1. Introduction
The paucity of molecular diagnostic tools suitable for monitoring treatment outcomes is a major hurdle for the successful management of cancer patients [
1]. There is a great need of tools that allow measuring the molecular response of tumors to cancer therapy in real time throughout the course of treatment [
2]. Optimally, these tools should be non-invasive and they should reflect not only the size of the lesions but also the molecular response [
3,
4]. An early molecular response to treatment would allow clinicians to know whether a particular therapeutic choice is actually working. A treatment that does not work anymore not only does not help the patients, but it might also be contributing to the development of acquired resistance to the drug administered and probably to other cancer drugs. The earlier it is known that a treatment does not work, the faster the treatment can be stopped so the tumor cannot develop additional protective therapy escape mechanisms.
Despite most cancer diagnostic tools beyond imaging tests being based on measuring circulating proteins, the modern identification of useful circulating protein tumor biomarkers has been disappointing. In particular, mass spec proteomic efforts to identify useful tumor-derived circulating proteins using unbiased genome-wide approaches has demonstrated to be a daunting task plagued by limitations and controversy [
5]. On one hand, tumor biology is extremely complex, cancer is a heterogenous group of diseases, and tumor-specific biomarkers are in low abundance in biological fluids. On the other hand, analytical techniques such as mass spectrometry cannot properly resolve the large dynamic range of protein concentrations in biological fluids, since tumor-derived proteins can only be enriched but not amplified. Alternative approaches including the profiling of secretomes, where tumor-derived proteins are enriched, with a follow-up validation in clinical samples have been attempted for tumor biomarker discovery [
6,
7].
Most mass-spec-based proteomic discovery workflows are based on the ability of trypsin to proteolyze proteins into peptides, i.e., bottom-up approach [
8]. Subsequently, tryptic digests are analyzed by mass spec for different applications, including protein ID, quantification, and the characterization of post-translational modifications (PTM). Therefore, proteins that cannot be proteolyzed are basically undetectable by standard discovery-based proteomic approaches. However, there is no scientific reason why proteolysis-resistant proteins cannot be tumor biomarkers. In fact, the extreme stability of these proteins could contribute to their stability in plasma. Particularly, this group of resistant proteins could be amenable to the identification of biomarkers involving tumor cell death induced by cancer drugs.
In this work, we performed a screening of proteolytic resistant proteins in a breast cancer cell line secretome. The implementation of a modification of the standard proteomic workflow allowed us to profile a set of proteins that are extremely stable against proteolysis. We took one of them, manganese superoxide dismutase (MnSOD or SOD2), as a model protein for its validation as a candidate response biomarker for breast cancer neoadjuvant treatment. Neoadjuvant chemotherapy is becoming a preferred therapeutic option in breast cancer patients with locally advanced disease [
9,
10]. This clinical setting offers a homogenous group of patients and it is frequently used to assess new anti-cancer drugs and to evaluate candidate tumor biomarkers [
11]. SOD2 is part of the superoxide dismutase family of redox enzymes that metabolize superoxide radicals into hydrogen peroxide, and it is located in the mitochondrial matrix of cells [
12]. SOD2 is a protein overexpressed in cancer, probably because it alleviates the molecular damage induced by the increase in oxidative stress that tumor cells experience [
13,
14]. However, the expression of SOD2 seems to be stage-dependent. The expression of this protein is low in early-stage cancer while its expression increases in advanced cancer, particularly in metastasis [
15]. Here, we carried out the functional validation of SOD2 as a response biomarker for chemotherapy, first in vitro, and then using plasma from BC patients undergoing neoadjuvant chemotherapy.
2. Materials and Methods
2.1. Cell Culture, Treatments, and Cell Titer
The breast cancer cell lines used in this project were purchased from ATCC and maintained at 37 °C in 5% CO
2 and 95% humidified atmosphere air. Specifically, MDA-MB-231 and MCF7 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM/F12; Invitrogen, Waltham, MA, USA), and BT549 cells in RPMI medium (Invitrogen). All cell lines were supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 2 mM L-glutamine (Invitrogen). When needed, cell lines were treated with paclitaxel (4 nM) or doxorubicin (30 nM or 2 µM). To generate a BT549 subline resistant to paclitaxel (BT549-DR cells), BT549 parental cells were treated chronically with 4 nM paclitaxel until they became resistant to the drug and started proliferating (
Figure S2). The concentration to kill and/or inhibit growth of cells by 50% (IC
50) of these drugs was quantified from drug–response curves by using CellTiter colorimetric assay analysis. The different cell lines were plated with four replicates for each condition (5000 cells/well) and after overnight incubation, drug treatment started. Specifically, the following drug concentrations were used: paclitaxel (1–8 nmol/L) and doxorubicin (1 nmol/L–2 μmol/L). Cell viability was measured after 72 h using the CellTiter assay (CellTiter-Blue Cell Viability Assay; Promega, Madison, WI, USA).
2.2. Molecular Cloning of SOD2 Mutants
His6-tagged human SOD2 cDNA sequence was cloned in the pcDNA 3.1 vector. The mutant Ile-58Thr-SOD2 was cloned in pcDNA3.1 and transfected in BT549-shSOD2 cells, specially designed for not silencing the mutant form. For the creation of shRNA-resistant versions of the cDNA, sequence 5′-CAGCCTGCACTGAAGTTCA-3′ was mutated to 5′-CAACCAGCTCTCAAATTTA-3′ to render an mRNA product insensitive to the shRNA expressed constitutively in the BT549 shSOD2 cells (target sequence CAGCCTGCACTGAAGTTCA). shRNA-resistant versions of SOD2 plasmids were transfected in BT549 cells expressing shSOD2 using FugeneHD (Roche, Basel, Switzerland) and following manufacturer recommendations. Briefly, 2.8 µg of DNA was added in 129 µL total volume of OptiMEM (Invitrogen). Next, 8.3 uL of FuGENE HD reagent was added and mixed by pipetting. The mix was incubated for 5–10 min at RT and 125 µL of complex was added to the cells seeded in a 100 mm plate containing 2 mL of fresh medium. After 48 h of transfection, medium was substituted with fresh growth medium containing 500 µg/mL of G418 (Gibco, Toronto, ON, Canada) to select for stably transfected clones. Cells were subsequently cultured for 12 days more, until non-transfected cells died, being subcultured at 1:5 ratio if needed.
2.3. Secretome Collection
Secretomes were prepared as previously described [
16]. Briefly, 4 × 10
6 cells in exponential phase were seeded in 150 mm tissue culture plates and allowed to grow for 48 h. After that, media was aspirated, and cells were washed five times: two times with PBS and the last three with serum-free media. After that, cells were maintained for the indicated time in the presence of serum-free media before collecting the conditioned media (secretome). The conditioned media were spun down at 200 g for 5 min, and the supernatants were collected and filtered through a Millex-GP 0.22 µM pore syringe driven filter (Millipore, Burlington, MA, USA). Then, secretomes were concentrated using a 10,000 MWCO Millipore Amicon Ultra (Millipore). Protein concentration was determined with a Pierce BCA protein assay kit (Thermo Scientific, Waltham, MA, USA).
2.4. In-Solution Digests
Trypsin digestion in the presence of urea: Secretome protein samples (15 µg of total protein), were taken to 40 µL of 6 M urea, and 50 mM ammonium bicarbonate by addition of the appropriate amount of lyophilized 8 M urea resulted in the 50 mM ammonium bicarbonate buffer. Samples were first reduced with DTT to a final concentration of 10 mM, for 1 h at RT, and then alkylated with 20 mM of iodoacetamide for 30 min at RT in the dark. Carbamidomethylation reaction was quenched by the addition of N-acetyl-L-cysteine to a final concentration of 35 mM followed by incubation for 15 min at RT in the dark. Samples were diluted with 50 mM ammonium bicarbonate to a final concentration of 1 M urea, modified porcine trypsin (Promega Gold) was added in a ratio of 1:10 (w/w), and the mixture was incubated overnight at 37 °C. The reaction was stopped with formic acid (FA) at a final concentration of 0.5% and the digest was kept at −20 °C until further analysis.
LysC/Trypsin digestion in the presence of guanidinium chloride: Secretome protein samples (15 µg of total protein), pretreated or not as detailed above, were taken from 44 µL of 6 M guanidinium chloride, 50 mM ammonium bicarbonate. Samples were first reduced with DTT to a final concentration of 10 mM, for 1 h at 60 °C, and then alkylated with 20 mM of iodoacetamide for 30 min at RT in the dark. Carbamidomethylation reaction was quenched by addition of N-acetyl-L-cysteine to final concentration of 35 mM followed by incubation for 15 min at RT in the dark. Samples were diluted with 50 mM ammonium bicarbonate to a final concentration of 2 M guanidinium chloride, endoproteinase LysC from Lysobacter enzymogenes (Sigma, Kawasaki, Japan) was added in a ratio of 1:10 (w/w), and the mixture was incubated at 37 °C for 6 h. The digest was then diluted with 50 mM ammonium bicarbonate to a final concentration of 0.9 M guanidinium chloride, modified porcine trypsin (Promega Gold) was added in a ratio of 1:10 (w/w), and the mixture was incubated overnight at 37 °C. The reaction was stopped with formic acid (FA) at a final concentration of 0.5% and the digest was kept at −20 °C until further analysis.
2.5. Liquid Chromatography-Mass Spectrometry Analysis (LC-MS) and Protein Identification
Protein tryptic digests were analyzed using a linear ion trap Velos-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Xcalibur software package was used to control the mass spectrometer, version 2.2.0 (Thermo Fisher Scientific, Bremen, Germany). Peptide mixtures were separated by on-line nanoflow liquid chromatography using an EASY-nLC 1000 system (Proxeon Biosystems, Thermo Fisher Scientific) with a two-linear-column system. Tryptic digests were first loaded on a trapping chromatographic column (Acclaim PepMap 100 nanoviper, 2 cm long, ID 75 μM and packed with C18, 3 μM particle size from Thermo Fisher Scientific) at 4 uL/min. Then, the peptide mixture was analyzed on the reversed-phase analytical column (Dr Maisch, 25 cm long, ID 75 μM, packed with Reprosil Pur C18-AQ, 3 μM particle size). Elution of peptides was carried out using 0.1% formic acid in water (mobile phase A) and acetonitrile with 0.1% formic acid (mobile phase B), with a linear gradient from 0 to 35% of mobile phase B for 120 min at a flow rate of 300 nL/min. Ions were produced by applying a voltage of 1.9kV to a stainless-steel nano-bore emitter (Proxeon, Thermo Fisher Scientific), attached to the end of the chromatographic column, on a Proxeon nano-spray flex ion source.
Data-dependent mode was used to operate the LTQ Orbitrap Velos mass spectrometer. A scan cycle started with a full-scan MS spectrum (from m/z 300 to 1600) obtained in the Orbitrap with setting a resolution of 30,000. The 20 most intense ions were targeted for collision-induced dissociation fragmentation in the linear ion trap when their intensity exceeded 1000 counts, dismissing singly charged ions. Collection of ions for both MS and MS/MS scans was achieved in the linear ion trap, and the AGC target values were set to 1 × 106 ions for survey MS and 5000 ions for MS/MS scans. A maximum ion accumulation time was set to 500 and 200 ms in the MS and MS/MS modes, respectively. The normalized collision energy was set to 35%, and one microscan was obtained per each spectrum. Ions subjected to MS/MS with a relative mass window of 10 ppm were excluded from further sequencing for 20 s. A window of 20 ppm and isolation width of 2 Da was defined for all precursor masses. The lock mass option (m/z 445.120024) for survey scans was enabled in the Orbitrap measurements to improve mass accuracy.
2.6. Protein Identification and Quantitative Differential Analysis
Proteomic data were analyzed using the Proteome Discoverer v. 2.1 software (Thermo Fisher Scientific). Protein identification was performed using Mascot v. 2.5 (Matrix Science, London, UK) using the SwissProt database (2018_11, taxonomy limited to human proteins, 20,413 sequences). A precursor mass tolerance of 10 ppm was used to search the MS/MS spectra, fragment tolerance was set to 0.7Da, trypsin specificity was controlled by setting a maximum of 2 missed cleavages, cysteine carbamidomethylation was set as fixed peptide modification, and methionine oxidation as variable peptide modification.
Protein identification files generated from Mascot (DAT files) were then loaded into the Scaffold software (version 3.00.07; Proteome software, Inc., Portland, OR, USA), resulting in a non-redundant list of identified proteins for each LC-MS/MS run per sample. Peptide identifications were validated whenever a PeptideProphet probability greater than 95% was achieved. Proteins identified with a probability higher than 95% and that contained at least two validated MS/MS spectra were accepted for further analysis. A false protein discovery rate (FDR) below 1.0%, as estimated by a database search, was achieved by using the described PeptideProphet and ProteinProphet tools. Using these filters, the files generated by the “Scaffold software” containing “spectral counts” (SpC) for each sample and their replicates were transferred to the R statistical environment. Exploratory data analysis was carried out by using principal component analysis (PCA) and hierarchical clustering of the SpC matrices to detect sample outliers and visualize patterns in the dataset. A GLM model based on the Poisson distribution was used to perform statistical modeling. During statistical analysis, an adjusted
p-value < 0.05, a fold change >0.8 and minimum number spectral counts (SpC) >2 were set as thresholds [
17,
18].
2.7. Proteinase K Treatments
For Western blot analysis, 100 µg of secretome or 10 ng of recombinant protein (marca commercial) were digested with 20 µg/mL of Proteinase K (ThermoFisher) in a final volume of 20 µL. Samples were incubated at 37 °C with agitation O/N. Then, 1 µL of PMSF 100 nM was added to inactivate Proteinase K. For LC-MS/MS proteomic analysis, 100 µg of secretome was digested following the same protocol. Samples were filtered with 10 kDa amicon (Millipore) to remove the digested peptides since we were interested in analyzing the proteolytic-resistant proteins.
2.8. Western Blot
Cells were seeded in complete growth medium and allowed to grow at the specified times and conditions. Total protein extraction was performed using an NP-40-based lysis buffer. Protein quantitation and electrophoresis were performed as described elsewhere. Western blot analysis was performed with the following primary antibodies: mouse anti-alpha-tubulin (clone B-5-1-2, Sigma-Aldrich, St. Louis, MO, USA) used at 1:10,000, mouse anti-fibronectin (BD Cell Signalling, San Jose, CA, USA), SOD2 (sc-FL-222, Santa Cruz, Dallas, TX, USA) used at 1:1000, Histag (Cell Signaling), and cParp (Cell Signaling). Horseradish-peroxidase-conjugated secondary antibodies were sheep anti-mouse and donkey anti-rabbit IgG (GE Healthcare, Chicago, IL, USA) and used at 1:5000. All antibodies, unless otherwise stated, were used at 1:1000. Immunodetection was followed by visualization and densitometry using Image J software (Rasband, W.S., ImageJ 1.46r, U. S. National Institutes of Health, Bethesda, MD, USA). To evaluate the quaternary structure of SOD2, a native PAGE gel was performed, following the same protocol mentioned but without SDS.
2.9. SOD2 ELISA
SOD2 levels were measured by ELISA using the commercial kit (KA0528-Abnova) and following the manufacturer’s indicated protocol.
2.10. SOD2 Stability in Human Active Serum
An amount of 7.5 µg of the BT549 secretome was mixed with human active serum in a final concentration of 4 µg/mL. The mix was split into 5 aliquots and incubated at 37 °C for 24 h, 48 h, 72 h, or 96 h. One sample of the mix without incubation was kept as the 0 h time point, as well as a sample of serum without secretome and the secretome as input. All samples were stored at −20 °C, and then SOD2 levels were analyzed by ELISA.
2.11. SOD2 Immunoprecipitation
An amount of 100 µg of BT549 secretomes expressing SOD2-WT or SOD2-I58T with a HisTag were incubated with human active serum. The mix was split into 6 aliquots and incubated at 37 °C for 15′, 2 h, 7 h, 24 h, and 48 h. One sample of the mix was kept without any incubation and used as the 0 h control. Then, 20 µL of nickel beads/aliquot (Millipore), with a binding capacity of 1–5.5 μg of His-tagged protein per μL of bead suspension, were incubated with 500 µL of equilibration buffer (phosphate-buffered saline, 0.05% Tween-20, 10 mM imidazole, pH 8.0) for 1 min at RT with gentle mixing. The tubes were placed in the magnet rack, and after allowing the beads to migrate to the magnet the buffer was removed. The equilibrated beads were then added to each serum time point and incubated for 30 min at RT and afterwards 2 h at 4 °C, both with rotation. Then, samples were placed in the magnetic rack and serum was removed. The magnetic beads were then washed 10 times in 500 µL of Washing Buffer (PBS, 0.05% Tween-20, 20 mM imidazole, pH 8.0) with gentle mixing for 1′ at RT. Finally, proteins attached to the magnetic beads were eluted incubating the beads with Elution Buffer (PBS, 0.05% Tween-20, 300 mM imidazole, pH 8.0) for 15 min at RT with rotation. Tubes were then placed in the magnetic rack and the eluted fraction was collected, run though SDS-PAGE, and immunoblotted against SOD2.
2.12. Patient Sample Collection
All plasma samples were obtained from Vall d’Hebron University Hospital (Barcelona, Spain). The study was approved by the hospital ethical committee, including a waiver of informed consent for the use of the samples for this project. An amount of 8 mL of venous blood was collected in an EDTA-BD Vacutainer (Beckton Dickinson, Franklin Lakes, NJ, USA). Blood was gently mixed by inverting 8 times to prevent clotting and immediately refrigerated in a vertical position. Tubes were then centrifuged at 1500 G for 15 min at 4 °C. Plasma was aliquoted in previously labeled and prechilled cryovials (Fisherbrand), and then stored at −80 °C. Repeated freeze–thaw cycles were avoided. Mammary tumors were measured using a caliper every three weeks. Tumor volume was calculated using the formula V = (W
2 × L)/2 [
19].
4. Discussion
Here, we tested the hypothesis that proteolytically resistant proteins could be candidate tumor biomarkers. Proteins resistant to proteolysis are likely to be relevant for tumor biomarker discovery and are drastically under-sampled by current proteomic workflows. The rationale of our approach is that upon tumor cell death, robust proteins will stay longer in circulation and provide a reliable sensor for the response to therapy. Most tumor-derived proteins are likely to be removed quickly from circulation, since they will not endure the physicochemical environment either by losing folding stability or being proteolyzed. Identifying highly stable tumor-derived proteins that enter into circulation could be a valuable approach to, for example, predict responses to cancer therapy. Our approach could complement other strategies relying on the tumor cell death induced by cancer drugs that are being tested to monitor the response to cancer drug therapy [
25,
26,
27].
Several of the proteins enriched by our proteolytic assay are unconventionally secreted proteins, since their sequences do not have signal peptides directing them to the ER–Golgi secretory pathway. Unconventional secretion of theoretically intracellular proteins has been recently proven far more common than previously expected. Different unconventional secretion pathways relying on either extracellular vesicles or soluble proteins have been described [
16,
28,
29]. Here, we chose to functionally validate SOD2 as a tumor biomarker. While thought to be a strict mitochondrial protein, SOD2 has been recently reported to be secreted in renal carcinoma [
30]. Likewise, the other intracellular superoxide dismutase, SOD1, has also been described to be non-classically secreted in yeast [
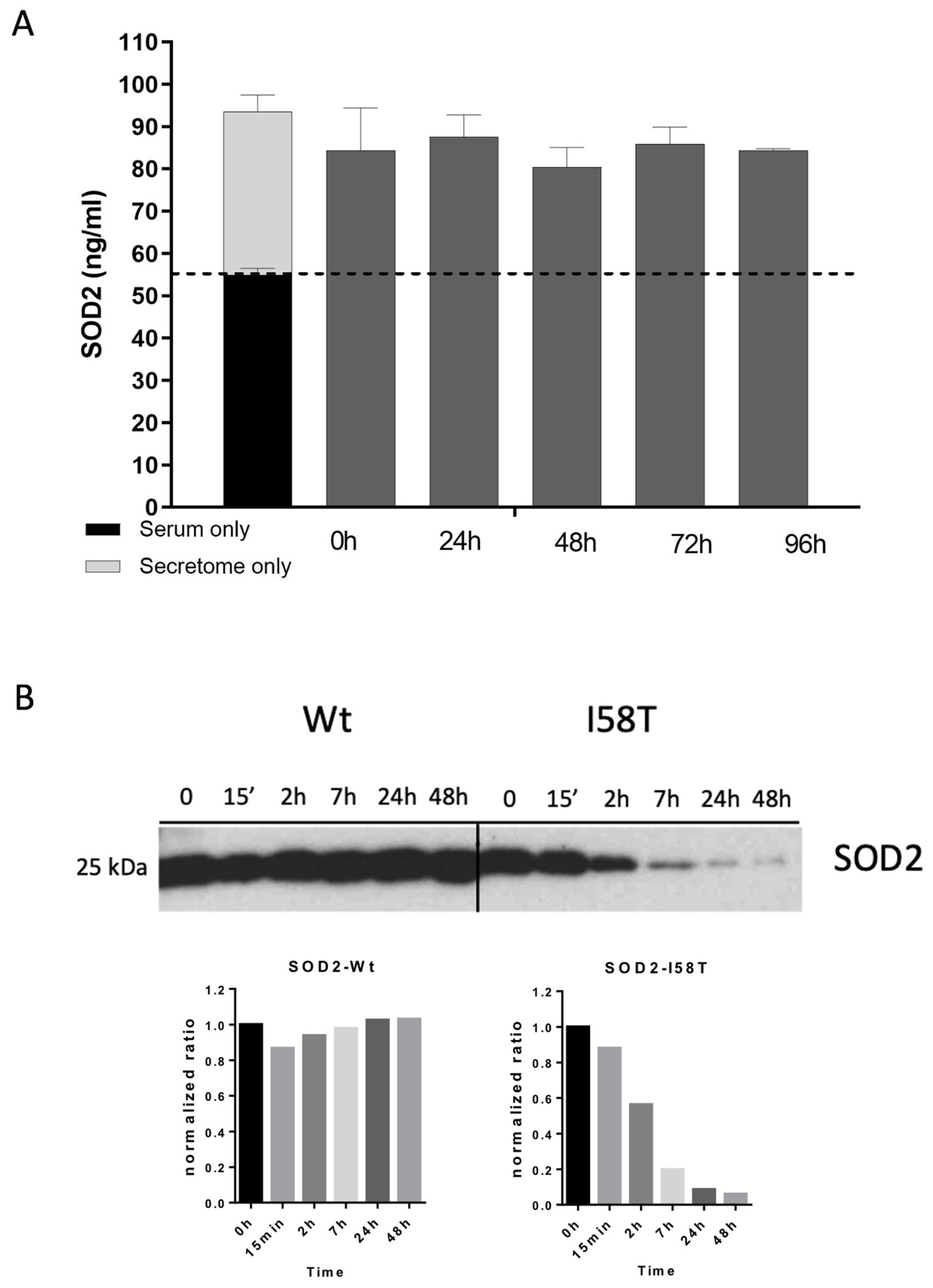
31]. Here, we suggest that SOD2 is also non-classically secreted in breast cancer cells. However, our current data suggest that the large increase in SOD2 levels measured by ELISA both in vitro (in tumor cells) and in vivo (on BC patients) would originate mostly from the mitochondrial form liberated upon cell death rather than from secretion.
Proteolytic activities have been previously proposed to generate new tumor biomarkers, for example in the form of patterns of the serum peptidome amplified by the proteolytic activities of tumor-derived proteases or by the proteolytic degradation of cytokeratin-18 by caspases during apoptosis [
27,
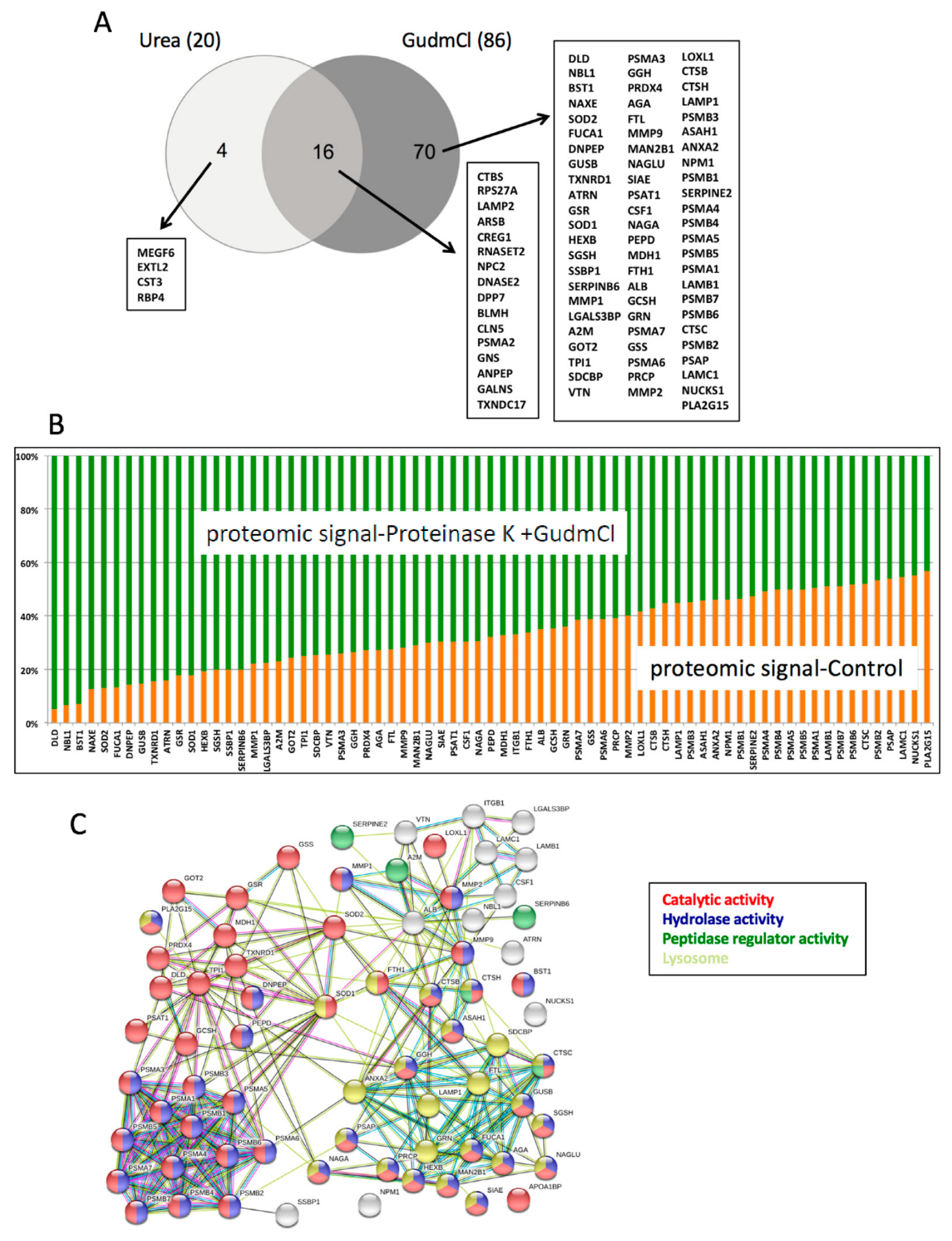
32]. In our case, we propose an in vitro approach to predict which tumor-derived proteins could be proteolytically resistant and hence stable in circulation during cancer therapy. The highly resistant proteins identified in this work are circumscribed to very specific cellular biochemistries. Most of these proteins are enzymes, and among them the majority are proteases, including proteasomal proteins, lysosomal proteases, cathepsins, and matrix metalloproteases. The second group of enzymes enriched by our proteolytic assay is made of redox enzymes functioning in different cellular organelles. A provocative idea derived from this work is that several of the stable proteins identified could potentially be candidate circulating cell death biomarkers. We believe that the dataset provided with this work contains other potential tumor biomarkers. However, parameters including protein solubility in plasma and clearance kinetics from circulation will play a role in the biomarker potential of each protein assayed.
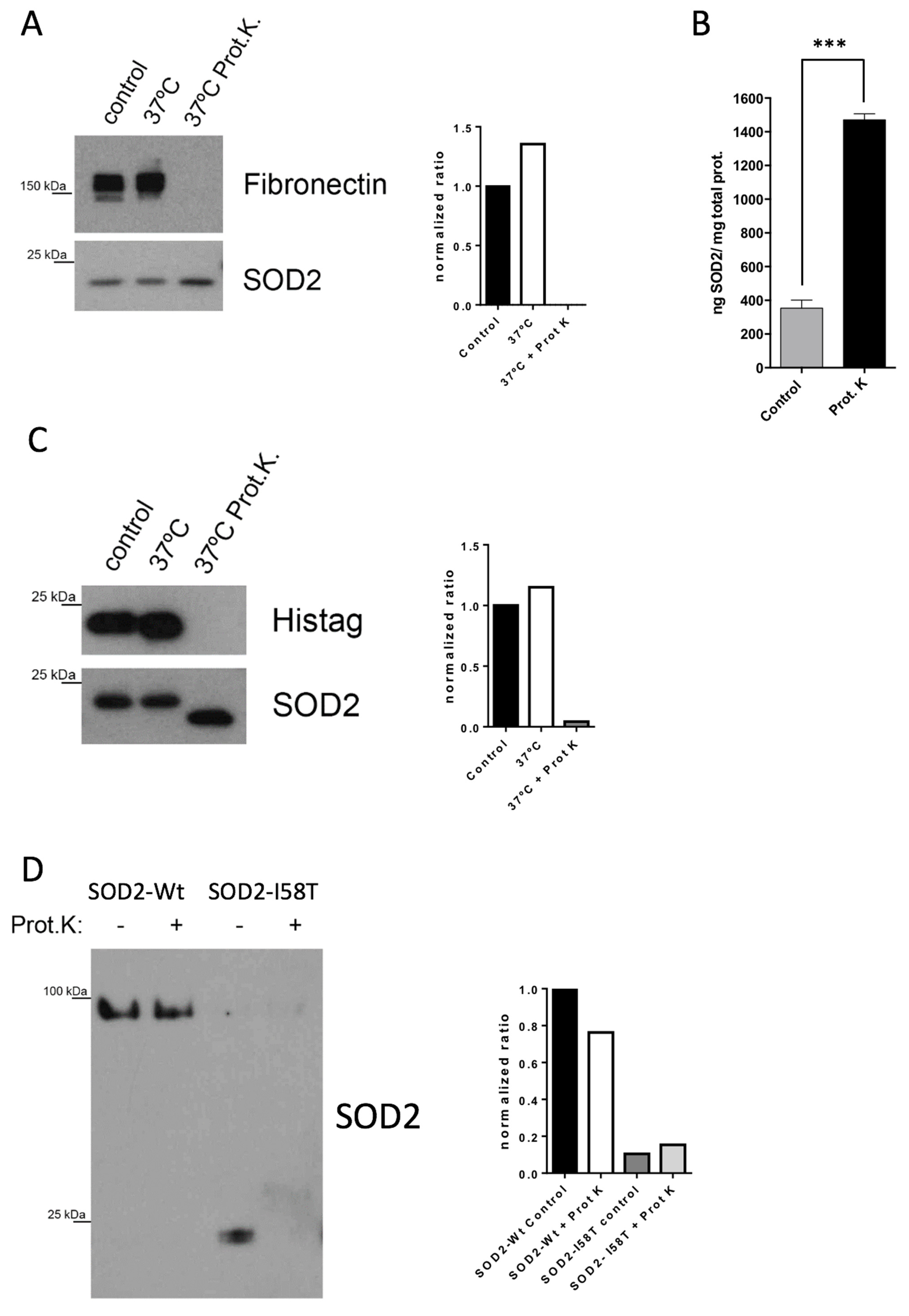
We have demonstrated that the extreme proteolytic stability shown by SOD2 in tumor cells is due to its quaternary structure. The disruption of the tetramer structure greatly diminishes the stability of the protein both in cell culture and in plasma. The initial in vitro validation studies prove that extracellular levels of SOD2 in cell lines correlate with the tumor cell death induced by chemotherapy. Our results show that regardless of whether a tumor cell is sensitive or resistant to chemotherapy, the changes in the slope of the curve describing the levels of SOD2 correlate well with tumor cell death. In fact, the biomarker is the increase in the levels of SOD2, independent of the baseline levels. This hypothesis is demonstrated using BT549 and MDA-MB-231 cells (
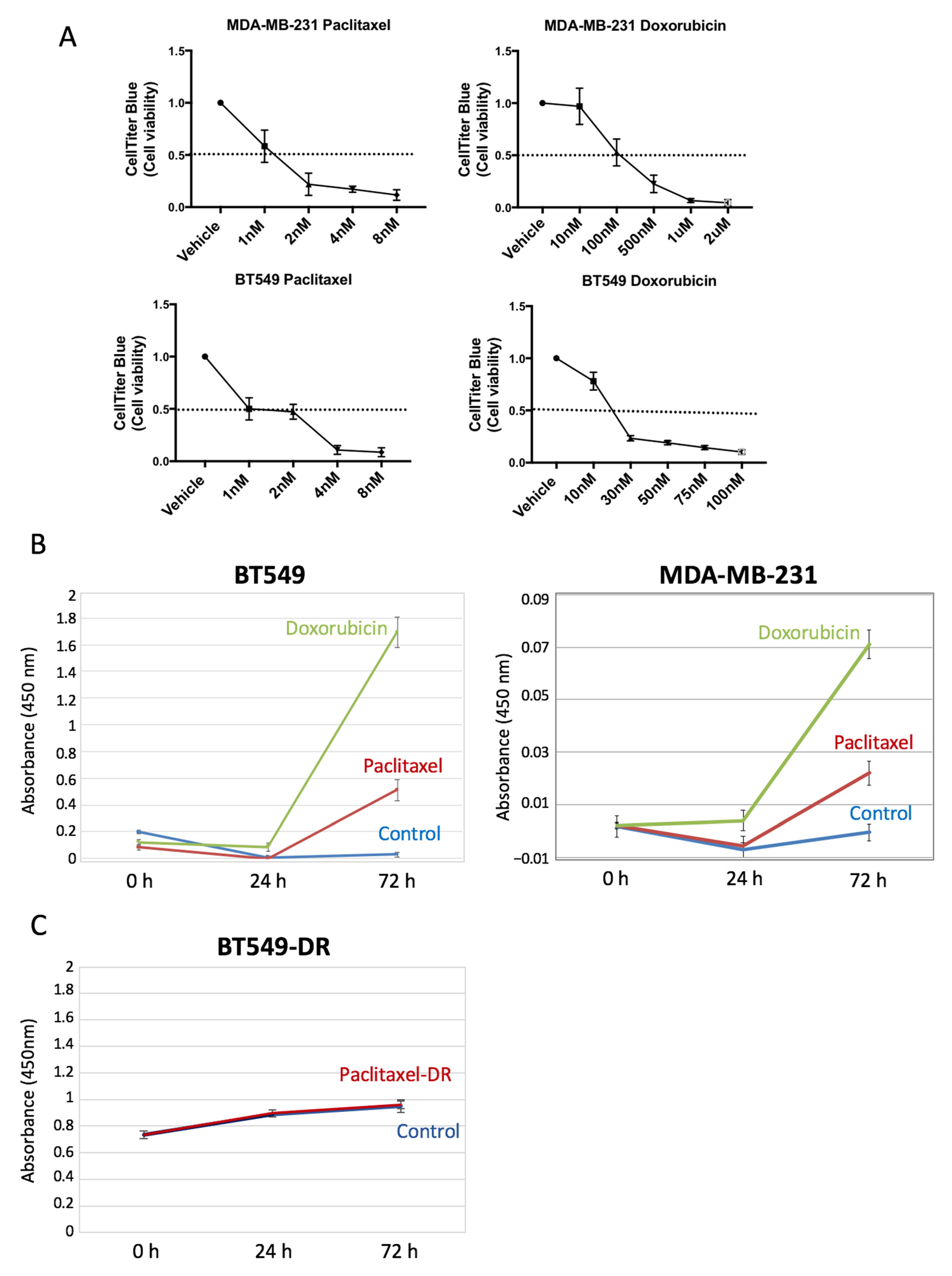
Figure 3B), which despite having different baseline levels of SOD2, show a significant increase in the SOD2 levels when treated with chemotherapeutic drugs. Furthermore, tumor cells resistant to paclitaxel do not show a change in the curve slope compared to their sensitive counterparts when treated, despite having higher baseline levels.
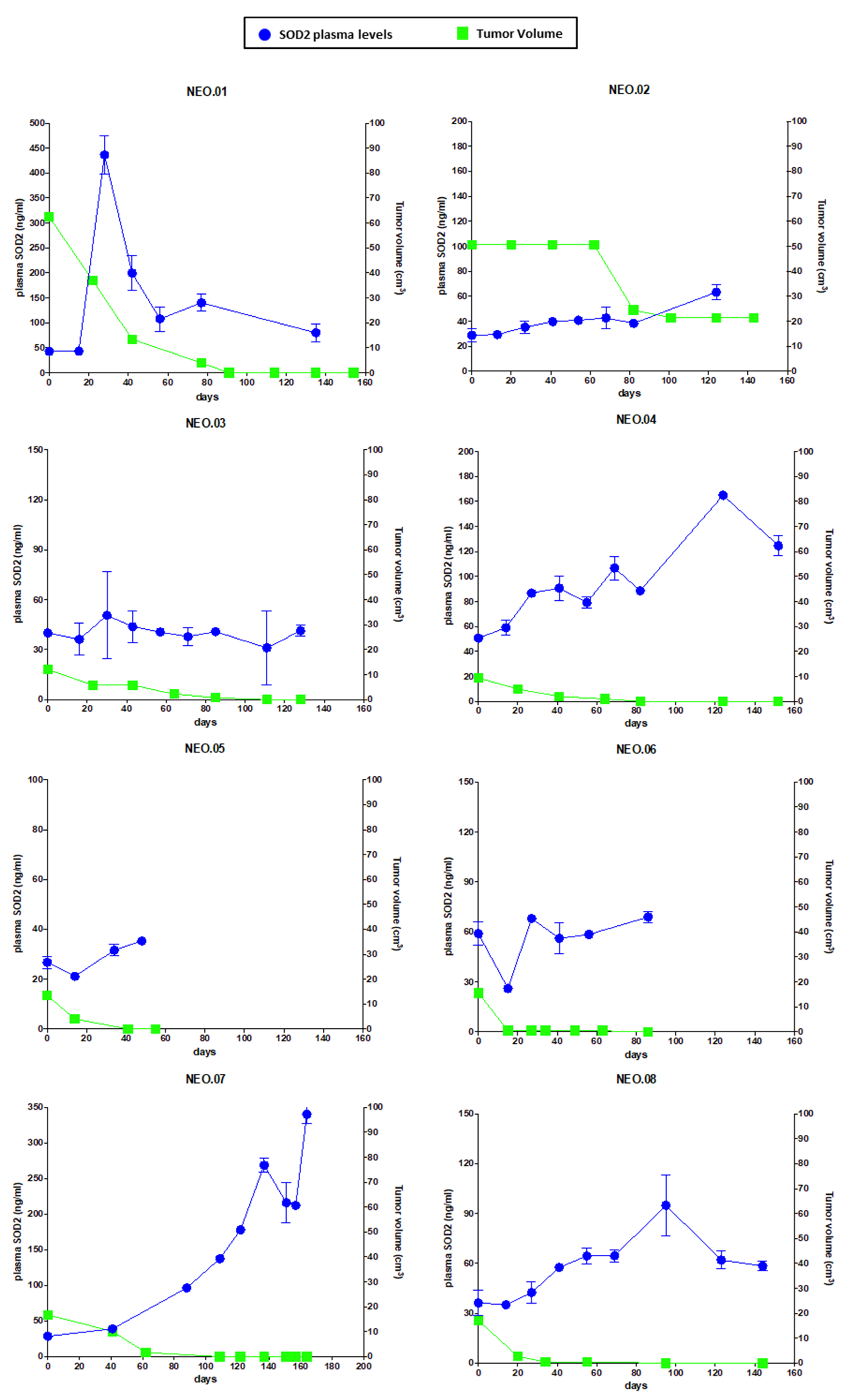
Next, we moved to perform a preliminary clinical validation of SOD2 as a response biomarker for chemotherapy treatment in BC patients. A remarkable finding in this work is that there is a correlation relationship between SOD2 plasma levels and tumor volume in a group of BC patients (TNBC and Luminal B HER2-negative) during neoadjuvant treatment. As shown in
Figure 5, the reduction in tumor size in patients undergoing neoadjuvant therapy correlates with the changes in the circulating levels of SOD2, despite their different SOD2 baseline levels. Therefore, the measurement of SOD2 levels could improve the non-invasive monitoring of the therapeutic treatment in BC patients. Furthermore, beyond our proof-of-concept study, we believe that SOD2 levels should be explored in the treatment of patients with advanced cancer. A drug able to kill tumor cells in the metastatic setting would most likely cause an increase in the levels of circulating SOD2 as long as there is tumor cell death. Therefore, a circulating biomarker that correlates with response to therapy in advanced disease could improve the response evaluation using a quick and non-invasive assessment collected at the time of treatment administration, complementing standard imaging techniques such as computed tomography (CT) scans. Moreover, SOD2 levels could provide a cost-effective biomarker of response to the treatment in the first 2–3 months as a cell death BM, because these levels could identify whether the tumor is responding to this therapy, information which is not available through imaging techniques in this early phase of the treatment.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




