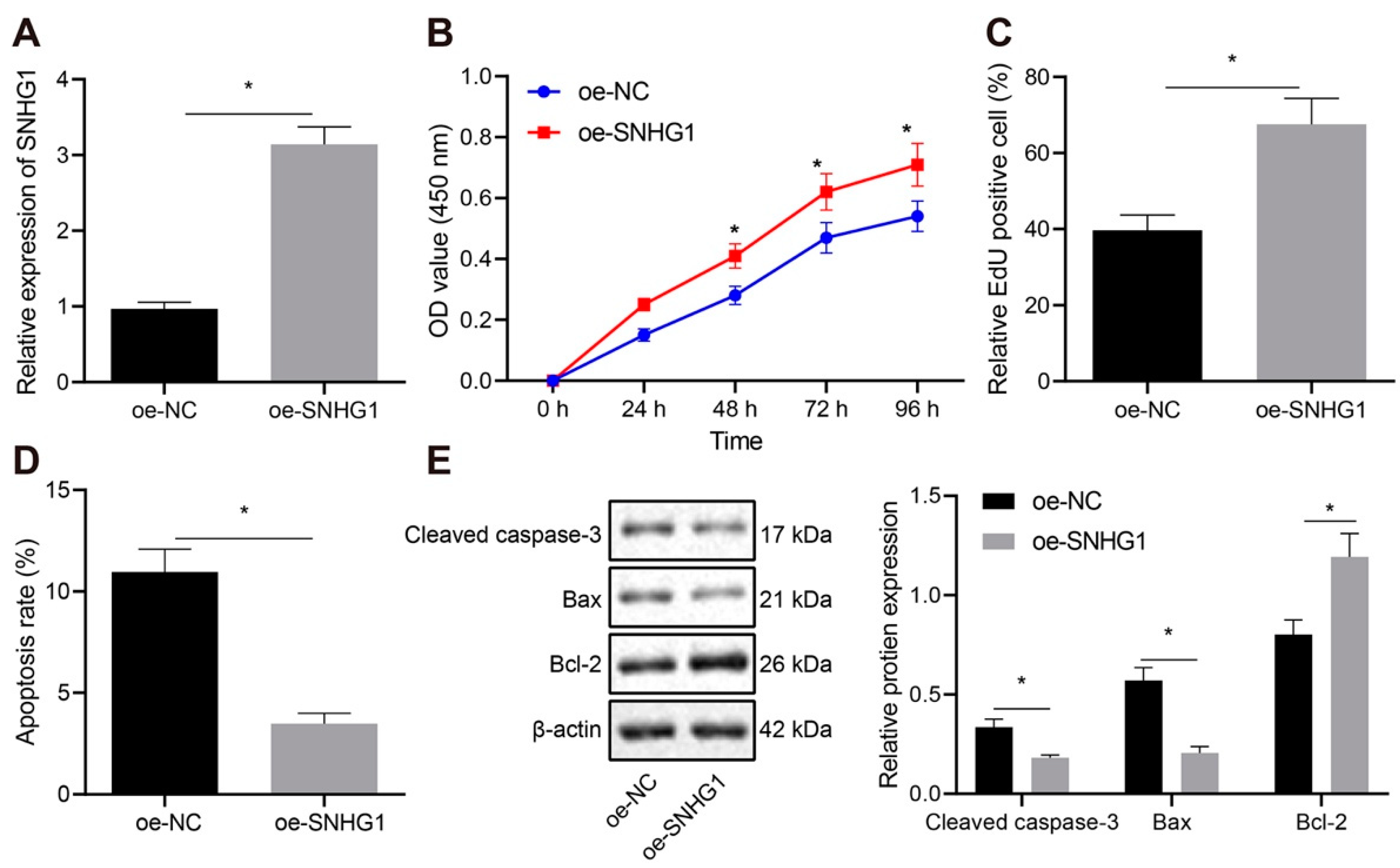
The transfected T24 and 5367 cells (the expression of SNHG1 was clearly highest in T24 and compared with sw780 cells, 5637 cells had better culture characteristics in our experiment) were resuspended and seeded in 96-well plates at 2 × 103/100 µL/well. Cell viability was evaluated by CCK-8 (Dojindo Laboratories, Kumamoto, Japan) method at 0, 24, 48, 72 and 96 h after seeding. The 10 µL CCK-8 solution was supplemented in each test for 4-h incubation before absorbance measurement at 450 nm with a microplate reader.
2.9. 5-Ethynyl-2′-Deoxyuridine (EdU) Assay
The cells to be tested were seeded in 24-well plates with three duplicated wells set for cells in each group. EdU (Invitrogen) was supplemented to the medium to achieve a concentration of 10 µmol/L. The medium was discarded subsequent to 2-h culture. Cells received 15-min phosphate buffer saline (PBS) encompassing 4% paraformaldehyde fixing at ambient temperature before 20-min incubation at ambient temperature with PBS encompassing 0.5% Triton-100. Each well was supplemented with 100 µL dye solution before 30-min culture in the dark at ambient temperature. DAPI was added for 5-min nuclear staining. After sealing, 6–10 fields of view were randomly observed under a fluorescence microscope (FM-600, Shanghai Pudan Optical Instrument Co., Ltd., Shanghai, China), and the number of positive cells in each field was recorded.
2.10. Flow Cytometry
Subsequent to 48-h transfection, the cell concentration was changed to 1 × 106 cells/mL. After cell fixing with 70% precooled ethanol solution at 4 °C, 100 μL cell suspension (no less than 1 × 106 cells/mL) was resuspended in 200 μL binding buffer. Subsequently, 15-min cells staining was implemented with 10 μL Annexin V-fluoresceinisothiocyanat and 5 μL propidium iodide at ambient temperature under dark conditions. After 300 μL of binding buffer was added, apoptosis (T24 and 5367 cell lines) was assessed on a flow cytometer at excitation wavelength of 488 nm (2 × 104 cells each time).
2.11. Western Blot Analysis
Subsequent to trypsin treatment, cells were lysed with enhanced radio-immunoprecipitation assay (RIPA) lysis encompassing protease inhibitors (BOSTER, Wuhan, Hubei, China), followed by estimation of protein concentration using Bicinchoninic Acid (BCA) Protein Quantification Kit (BOSTER). Proteins underwent separation by 10% sodium dodecyl sulfate polyacrylamide gel electropheresis (SDS-PAGE). Then, the separated proteins were electroblotted into a polyvinylidene fluoride (PVDF) membrane which was sealed by 5% bovine serum albumin to block nonspecific binding. Overnight cell incubation was conducted at 4 °C after supplementation with primary rabbit antibodies (Abcam, Cambridge, UK) to Cleaved caspase-3 (ab49822, 1:500), Bcl-2-Associated X (Bax, ab32503, 1:1000), B-cell lymphoma-2 (Bcl-2, ab196495, 1:500), MDM2 (ab226939, 1:3000), PPARγ (ab45036, 1:500), Ubiquitin (ab7780, 1:2000), and β-actin (ab8227, 1:500). Then, horseradish peroxidase-tagged goat anti-rabbit secondary antibodies (ab205719, 1:2000, Abcam) were supplemented for 1-h membrane incubation at 4 °C. After development in ECL working fluid (EMD Millipore Corporation, Billerica, MA, USA), the bands in Western blot images were quantified by Image J analysis software by normalizing to β-actin.
2.12. RNA Pull Down
Cells were transfected with biotinylated wild type (WT) miR-9-3p and mutant type (MUT) miR-9-3p (50 nM each). After 48 h of transfection, 10-min cell incubation was implemented with specific cell lysis (Ambion, Austin, Texas, TX, USA). Then, 3-h lysate incubation was conducted with M-280 streptavidin magnetic beads (Sigma, St. Louis, MO, USA) pre-coated with RNase-free and yeast tRNA at 4 °C before two cell washes in cold lysis and qRT-PCR detection of SNHG1 expression.
2.13. RNA Immunoprecipitation (RIP) Assay
The binding of miR-9-3p to MDM2 was detected by RIP kit (Millipore, Temecula, CA, USA). Briefly, 5-min cell lysing was implemented in an ice bath with equal volume of RIPA lysis (P0013B, Beyotime, Shanghai, China), and supernatant was removed subsequent to 10-min centrifugation at 14,000× g rpm and 4 °C. A portion of the cell extract was applied as input, and a portion was co-precipitated with antibody. RNA extraction was implemented by treating samples with proteinase K for subsequent qRT-PCR detection of MDM2. Antibodies used for RIP were as follows: rabbit anti-Argonaute 2 (AGO2) (1:100, ab32381, Abcam) was mixed at ambient temperature for 30 min, and rabbit anti-human Immunoglobulin G (IgG; 1:100, ab109489, Abcam) was applied as a normal control (NC).
2.14. Dual Luciferase Reporter Gene Assay
The synthesized MDM2 3′ untranslated region (UTR) gene fragment MDM2-WT and the MDM2-MUT mutated at the binding site were constructed into a pMIR-reporter plasmid (Beijing Huayueyang Biotechnology, Beijing, China). Luciferase reporter plasmids were co-transfected with miR-9-3p into HEK293T cells (Shanghai Beinuo Biotechnology, Shanghai, China). Then 48 h subsequent to transfection, cells were lysed, and detected using a luciferase detection kit (K801-200; Biovision, Mountain View, CA, USA).
2.15. Immunoprecipitation (IP)
T24 cells were lysed in lysis buffer [mixture of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, 1 mM ethylene diamine tetraacetic acid, 0.5% NP-40, and protease inhibitor], and cell debris was cleared by centrifugation. After the concentration of lysis was measured by BCA, the same amount of protein was taken from oe-NC/OE-MDM2 group and replenished to the same volume with cell lysate. Afterwards, 1 μg anti-MDM2 (ab226939, 1:100, Abcam), PPARγ (ab45036, 1:100, Abcam) and 15 μL protein A/G beads (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were added for 2-h incubation. Subsequent to three washes with cell lysis, beads were collected by centrifugation, added into an equal volume of reductive loading buffer, and boiled at 100 °C for 5 min. Subsequent to SDS-PAGE, samples were electroblotted to PVDF membranes (Millipore), and then analyzed by immunoblotting.
2.16. Subcutaneous Tumorigenesis Model in Nude Mice
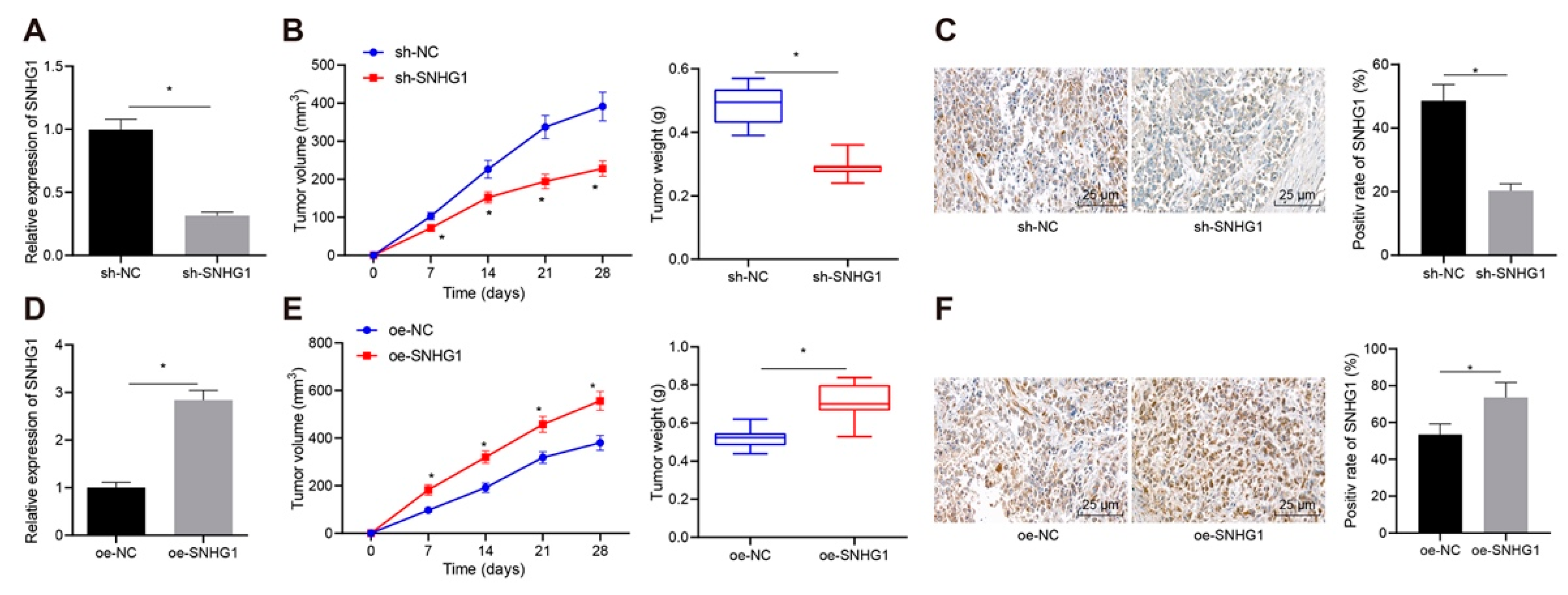
Healthy nude mice aged 6–8 weeks (Beijing Institute of Pharmacology, Chinese Academy of Medical Sciences, Beijing, China) were bred in a specific pathogen-free animal laboratory with 60–65% humidity at 22–25 °C. They were fed in separate cages under 12:12-h light-dark cycle with food and water available ad libitum. The experiment was started one week after acclimation, and the health status of nude mice was observed before the experiment. Approximately 2 × 106 5637 cells were suspended in 200 μL PBS, and then subcutaneously injected into the left or right hindlimbs of nude mice (antagomir used). At 28 days subsequent to injection, mice were euthanized, followed by measurement and weighing of tumors.
2.17. Statistical Analysis
All measurement data were manifested as mean ± standard deviation and analyzed by SPSS 21.0 software (IBM, Armonk, NY, USA), with p < 0.05 as a level of statistical significance. If data conformed to normal distribution and homogeneity of variance, data within groups were compared by paired t test, while data between two groups were compared by unpaired t test. Comparisons among multiple groups were performed using one-way analysis of variance (ANOVA) or repeated measures ANOVA. Intra-group pairwise comparison was performed using a post-hoc test. Rank sum test was performed if data did not conform to normal distribution or homogeneity of variance. Kaplan–Meier was adopted to calculate patient survival curves, and log-rank was utilized to analyze patient survival differences.
4. Discussion
As one of the most prevalent genitourinary cancers with high mortality on a global scale, bladder cancer currently can be treated by local or systemic immunotherapy, radiotherapy, chemotherapy, and endoscopic and open surgery [
19]. However, the curative effect of such therapies is limited because of recurrence or distant spread [
20]. Moreover, lncRNAs has emerged as a modulator in the complexity of bladder cancer [
21]. Consequently, this research investigated the mechanism of SNHG1 in bladder cancer with the involvement of miR-9-3p. Notably, the present study provided evidence that SNHG1 promotes MDM2 expression by binding to miR-9-3p to promote PPARγ ubiquitination and downregulate PPARγ expression, thereby resulting in elevation of bladder cancer cell proliferation in vitro and tumorigenesis in vivo.
Initially, data from our project and online database unraveled that SNHG1 was highly expressed in bladder cancer tissues and cells. Additionally, when SNHG1 was silenced in bladder cancer cells and mice, cell proliferative capacity was depressed but cell apoptosis was accelerated in vitro, and tumorigenesis was inhibited in vivo. Importantly, SNHG1 has emerged as a novel oncogenic lncRNA in various cancers, including esophageal, colorectal, prostate, gastric, liver, and lung cancers, inducing cell proliferative, metastatic, migratory and invasive capacities of cancer cells [
6]. Consistently, Lu et al. observed that SNHG1 expression was strikingly high in NSCLC tissues and cells, and that SNHG1 silencing decreased tumor volumes in mice and reduced NSCLC cell proliferation, invasion and migration [
22]. Meanwhile, data collected by Bai et al. found that SNHG1 expression was upregulated in colorectal cancer cells, and that ectopically expressed SNHG1 could enhance cell migratory, proliferative, and invasive capacities in vitro and led to tumor growth [
23,
24]. Another study uncovered that SNHG1 silencing contributed to decline of tumor growth of breast cancer in vivo [
25]. These findings indirectly supported the tumor-promoting potential of SNHG1 in bladder cancer by enhancing cell proliferation and tumor growth and reducing apoptosis.
It is well-recognized that lncRNAs may function as endogenous sponges to regulate miRNA function in diseases [
26]. For example, prior research showed that SNHG1 could bind to miR-204 to inhibit it, thus promoting migratory, and invasive abilities but repressing apoptosis in esophageal squamous cell cancer [
27]. These findings indirectly confirmed the binding relationship between SNHG1 and miR-9-3p in bladder cancer cells which was observed by our study. Further investigations of our study identified that miR-9-3p inhibition led to increase of cell proliferation and decrease of apoptosis in vitro and promotion of tumorigenesis in vivo and reversed the effect of SNHG1 silencing in bladder cancer. Similarly, the research conducted by Cai et al. clarified that in bladder cancer, miR-9-3p overexpression triggered repression of cell viability, migration, and invasion, induction of cell apoptosis in vitro, and inhibition in vivo tumor growth and metastasis [
10]. Notably, another study illustrated that miR-9-3p exerted tumor-suppressive effect on hepatocellular carcinoma by depressing hepatocellular carcinoma cell proliferation [
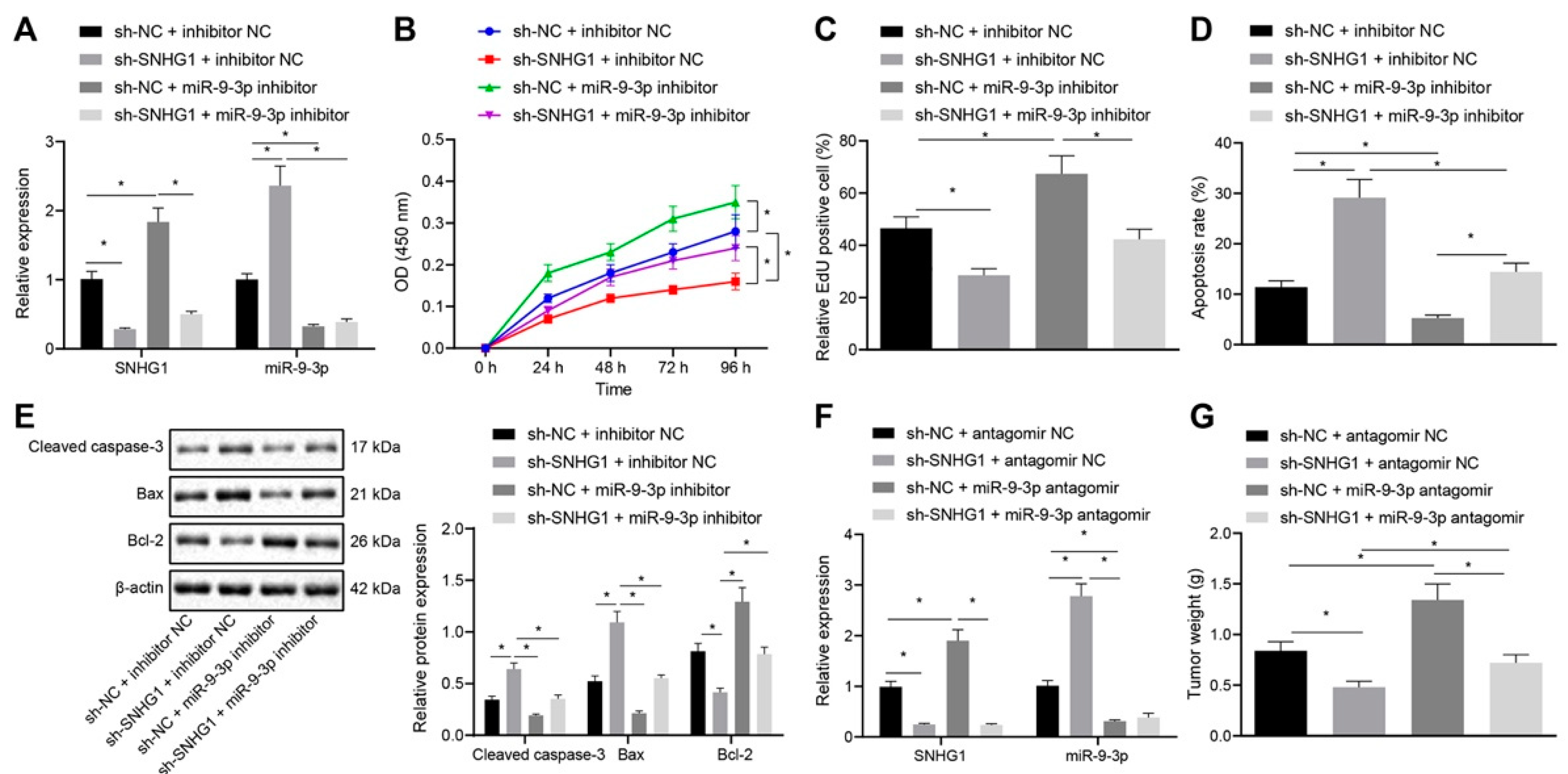
28], which was in line with our results. Hence, these results confirmed that SNHG1 overexpression promoted bladder cancer progression by binding to miR-9-3p.
It is well-established that miRs inhibit expression of target genes at post-transcriptional level by targeting the 3′UTR of mRNA [
29]. As previously reported, miR-9-3p targeted HBGF-5 to function as a tumor suppressor in hepatocellular carcinoma [
30]. Moreover, in our study, TargetScan website predicted the binding sites between miR-9-3p and MDM2 3′UTR, and then the targeting relationship between miR-9-3p and MDM2 was verified by dual luciferase reporter gene assay. In the subsequent experiments, we found that MDM2 bound to PPARγ and downregulated PPARγ by inducing PPARγ ubiquitination, which was similar to the results observed by Xu et al. [
12]. Our experiments show that Bcl-2 and Bax were also related to a certain extent, which is worth further in-depth study. Furthermore, our data elaborated that MDM2 ectopic expression neutralized the inhibitory effect of SNHG1 silencing on cell proliferation in vitro and tumor growth in vivo and the promoting effect of SNHG1 silencing on cell apoptosis in vitro in bladder cancer, suggesting the oncogenic role of MDM2 in bladder cancer. Similarly, a prior study uncovered that inhibition of MDM2 exerted tumor-suppressive effects on bladder cancer by decreasing cell invasive, proliferative, and migratory capacities [
11]. Accordingly, PPARγ activation gave rise to inhibition of proliferation of 9 bladder cancer cell lines [
31] All in all, the SNHG1/miR-9-3p/MDM2/PPARγ axis was involved in bladder cancer progression.
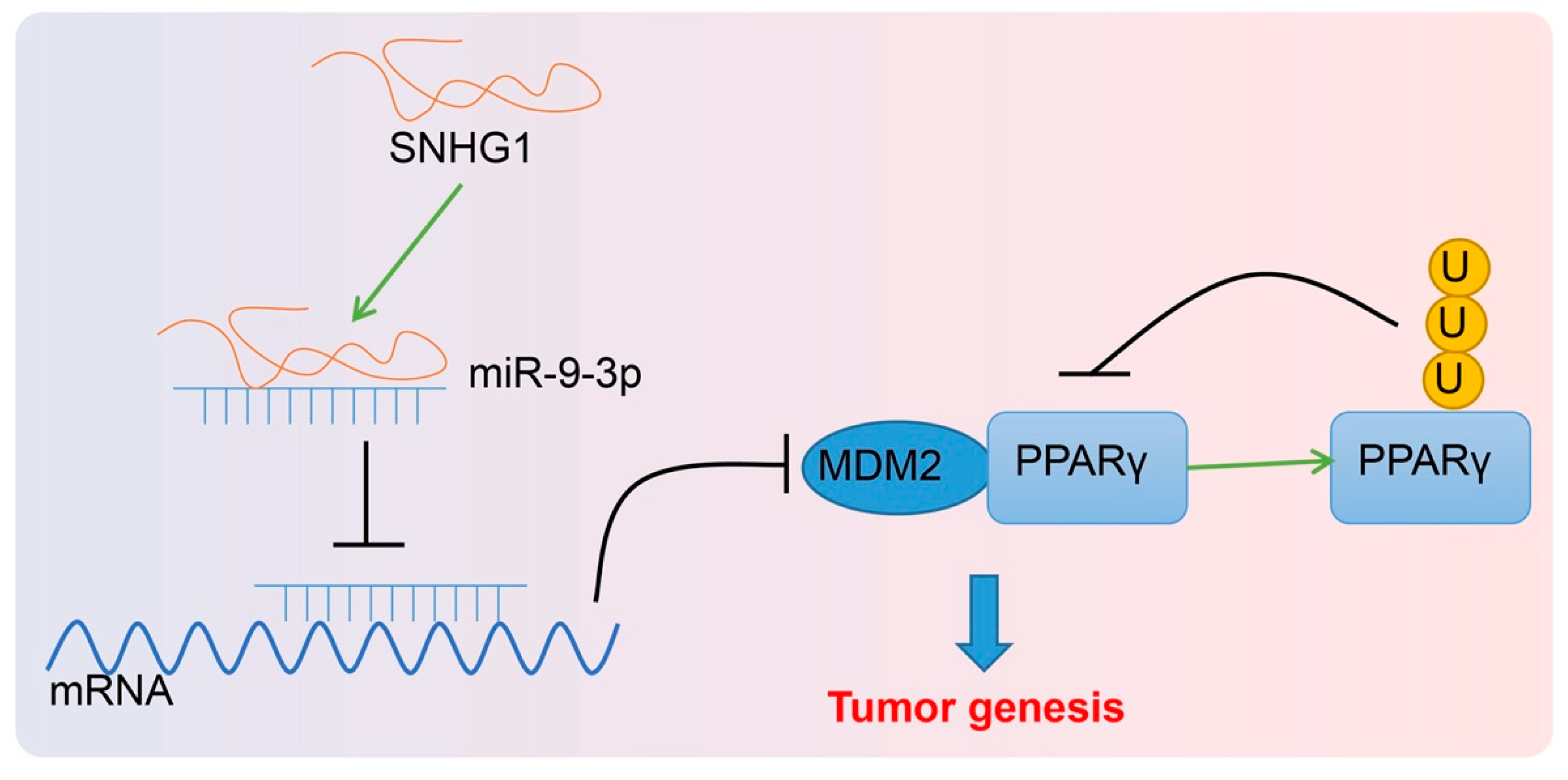
Collectively, this study provides evidence that SNHG1 upregulation promoted cell proliferation but depressed cell apoptosis in bladder cancer via MDM2-inhibited PPARγ by binding to miR-9-3p (
Figure 9). Thus, this finding offers a fresh molecular insight that might be utilized in new therapy development for bladder cancer. However, further studies are required on the mechanism of PPARγ in bladder cancer.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}