
Current Progress of CAR-NK Therapy in Cancer Treatment


Abstract
:Simple Summary
Abstract
1. T-Cell Therapy in Cancer Treatment
2. Adverse Events of CAR-T Therapy
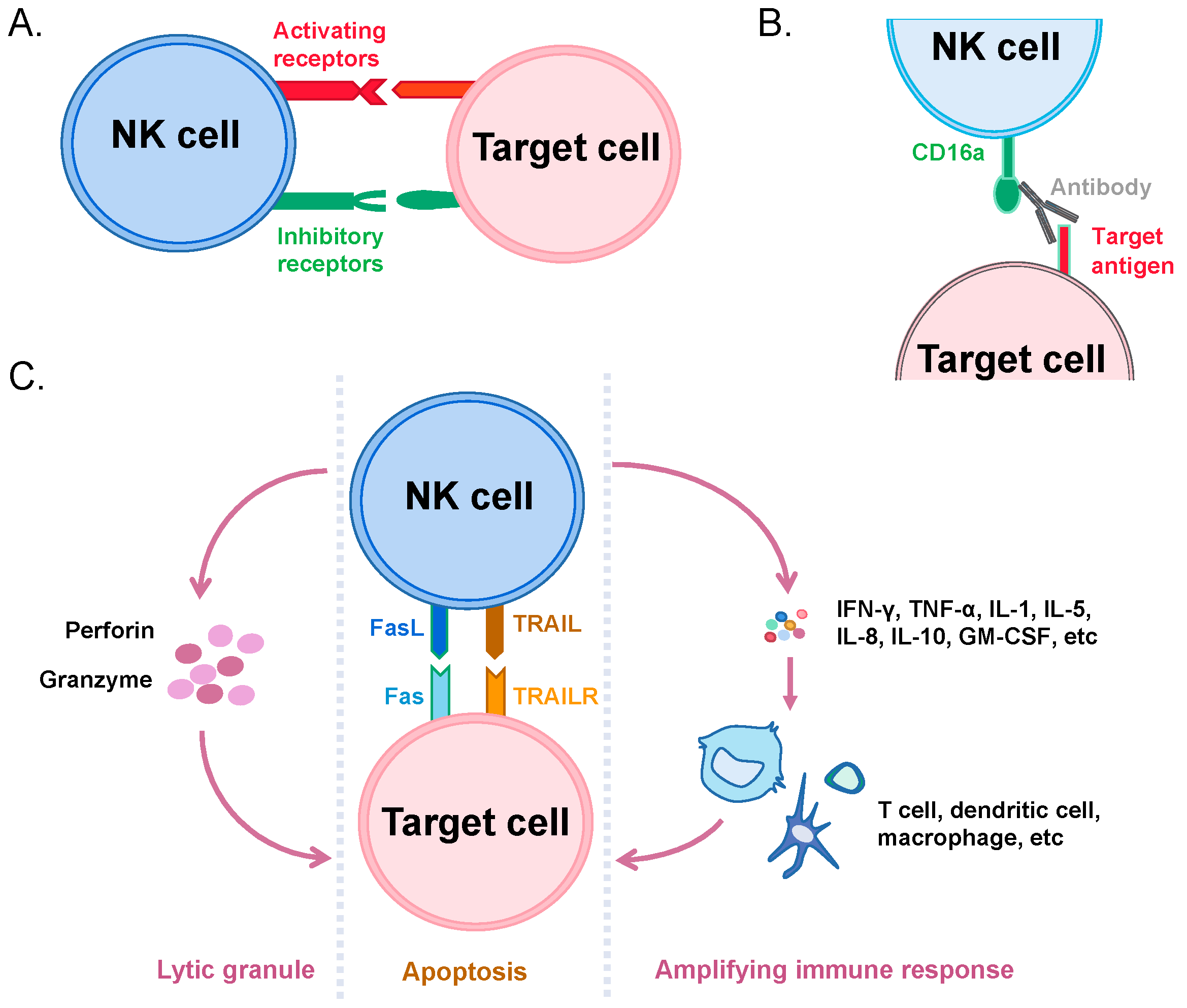
3. Recognition and Killing of Cancer Cells by NK Cells
4. Immunotherapeutic Applications of NK Cells
5. Comparison of Biology in CAR-NK and CAR-T
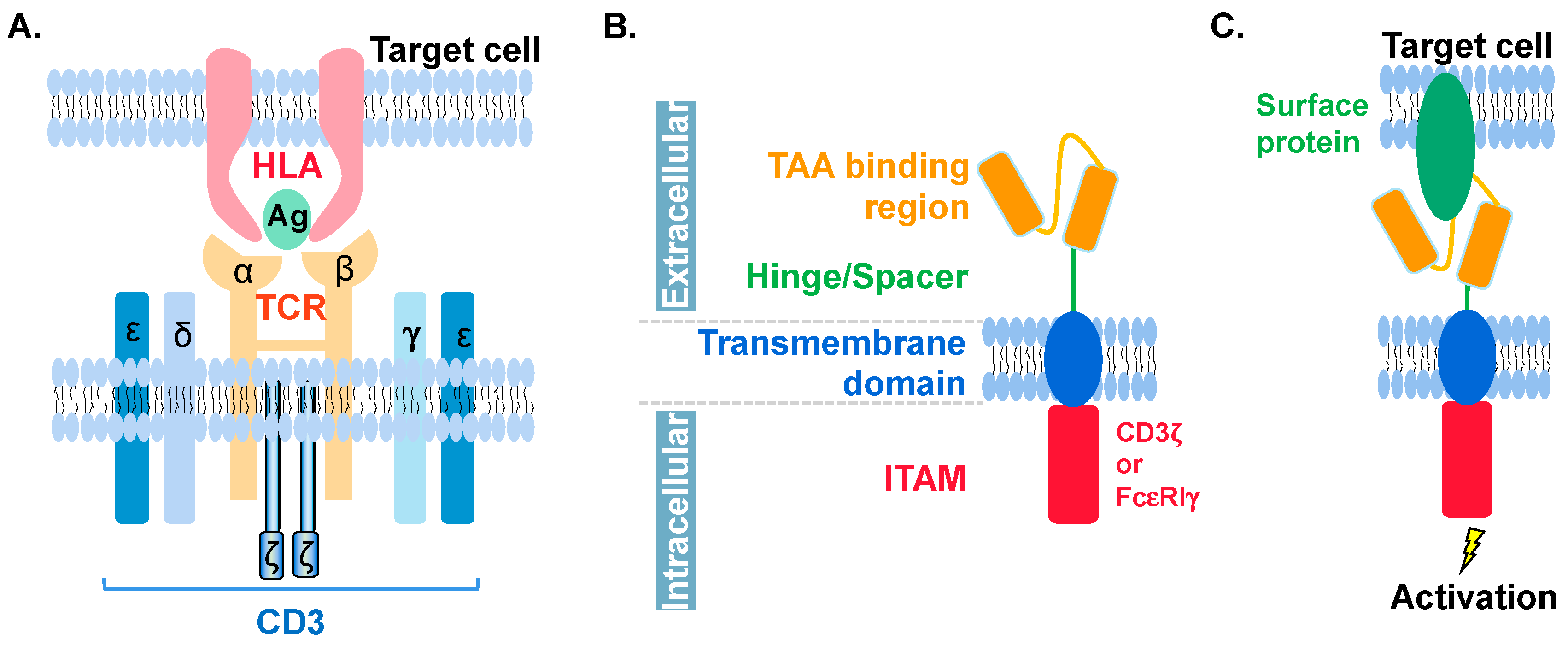
6. Molecular Features of CAR in CAR-NK and CAR-T
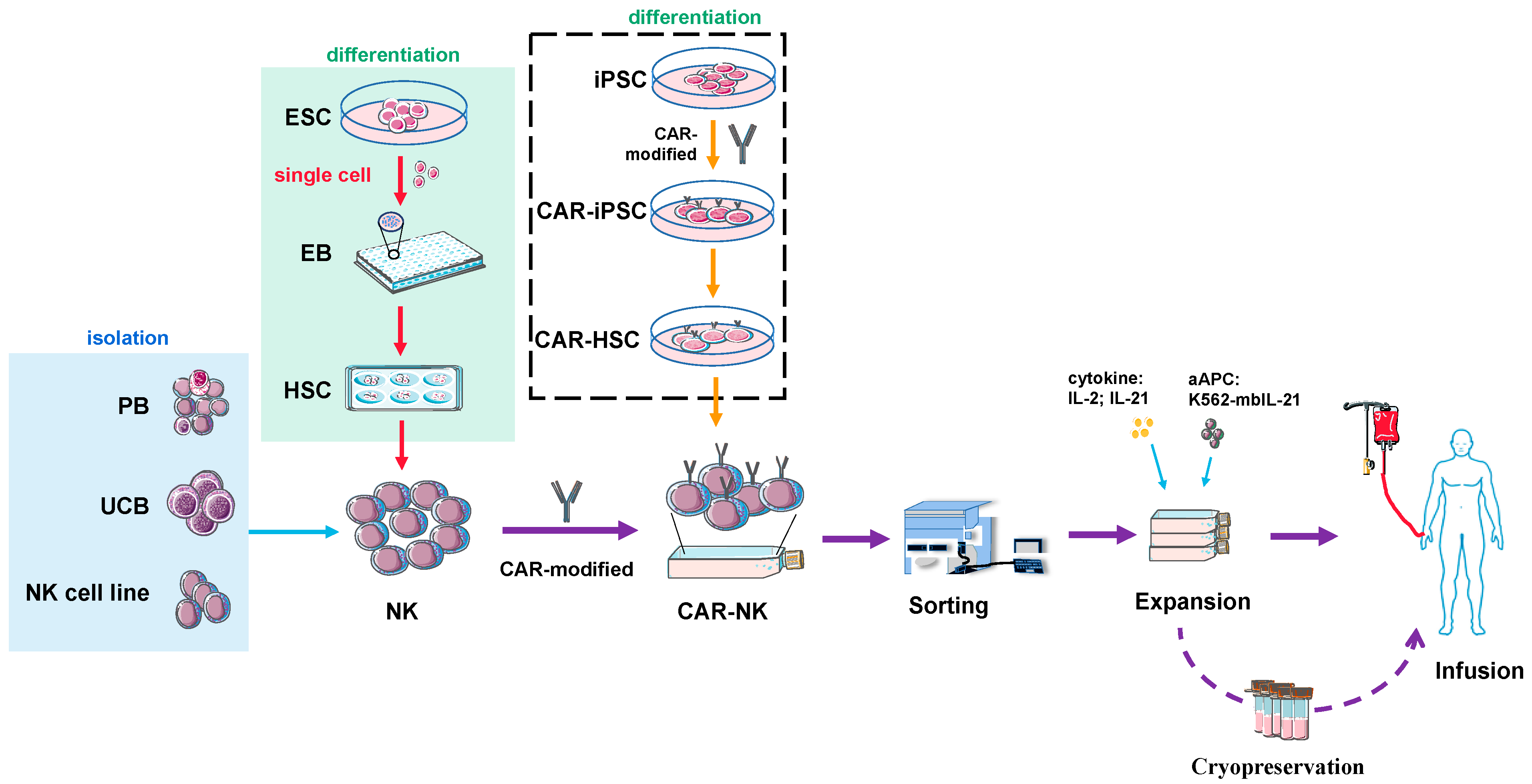
7. Source and Preparation of NK or CAR-NK Cells for Clinical Use
8. Current Clinical Trials of CAR-NK
9. Conclusions and Prospects
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | amino acid; |
| aAPC | artificial antigen-presenting cell; |
| AAV | adeno-associated virus |
| ACT | adoptive cell therapy; |
| ADCC | antibody-dependent cell-mediated cytotoxicity; |
| AICD | activation-induced cell death; |
| ALL | acute lymphoblastic leukemia; |
| BBB | blood-brain barrier; |
| CAR | chimeric antigen receptor; |
| CAR-NK | CAR-modified NK cells; |
| CHL | classical Hodgkin’s lymphoma; |
| CLL | chronic lymphocytic leukemia; |
| CRS | cytokine release syndrome; |
| CTL | cytotoxic T lymphocytes; |
| DLBCL | diffuse large B-cell lymphoma; |
| DLL1 | Delta-like-1; |
| EBs | embryoid bodies; |
| EGFR | epidermal growth factor receptor; |
| ES | embryonic stem cells; |
| FasL | Fas ligand; |
| GM-CSF | granulocyte macrophage colony stimulating factor; |
| GMP | good manufacturing practice; |
| GvHD | graft-versus-host disease; |
| HLA-I | human leukocyte antigen-I; |
| HSC | hematopoietic stem cells; |
| ICANS | immune effector cell-associated neurotoxicity syndrome; |
| IFN-γ | interferon-γ; |
| IL | interleukin; |
| iPSCs | induced pluripotent stem cells; |
| ITAM | immunoreceptor tyrosine-based activation motif; |
| KIRs | killer cell immunoglobulin-like receptors; |
| KLRs | killer lectin-like receptors; |
| LILRs | leukocyte immunoglobulin-like receptors; |
| mbIL-21 | membrane-bound IL-21; |
| MDSC | myeloid-derived suppresive cell; |
| MNC | mononuclear cell; |
| NHL | non-Hodgkin’s lymphoma; |
| PB | peripheral blood; |
| PBMCS | peripheral blood mononuclear cells; |
| PB-NKs | peripheral blood-derived NK cells; |
| PD-1 | programmed cell death protein 1; |
| scFv | single-chain fragment variable; |
| sgp130 | soluble glycoprotein130; |
| sIL-2Rα | soluble interleukin 2 receptor-α; |
| TAA | tumor-associated antigen; |
| TAM | tumor-associated macrophage; |
| TCM | central memory T cell; |
| TCRs | T cell receptors; |
| TEM | effector memory T cells; |
| Th1 | T helper 1; |
| TM | transmembrane domain; |
| TME | tumor microenvironment; |
| TN | naive T cells; |
| TNF-α | tumor necrosis factor-α; |
| TRAIL | TNF-related apoptosis-inducing ligand; |
| TRAILR | TRAIL receptor; |
| Treg | regulatory T cells; |
| UCB | umbilical cord blood; |
| UCB-NKs | Umbilical cord blood-derived NK cells |
References
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, B.; Mensurado, S.; Coffelt, S.B. Γδ T Cells: Pleiotropic Immune Effectors with Therapeutic Potential in Cancer. Nat. Rev. Cancer 2019, 19, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Riond, J.; Rodriguez, S.; Nicolau, M.-L.; al Saati, T.; Gairin, J.E. In Vivo Major Histocompatibility Complex Class I (MHCI) Expression on MHCIlow Tumor Cells Is Regulated by Γδ T and NK Cells during the Early Steps of Tumor Growth. Cancer Immun. 2009, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Yang, J.C.; Langan, R.C.; Dudley, M.E.; Nathan, D.-A.N.; Feldman, S.A.; Davis, J.L.; Morgan, R.A.; Merino, M.J.; Sherry, R.M.; et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol. Ther. 2011, 19, 620–626. [Google Scholar] [CrossRef]
- Beckermann, K.E.; Dudzinski, S.O.; Rathmell, J.C. Dysfunctional T Cell Metabolism in the Tumor Microenvironment. Cytokine Growth Factor Rev. 2017, 35, 7–14. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T Cells Are Abundant and Phenotypically Distinct in Human Tumour Infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Garrido, F. HLA Class-I Expression and Cancer Immunotherapy. In MHC Class-I Loss and Cancer Immune Escape; Garrido, F., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2019; pp. 79–90. ISBN 978-3-030-17864-2. [Google Scholar]
- Dong, D.; Zheng, L.; Lin, J.; Zhang, B.; Zhu, Y.; Li, N.; Xie, S.; Wang, Y.; Gao, N.; Huang, Z. Structural Basis of Assembly of the Human T Cell Receptor–CD3 Complex. Nature 2019, 573, 546–552. [Google Scholar] [CrossRef]
- Lozano, A.X.; Chaudhuri, A.A.; Nene, A.; Bacchiocchi, A.; Earland, N.; Vesely, M.D.; Usmani, A.; Turner, B.E.; Steen, C.B.; Luca, B.A.; et al. T Cell Characteristics Associated with Toxicity to Immune Checkpoint Blockade in Patients with Melanoma. Nat. Med. 2022, 28, 353–362. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric Antigen Receptors for T Cell Immunotherapy: Current Understanding and Future Directions. J. Gene Med. 2012, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New Development in CAR-T Cell Therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.T.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Singh, N.; Frey, N.V.; Grupp, S.A.; Maude, S.L. CAR T Cell Therapy in Acute Lymphoblastic Leukemia and Potential for Chronic Lymphocytic Leukemia. Curr. Treat. Options Oncol. 2016, 17, 28. [Google Scholar] [CrossRef]
- Horowitz, M.; Gale, R.; Sondel, P.; Goldman, J.; Kersey, J.; Kolb, H.; Rimm, A.; Ringden, O.; Rozman, C.; Speck, B. Graft-versus-Leukemia Reactions after Bone Marrow Transplantation. Blood 1990, 75, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-Derived CD19-Targeted T Cells Cause Regression of Malignancy Persisting after Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Somerville, R.P.T.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef]
- Jacoby, E.; Yang, Y.; Qin, H.; Chien, C.D.; Kochenderfer, J.N.; Fry, T.J. Murine Allogeneic CD19 CAR T Cells Harbor Potent Antileukemic Activity but Have the Potential to Mediate Lethal GVHD. Blood 2016, 127, 1361–1370. [Google Scholar] [CrossRef]
- Yang, L.; Baltimore, D. Long-Term in Vivo Provision of Antigen-Specific T Cell Immunity by Programming Hematopoietic Stem Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 4518–4523. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewski, J.L.; Suh, D.; Markley, J.C.; Smith, O.M.; King, C.; Goldberg, G.L.; Jenq, R.; Holland, A.M.; Grubin, J.; Cabrera-Perez, J.; et al. Tumor Immunotherapy across MHC Barriers Using Allogeneic T-Cell Precursors. Nat. Biotechnol. 2008, 26, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, A.; Cui, H.; Caligiuri, M.A.; Yu, J. Chimeric Antigen Receptor-Engineered Natural Killer Cells for Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 168. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. ‘Off-the-Shelf’ Allogeneic CAR T Cells: Development and Challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Turtle, C.J. Toxicities of CD19 CAR-T Cell Immunotherapy. Am. J. Hematol. 2019, 94, S42–S49. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Rabilloud, T.; Potier, D.; Pankaew, S.; Nozais, M.; Loosveld, M.; Payet-Bornet, D. Single-Cell Profiling Identifies Pre-Existing CD19-Negative Subclones in a B-ALL Patient with CD19-Negative Relapse after CAR-T Therapy. Nat. Commun. 2021, 12, 865. [Google Scholar] [CrossRef]
- Li, N.; Wang, S.A.; Lin, P.; Jabbour, E.; Thompson, P.; Chen, Z.; Li, S.; Xu, J.; You, M.J.; Bueso-Ramos, C.E.; et al. Relapsed B-Acute Lymphoblastic Leukemia with Aberrant Myeloperoxidase Expression Following CAR T-Cell Therapy: A Diagnostic Challenge. Am. J. Hematol. 2019, 94, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, I.; Prasad, V.; Gellad, W.F. Total Costs of Chimeric Antigen Receptor T-Cell Immunotherapy. JAMA Oncol. 2018, 4, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, R.; Klein, E.; Wigzell, H. „Natural” Killer Cells in the Mouse. I. Cytotoxic Cells with Specificity for Mouse Moloney Leukemia Cells. Specificity and Distribution According to Genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Mingari, M.C.; Biassoni, R.; Moretta, L. What Is a Natural Killer Cell? Nat. Immunol. 2002, 3, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The Biology of Human Natural Killer-Cell Subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Wang, J.; Li, C.-D.; Sun, L. Recent Advances in Molecular Mechanisms of the NKG2D Pathway in Hepatocellular Carcinoma. Biomolecules 2020, 10, 301. [Google Scholar] [CrossRef]
- Cai, X.; Caballero-Benitez, A.; Gewe, M.M.; Jenkins, I.C.; Drescher, C.W.; Strong, R.K.; Spies, T.; Groh, V. Control of Tumor Initiation by NKG2D Naturally Expressed on Ovarian Cancer Cells. Neoplasia 2017, 19, 471–482. [Google Scholar] [CrossRef]
- Hara, R.; Onizuka, M.; Matsusita, E.; Kikkawa, E.; Nakamura, Y.; Matsushita, H.; Ohgiya, D.; Murayama, H.; Machida, S.; Ohmachi, K.; et al. NKG2D Gene Polymorphisms Are Associated with Disease Control of Chronic Myeloid Leukemia by Dasatinib. Int. J. Hematol. 2017, 106, 666–674. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the Tightrope: Natural Killer Cell Activation and Inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Moretta, L.; Bottino, C.; Pende, D.; Castriconi, R.; Mingari, M.C.; Moretta, A. Surface NK Receptors and Their Ligands on Tumor Cells. Semin. Immunol. 2006, 18, 151–158. [Google Scholar] [CrossRef]
- Parham, P. MHC Class I Molecules and Kirs in Human History, Health and Survival. Nat. Rev. Immunol. 2005, 5, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Maskalenko, N.A.; Zhigarev, D.; Campbell, K.S. Harnessing Natural Killer Cells for Cancer Immunotherapy: Dispatching the First Responders. Nat. Rev. Drug Discov. 2022, 21, 559–577. [Google Scholar] [CrossRef]
- Bruhns, P. Properties of Mouse and Human IgG Receptors and Their Contribution to Disease Models. Blood 2012, 119, 5640–5649. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Liesche, C.; van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandström, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK Cells Switch from Granzyme B to Death Receptor–Mediated Cytotoxicity during Serial Killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of Natural Killer Cell-Mediated Cellular Cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; González, S.; López-Soto, A. Chapter Eighteen—A Cytofluorimetric Assay to Evaluate Intracellular Cytokine Production by NK Cells. In Methods in Enzymology; Galluzzi, L., Rudqvist, N.-P., Eds.; Tumor Immunology and Immunotherapy—Cellular Methods Part A; Academic Press: San Diego, CA, USA, 2020; Volume 631, pp. 343–355. [Google Scholar]
- Ishii, H.; Tanabe, S.; Ueno, M.; Kubo, T.; Kayama, H.; Serada, S.; Fujimoto, M.; Takeda, K.; Naka, T.; Yamashita, T. Ifn-γ-Dependent Secretion of IL-10 from Th1 Cells and Microglia/Macrophages Contributes to Functional Recovery after Spinal Cord Injury. Cell Death Dis. 2013, 4, e710. [Google Scholar] [CrossRef]
- Crinier, A.; Narni-Mancinelli, E.; Ugolini, S.; Vivier, E. SnapShot: Natural Killer Cells. Cell 2020, 180, 1280–1280.e1. [Google Scholar] [CrossRef]
- Storkus, W.J.; Howell, D.N.; Salter, R.D.; Dawson, J.R.; Cresswell, P. NK Susceptibility Varies Inversely with Target Cell Class I HLA Antigen Expression. J. Immunol. 1987, 138, 1657–1659. [Google Scholar]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural Killer Cells and Other Innate Lymphoid Cells in Cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- van Vliet, A.A.; Georgoudaki, A.-M.; Raimo, M.; de Gruijl, T.D.; Spanholtz, J. Adoptive NK Cell Therapy: A Promising Treatment Prospect for Metastatic Melanoma. Cancers 2021, 13, 4722. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Juliá, E.P.; Mordoh, J.; Levy, E.M. Cetuximab and IL-15 Promote NK and Dendritic Cell Activation In Vitro in Triple Negative Breast Cancer. Cells 2020, 9, 1573. [Google Scholar] [CrossRef] [PubMed]
- van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the Novel Immune Checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A Phase 1 Study of Lirilumab (Antibody against Killer Immunoglobulin-like Receptor Antibody KIR2D; IPH2102) in Patients with Solid Tumors and Hematologic Malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [PubMed]
- Symons, H.J.; Leffell, M.S.; Rossiter, N.D.; Zahurak, M.; Jones, R.J.; Fuchs, E.J. Improved Survival with Inhibitory Killer Immunoglobulin Receptor (KIR) Gene Mismatches and KIR Haplotype B Donors after Nonmyeloablative, HLA-Haploidentical Bone Marrow Transplantation. Biol. Blood Marrow Transplant. 2010, 16, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Frey, N.; Porter, D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant. 2019, 25, e123–e127. [Google Scholar] [CrossRef]
- Siegler, E.L.; Zhu, Y.; Wang, P.; Yang, L. Off-the-Shelf CAR-NK Cells for Cancer Immunotherapy. Cell Stem Cell 2018, 23, 160–161. [Google Scholar] [CrossRef]
- Pahl, J.H.W.; Koch, J.; Götz, J.-J.; Arnold, A.; Reusch, U.; Gantke, T.; Rajkovic, E.; Treder, M.; Cerwenka, A. CD16A Activation of NK Cells Promotes NK Cell Proliferation and Memory-Like Cytotoxicity against Cancer Cells. Cancer Immunol. Res. 2018, 6, 517–527. [Google Scholar] [CrossRef]
- Xia, J.; Minamino, S.; Kuwabara, K. CAR-Expressing NK Cells for Cancer Therapy: A New Hope. Biosci. Trends 2020, 14, 354–359. [Google Scholar] [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef]
- Hu, B.; Jacobs, R.; Ghosh, N. Checkpoint Inhibitors Hodgkin Lymphoma and Non-Hodgkin Lymphoma. Curr. Hematol. Malig. Rep. 2018, 13, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The Diverse Functions of the PD1 Inhibitory Pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Guo, C.; Chen, H.; Zhang, H.; Zhi, L.; Lv, T.; Li, M.; Niu, Z.; Lu, P.; Zhu, W. A Novel Chimeric PD1-NKG2D-41BB Receptor Enhances Antitumor Activity of NK92 Cells against Human Lung Cancer H1299 Cells by Triggering Pyroptosis. Mol. Immunol. 2020, 122, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jiang, J.; Wu, C. CAR-NK for Tumor Immunotherapy: Clinical Transformation and Future Prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK Cells: A Promising Cellular Immunotherapy for Cancer. eBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Billadeau, D.D.; Upshaw, J.L.; Schoon, R.A.; Dick, C.J.; Leibson, P.J. NKG2D-DAP10 Triggers Human NK Cell–Mediated Killing via a Syk-Independent Regulatory Pathway. Nat. Immunol. 2003, 4, 557–564. [Google Scholar] [CrossRef]
- Lanier, L.L.; Corliss, B.C.; Wu, J.; Leong, C.; Phillips, J.H. Immunoreceptor DAP12 Bearing a Tyrosine-Based Activation Motif Is Involved in Activating NK Cells. Nature 1998, 391, 703–707. [Google Scholar] [CrossRef]
- Nakajima, H.; Colonna, M. 2B4: An NK Cell Activating Receptor with Unique Specificity and Signal Transduction Mechanism. Hum. Immunol. 2000, 61, 39–43. [Google Scholar] [CrossRef]
- Fujiwara, K.; Masutani, M.; Tachibana, M.; Okada, N. Impact of ScFv Structure in Chimeric Antigen Receptor on Receptor Expression Efficiency and Antigen Recognition Properties. Biochem. Biophys. Res. Commun. 2020, 527, 350–357. [Google Scholar] [CrossRef]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric Antigen Receptor Natural Killer (CAR-NK) Cell Design and Engineering for Cancer Therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Thokala, R.; Olivares, S.; Mi, T.; Maiti, S.; Deniger, D.; Huls, H.; Torikai, H.; Singh, H.; Champlin, R.E.; Laskowski, T.; et al. Redirecting Specificity of T Cells Using the Sleeping Beauty System to Express Chimeric Antigen Receptors by Mix-and-Matching of VL and VH Domains Targeting CD123+ Tumors. PLoS ONE 2016, 11, e0159477. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Almåsbak, H.; Walseng, E.; Kristian, A.; Myhre, M.R.; Suso, E.M.; Munthe, L.A.; Andersen, J.T.; Wang, M.Y.; Kvalheim, G.; Gaudernack, G.; et al. Inclusion of an IgG1-Fc Spacer Abrogates Efficacy of CD19 CAR T Cells in a Xenograft Mouse Model. Gene Ther. 2015, 22, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human IPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-Tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef] [PubMed]
- MacKay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L.; et al. The Therapeutic Landscape for Cells Engineered with Chimeric Antigen Receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Q.J.; Yang, S.; Kochenderfer, J.N.; Zheng, Z.; Zhong, X.; Sadelain, M.; Eshhar, Z.; Rosenberg, S.A.; Morgan, R.A. A Herceptin-Based Chimeric Antigen Receptor with Modified Signaling Domains Leads to Enhanced Survival of Transduced T Lymphocytes and Antitumor Activity. J. Immunol. 2009, 183, 5563–5574. [Google Scholar] [CrossRef]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR Activation Potential Directs Alternative T Cell Fates and Therapeutic Potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic Analysis of Chimeric Antigen Receptor Signaling Reveals Kinetic and Quantitative Differences That Affect Cell Function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-Engineered T Cells for Cancer Therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Jing, R.; Qian, L.; Zhou, C.; Sun, J. Engineering Cytoplasmic Signaling of CD28ζ CARs for Improved Therapeutic Functions. Front. Immunol. 2020, 11, 1046. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, E.J.; Sheard, V.; Hombach, A.A.; Chmielewski, M.; Riet, T.; Berrevoets, C.; Schooten, E.; Lamers, C.; Abken, H.; Debets, R.; et al. Chimeric Antigen Receptors for T-Cell Based Therapy. In Antibody Engineering: Methods and Protocols, 2nd ed.; Chames, P., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 645–666. ISBN 978-1-61779-974-7. [Google Scholar]
- Shimasaki, N.; Jain, A.; Campana, D. NK Cells for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Jawdat, D.; Madrigal, J.A.; Saudemont, A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front. Immunol. 2017, 8, 329. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Xiao, W.; Tian, Z. NK Cell-Based Immunotherapy for Cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a Human Cell Line (NK-92) with Phenotypical and Functional Characteristics of Activated Natural Killer Cells. Leukemia 1994, 8, 652–658. [Google Scholar]
- Boissel, L.; Betancur, M.; Lu, W.; Krause, D.; Van Etten, R.; Wels, W.; Klingemann, H. Retargeting NK-92 Cells by Means of CD19- and CD20-Specific Chimeric Antigen Receptors Compares Favorably with Antibody-Dependent Cellular Cytotoxicity. OncoImmunology 2013, 2, e26527. [Google Scholar] [CrossRef]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef]
- Srivastava, S.; Riddell, S.R. Chimeric Antigen Receptor T Cell Therapy: Challenges to Bench-to-Bedside Efficacy. J. Immunol. 2018, 200, 459–468. [Google Scholar] [CrossRef]
- Goldenson, B.H.; Hor, P.; Kaufman, D.S. IPSC-Derived Natural Killer Cell Therapies—Expansion and Targeting. Front. Immunol. 2022, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Smith, M.; James, S.E.; Davila, M.L.; Velardi, E.; Argyropoulos, K.V.; Gunset, G.; Perna, F.; Kreines, F.M.; Levy, E.R.; et al. Donor CD19 CAR T Cells Exert Potent Graft-versus-Lymphoma Activity with Diminished Graft-versus-Host Activity. Nat. Med. 2017, 23, 242–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, P.S.A.; Suck, G.; Nowakowska, P.; Ullrich, E.; Seifried, E.; Bader, P.; Tonn, T.; Seidl, C. Selection and Expansion of Natural Killer Cells for NK Cell-Based Immunotherapy. Cancer Immunol. Immunother. CII 2016, 65, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Lim, O.; Lee, Y.; Chung, H.; Her, J.H.; Kang, S.M.; Jung, M.; Min, B.; Shin, H.; Kim, T.M.; Heo, D.S.; et al. GMP-Compliant, Large-Scale Expanded Allogeneic Natural Killer Cells Have Potent Cytolytic Activity against Cancer Cells in Vitro and in Vivo. PLoS ONE 2013, 8, e53611. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.X.; Zhao, Y.X.; Li, B.Y.; Bao, H.J.; Jiang, H.; Qi, X.L.; Bai, L.Y.; Wang, Y.H.; Ma, Z.J.; Wu, X.Y. A Simple Method for in Vitro Preparation of Natural Killer Cells from Cord Blood. BMC Biotechnol. 2019, 19, 80. [Google Scholar] [CrossRef]
- Fuchs, E.; Tumbar, T.; Guasch, G. Socializing with the Neighbors: Stem Cells and Their Niche. Cell 2004, 116, 769–778. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.-C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of Allogeneic HSC-Engineered INKT Cells for off-the-Shelf Cancer Immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]
- Pfaff, N.; Lachmann, N.; Kohlscheen, S.; Sgodda, M.; Araúzo-Bravo, M.J.; Greber, B.; Kues, W.; Glage, S.; Baum, C.; Niemann, H.; et al. Efficient Hematopoietic Redifferentiation of Induced Pluripotent Stem Cells Derived from Primitive Murine Bone Marrow Cells. Stem Cells Dev. 2012, 21, 689–701. [Google Scholar] [CrossRef]
- Woll, P.S.; Grzywacz, B.; Tian, X.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human Embryonic Stem Cells Differentiate into a Homogeneous Population of Natural Killer Cells with Potent in Vivo Antitumor Activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef]
- Lim, W.F.; Inoue-Yokoo, T.; Tan, K.S.; Lai, M.I.; Sugiyama, D. Hematopoietic Cell Differentiation from Embryonic and Induced Pluripotent Stem Cells. Stem Cell Res. Ther. 2013, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.N.; Lee, D.A.; Kaufman, D.S. Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Lupo, K.B.; Moon, J.-I.; Chambers, A.M.; Matosevic, S. Differentiation of Natural Killer Cells from Induced Pluripotent Stem Cells under Defined, Serum- and Feeder-Free Conditions. Cytotherapy 2021, 23, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.S.; Davis, R.P.; Azzola, L.; Stanley, E.G.; Elefanty, A.G. Forced Aggregation of Defined Numbers of Human Embryonic Stem Cells into Embryoid Bodies Fosters Robust, Reproducible Hematopoietic Differentiation. Blood 2005, 106, 1601–1603. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.L.; Ni, Z.; Kaufman, D.S. Human Pluripotent Stem Cells as a Renewable Source of Natural Killer Cells. In Hematopoietic Differentiation of Human Pluripotent Stem Cells; Cheng, T., Ed.; SpringerBriefs in Stem Cells; Springer: Dordrecht, The Netherlands, 2015; pp. 69–79. ISBN 978-94-017-7312-6. [Google Scholar]
- Kaneko, S. In Vitro Differentiation of T-Cells; Springer: Dordrecht, The Netherlands, 2019; ISBN 1-4939-9727-0. [Google Scholar]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells 2016, 34, 93–101. [Google Scholar] [CrossRef]
- Zhu, H.; Kaufman, D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. In In Vitro Differentiation of T-Cells: Methods and Protocols; Kaneko, S., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 107–119. ISBN 978-1-4939-9728-2. [Google Scholar]
- van der Ploeg, K.; Chang, C.; Ivarsson, M.A.; Moffett, A.; Wills, M.R.; Trowsdale, J. Modulation of Human Leukocyte Antigen-C by Human Cytomegalovirus Stimulates KIR2DS1 Recognition by Natural Killer Cells. Front. Immunol. 2017, 8, 298. [Google Scholar] [CrossRef]
- Zeng, J.; Tang, S.Y.; Toh, L.L.; Wang, S. Generation of “Off-the-Shelf” Natural Killer Cells from Peripheral Blood Cell-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 1796–1812. [Google Scholar] [CrossRef]
- Jaleco, A.C.; Neves, H.; Hooijberg, E.; Gameiro, P.; Clode, N.; Haury, M.; Henrique, D.; Parreira, L. Differential Effects of Notch Ligands Delta-1 and Jagged-1 in Human Lymphoid Differentiation. J. Exp. Med. 2001, 194, 991–1002. [Google Scholar] [CrossRef]
- Sung, T.-C.; Li, H.-F.; Higuchi, A.; Kumar, S.S.; Ling, Q.-D.; Wu, Y.-W.; Burnouf, T.; Nasu, M.; Umezawa, A.; Lee, K.-F.; et al. Effect of Cell Culture Biomaterials for Completely Xeno-Free Generation of Human Induced Pluripotent Stem Cells. Biomaterials 2020, 230, 119638. [Google Scholar] [CrossRef]
- Low, P.T.; Lai, M.I.; Ngai, S.C.; Abdullah, S. Transgene Expression from CpG-Reduced Lentiviral Gene Delivery Vectors in Vitro. Gene 2014, 533, 451–455. [Google Scholar] [CrossRef]
- Perry, C.; Rayat, A.C.M.E. Lentiviral Vector Bioprocessing. Viruses 2021, 13, 268. [Google Scholar] [CrossRef]
- Li, L.; Liu, L.N.; Feller, S.; Allen, C.; Shivakumar, R.; Fratantoni, J.; Wolfraim, L.A.; Fujisaki, H.; Campana, D.; Chopas, N.; et al. Expression of Chimeric Antigen Receptors in Natural Killer Cells with a Regulatory-Compliant Non-Viral Method. Cancer Gene Ther. 2010, 17, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimasaki, N.; Fujisaki, H.; Cho, D.; Masselli, M.; Lockey, T.; Eldridge, P.; Leung, W.; Campana, D. A Clinically Adaptable Method to Enhance the Cytotoxicity of Natural Killer Cells against B-Cell Malignancies. Cytotherapy 2012, 14, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Song, D.; Li, Z.; Guo, B.; Xiao, Y.; Liu, W.; Liang, L.; Feng, C.; Gao, T.; Chen, Y.; et al. Immunity-and-Matrix-Regulatory Cells Derived from Human Embryonic Stem Cells Safely and Effectively Treat Mouse Lung Injury and Fibrosis. Cell Res. 2020, 30, 794–809. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. IPSC-Derived NK Cells Maintain High Cytotoxicity and Enhance in Vivo Tumor Control in Concert with T Cells and Anti–PD-1 Therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef] [PubMed]
- Valamehr, B.; Abujarour, R.; Robinson, M.; Le, T.; Robbins, D.; Shoemaker, D.; Flynn, P. A Novel Platform to Enable the High-Throughput Derivation and Characterization of Feeder-Free Human IPSCs. Sci. Rep. 2012, 2, 213. [Google Scholar] [CrossRef]
- Valamehr, B.; Robinson, M.; Abujarour, R.; Rezner, B.; Vranceanu, F.; Le, T.; Medcalf, A.; Lee, T.T.; Fitch, M.; Robbins, D.; et al. Platform for Induction and Maintenance of Transgene-Free HiPSCs Resembling Ground State Pluripotent Stem Cells. Stem Cell Rep. 2014, 2, 366–381. [Google Scholar] [CrossRef]
- Bai, Q.; Ramirez, J.-M.; Becker, F.; Pantesco, V.; Lavabre-Bertrand, T.; Hovatta, O.; Lemaître, J.-M.; Pellestor, F.; De Vos, J. Temporal Analysis of Genome Alterations Induced by Single-Cell Passaging in Human Embryonic Stem Cells. Stem Cells Dev. 2015, 24, 653–662. [Google Scholar] [CrossRef]
- Matsubara, H.; Niwa, A.; Nakahata, T.; Saito, M.K. Induction of Human Pluripotent Stem Cell-Derived Natural Killer Cells for Immunotherapy under Chemically Defined Conditions. Biochem. Biophys. Res. Commun. 2019, 515, 1–8. [Google Scholar] [CrossRef]
- Tosic, V.; Thomas, D.L.; Kranz, D.M.; Liu, J.; McFadden, G.; Shisler, J.L.; MacNeill, A.L.; Roy, E.J. Myxoma Virus Expressing a Fusion Protein of Interleukin-15 (IL15) and IL15 Receptor Alpha Has Enhanced Antitumor Activity. PLoS ONE 2014, 9, e109801. [Google Scholar] [CrossRef]
- Lin, J.-X.; Leonard, W.J. The Common Cytokine Receptor γ Chain Family of Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028449. [Google Scholar] [CrossRef] [PubMed]
- Phan, M.-T.T.; Lee, S.-H.; Kim, S.-K.; Cho, D. Expansion of NK Cells Using Genetically Engineered K562 Feeder Cells. In Natural Killer Cells: Methods and Protocols; Somanchi, S.S., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 167–174. ISBN 978-1-4939-3684-7. [Google Scholar]
- Romee, R.; Schneider, S.E.; Leong, J.W.; Chase, J.M.; Keppel, C.R.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Cytokine Activation Induces Human Memory-like NK Cells. Blood 2012, 120, 4751–4760. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, S.; Varchetta, S.; Mele, D.; Donadon, M.; Torzilli, G.; Soldani, C.; Franceschini, B.; Porta, C.; Chiellino, S.; Pedrazzoli, P.; et al. An Anti-MICA/B Antibody and IL-15 Rescue Altered NKG2D-Dependent NK Cell Responses in Hepatocellular Carcinoma. Cancers 2020, 12, 3583. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-N.; Cui, Y.-X.; Ruge, F.; Jiang, W.G. Interleukin 21 and Its Receptor Play a Role in Proliferation, Migration and Invasion of Breast Cancer Cells. Cancer Genom. Proteom. 2015, 12, 211–221. [Google Scholar]
- Kasaian, M.T.; Whitters, M.J.; Carter, L.L.; Lowe, L.D.; Jussif, J.M.; Deng, B.; Johnson, K.A.; Witek, J.S.; Senices, M.; Konz, R.F.; et al. IL-21 Limits NK Cell Responses and Promotes Antigen-Specific T Cell Activation: A Mediator of the Transition from Innate to Adaptive Immunity. Immunity 2002, 16, 559–569. [Google Scholar] [CrossRef]
- Skak, K.; Frederiksen, K.S.; Lundsgaard, D. Interleukin-21 Activates Human Natural Killer Cells and Modulates Their Surface Receptor Expression. Immunology 2008, 123, 575–583. [Google Scholar] [CrossRef]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-Bound IL-21 Promotes Sustained Ex Vivo Proliferation of Human Natural Killer Cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Hu, J.; He, X.; Xu, J.; Zhou, Y.; Yue, Y.; Gao, Y.; Church, G.; Yang, L. Abstract 6070: Functional Natural Killer Cells Derived from Engineered HiPSC with Hypoimmunity Gene Combo Demonstrate Hypoimmunity Features in Evading Host Attacks. Cancer Res. 2022, 82, 6070. [Google Scholar] [CrossRef]
- Elahi, R.; Heidary, A.H.; Hadiloo, K.; Esmaeilzadeh, A. Chimeric Antigen Receptor-Engineered Natural Killer (CAR NK) Cells in Cancer Treatment; Recent Advances and Future Prospects. Stem Cell Rev. Rep. 2021, 17, 2081–2106. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Melo Garcia, L.; Li, Y.; Rezvani, K. CAR-NK Cells: The next Wave of Cellular Therapy for Cancer. Clin. Transl. Immunol. 2021, 10, e1274. [Google Scholar] [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-Edited, Donor-Derived Allogeneic Anti-CD19 Chimeric Antigen Receptor T Cells in Paediatric and Adult B-Cell Acute Lymphoblastic Leukaemia: Results of Two Phase 1 Studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Faridi, R.M.; Kemp, T.J.; Dharmani-Khan, P.; Lewis, V.; Tripathi, G.; Rajalingam, R.; Daly, A.; Berka, N.; Storek, J.; Khan, F.M. Donor-Recipient Matching for KIR Genotypes Reduces Chronic GVHD and Missing Inhibitory KIR Ligands Protect against Relapse after Myeloablative, HLA Matched Hematopoietic Cell Transplantation. PLoS ONE 2016, 11, e0158242. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Portillo, A.L.; Hogg, R.; Ashkar, A.A. Production of Human CAR-NK Cells with Lentiviral Vectors and Functional Assessment in Vitro. STAR Protoc. 2021, 2, 100956. [Google Scholar] [CrossRef]
- Wang, Y.; Li, S.; Tian, Z.; Sun, J.; Liang, S.; Zhang, B.; Bai, L.; Zhang, Y.; Zhou, X.; Xiao, S.; et al. Generation of a Caged Lentiviral Vector through an Unnatural Amino Acid for Photo-Switchable Transduction. Nucleic Acids Res. 2019, 47, e114. [Google Scholar] [CrossRef]
- Clements, M.O.; Godfrey, A.; Crossley, J.; Wilson, S.J.; Takeuchi, Y.; Boshoff, C. Lentiviral Manipulation of Gene Expression in Human Adult and Embryonic Stem Cells. Tissue Eng. 2006, 12, 1741–1751. [Google Scholar] [CrossRef]
- Piersanti, S.; Sacchetti, B.; Funari, A.; Cesare, S.D.; Bonci, D.; Cherubini, G.; Peschle, C.; Riminucci, M.; Bianco, P.; Saggio, I. Lentiviral Transduction of Human Postnatal Skeletal (Stromal, Mesenchymal) Stem Cells: In Vivo Transplantation and Gene Silencing. Calcif. Tissue Int. 2006, 78, 372–384. [Google Scholar] [CrossRef]
- Lu, H.; Zhao, X.; Li, Z.; Hu, Y.; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 3151. [Google Scholar] [CrossRef]
- Hodgins, J.J.; Khan, S.T.; Park, M.M.; Auer, R.C.; Ardolino, M. Killers 2.0: NK Cell Therapies at the Forefront of Cancer Control. J. Clin. Investig. 2019, 129, 3499–3510. [Google Scholar] [CrossRef]
- Porter, M.D.; Shadbolt, B. Randomized Controlled Trial of Accelerated Rehabilitation versus Standard Protocol Following Surgical Repair of Ruptured Achilles Tendon. ANZ J. Surg. 2015, 85, 373–377. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domains | CAR-T | CAR-NK | |
|---|---|---|---|
| TAA binding region | VH-VL, VL-VH | VH-VL, VL-VH, VH-only | |
| Hinge domain/spacer | Non IgG-based: | CD8a, CD28 | CD8a, CD28, DAP12, IgG4, IgG2 CH2-CH3, IgG1 CH2-CH3, IgG4 CH2-CH3 |
| IgG-based: | IgG1, IgG4 | ||
| Transmembrane | CD3ζ, CD28, ICOS, CD8a, CD4 | CD3ζ, CD28, CD8a, 4-1BB, DAP12, TCR ab, CD28-CD3ζ, FceRIγ, murine CD3ζ | |
| ITAM | e.g., CD3ζ, CD28, 4-1BB, CD27, ICOS, OX-40, MYD88, CD40, KIR2DSS2 | e.g., CD3ζ, CD28, 4-1BB | |
| Region Name | China | United States | Japan | Europe | Other Countries and Regions |
|---|---|---|---|---|---|
| CAR-T | 447 | 240 | 10 | 59 | 35 |
| CAR-NK | 18 | 5 | 0 | 0 | 5 |
| Clinical Trial | NK Source | Interventions | Conditions | Status | Locations | Phase |
|---|---|---|---|---|---|---|
| NCT04887012 | unpublished | Biological: anti-CD19 CAR-NK | B-cell Non Hodgkin Lymphoma | Recruiting | 2nd Affiliated Hospital, School of Medicine, Zhejiang University Hangzhou, Zhejiang, China | Phase 1 |
| NCT05213195 | unpublished | Drug: NKG2D CAR-NK | Refractory Metastatic Colorectal Cancer | Recruiting | The First Affiliated Hospital, Zhejiang University Hangzhou, Zhejiang, China | Phase 1 |
| NCT05215015 | unpublished | Biological: Anti-CD33/CLL1 CAR-NK Cells | Acute Myeloid Leukemia | Recruiting | Wuxi People’s Hospital Wuxi, Jiangsu, China | Early Phase 1 |
| NCT05194709 | unpublished | Biological: Anti-CAR-NK Cells | Advanced Solid Tumors | Recruiting | Wuxi People’s Hospital Wuxi, Jiangsu, China | Early Phase 1 |
| NCT04639739 | unpublished | Biological: anti-CD19 CAR NK | NHL | Not yet recruiting | Department of Hematology, Xinqiao Hospital ChongQing, Chongqing, China | Early Phase 1 |
| NCT03692767 | unpublished | Biological: Anti-CD22 CAR NK Cells | Refractory B-Cell Lymphoma | Unknown | Early Phase 1 | |
| NCT03690310 | unpublished | Biological: Anti-CD19 CAR NK Cells | Refractory B-Cell Lymphoma | Unknown | Early Phase 1 | |
| NCT05008575 | unpublished | Biological: anti-CD33 CAR NK cells Drug: Fludarabine Drug: Cytoxan | Leukemia, Myeloid, Acute | Recruiting | Department of Hematology, Xinqiao Hospital Chongqing, Chongqing, China | Phase 1 |
| NCT04324996 | unpublished | Biological: NK cells, IL15-NK cells, NKG2D CAR-NK cells, ACE2 CAR-NK cells, NKG2D-ACE2 CAR-NK cells | COVID-19 | Recruiting | Chongqing Public Health Medical Center Chongqing, China | Phase 1 Phase 2 |
| NCT03692637 | peripheral blood | Biological: anti-Mesothelin Car NK Cells | Epithelial Ovarian Cancer | Unknown | Early Phase 1 | |
| NCT03415100 | peripheral blood | Biological: CAR-NK cells targeting NKG2D ligands | Solid Tumours | Unknown | Third Affiliated Hospital of Guangzhou Medical University Guangzhou, Guangdong, China | Phase 1 |
| NCT03692663 | unpublished | Biological: anti-PSMA CAR NK cells | Castration-resistant Prostate Cancer | Unknown | Early Phase 1 | |
| NCT05008536 | unpublished | Biological: Anti-BCMA CAR-NK Cells Drug: Fludarabine Drug: Cytoxan | Multiple Myeloma, Refractory | Recruiting | Department of Hematology, Xinqiao Hospital Chongqing, Chongqing, China | Early Phase 1 |
| NCT03940820 | unpublished | Biological: ROBO1 CAR-NK cells | Solid Tumor | Recruiting | Radiation Therapy Department, Suzhou Cancer Center, Suzhou Hospital Affiliated to Nanjing Medical University Suzhou, Jiangsu, China | Phase 1 Phase 2 |
| NCT03940833 | unpublished | Biological: BCMA CAR-NK 92 cells | Multiple Myeloma | Recruiting | Department of Hematology, Wuxi People’s Hospital, Nanjing Medical University Wuxi, Jiangsu, China | Phase 1 Phase 2 |
| NCT04847466 | unpublished | Drug: N-803 Drug: Pembrolizumab Biological: PD-L1 t-haNK | Gastroesophageal Junction (GEJ) Cancers Advanced HNSCC | Recruiting | National Institutes of Health Clinical Center Bethesda, MD, USA | Phase 2 |
| NCT03824964 | unpublished | Biological: Anti-CD19/CD22 CAR NK Cells | Refractory B-Cell Lymphoma | Unknown | Early Phase 1 | |
| NCT05020678 | peripheral blood | Biological: NKX019 | Lymphoma, Non-Hodgkin B-cell Acute Lymphoblastic Leukemia Large B-cell Lymphoma (and 7 more) | Recruiting | Colorado Blood Cancer InstituteDenver, CO, USA University of Chicago Chicago, IL, USA The Cleveland Clinic Foundation Cleveland, OH, USA (and 4 more…) | Phase 1 |
| NCT02944162 | NK-92 cell line | Biological: anti-CD33 CAR-NK cells | Acute Myelogenous Leukemia Acute Myeloid Leukemia Acute Myeloid Leukemia With Maturation (and 2 more) | Unknown | PersonGen BioTherapeutics (Suzhou) Co., Ltd. Suzhou, Jiangsu, China | Phase 1 Phase 2 |
| NCT03579927 | umbilical Cord Blood | Procedure: Autologous Hematopoietic Stem Cell Transplantation Drug: Carmustine Drug: Cytarabine (and 5 more…) | CD19 Positive Mantle Cell Lymphoma Recurrent Diffuse Large B-Cell Lymphoma (and 4 more) | Withdrawn | M D Anderson Cancer Center Houston, TX, USA | Phase 1 Phase 2 |
| NCT05182073 | peripheral blood | Drug: FT576 Drug: Cyclophosphamide Drug: Fludarabine Drug: Daratumumab | Multiple Myeloma Myeloma | Recruiting | Colorado Blood Cancer Institute Denver, CO, USA Tennessee Oncology—Nashville Nashville, TN, USA | Phase 1 |
| NCT05248048 | unpublished | Biological: CAR-T infusion | Refractory Metastatic Colorectal Cancer | Recruiting | The Third Affiliated Hospital of Guangzhou Medical University Guangzhou, Guangdong, China | Early Phase 1 |
| NCT05410717 | peripheral blood | Biological: Claudin6 targeting CAR-NK cell | Stage IV Ovarian Cancer Testis Cancer, Refractory Endometrial Cancer Recurrent CAR NK | Recruiting | The Second Affiliated Hospital of Guangzhou Medical University Guangzhou, Guangdong, China Guangzhou, Guangdong, China | Phase 1 Phase 2 |
| NCT05247957 | unpublished | Biological: CAR-NK cells | Safety and Efficacy | Recruiting | Hebei Yanda Lu Daopei HospitalSanhe, Hebei, China | Phase 1 |
| NCT05472558 | cord blood | Biological: anti-CD19 CAR-NK | B-cell Non Hodgkin Lymphoma | Not yet recruiting | 2nd Affiliated Hospital, School of Medicine, Zhejiang University Hanzhou, Zhejiang, China | Phase 1 |
| NCT04623944 | unpublished | Biological: NKX101—CAR NK cell therapy | Relapsed/Refractory AML AML, AdultMDS Refractory Myelodysplastic Syndromes | Recruiting | Colorado Blood Cancer Institute Denver, CO, USA Winship Cancer Institute, Emory University Atlanta, GA, USA University of Chicago Medical Center Chicago, IL, USA (and 4 more) | Phase 1 |
| NCT05410041 | unpublished | Biological: CAR-NK-CD19 Cells | Acute Lymphocytic Leukemia Chronic Lymphocytic Leukemia Non Hodgkin Lymphoma | Recruiting | Beijing Boren Hospital Beijing, Beijing, China | Phase 1 |
| NCT05336409 | unpublished | Biological: CNTY-101 Biological: IL-2 Drug: Lymphodepleting Chemotherapy | R/R CD19-Positive B-Cell Malignancies Indolent Non-Hodgkin LymphomaAggressive Non-Hodgkin Lymphoma | Not yet recruiting | Phase 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pang, Z.; Wang, Z.; Li, F.; Feng, C.; Mu, X. Current Progress of CAR-NK Therapy in Cancer Treatment. Cancers 2022, 14, 4318. https://doi.org/10.3390/cancers14174318
Pang Z, Wang Z, Li F, Feng C, Mu X. Current Progress of CAR-NK Therapy in Cancer Treatment. Cancers. 2022; 14(17):4318. https://doi.org/10.3390/cancers14174318
Chicago/Turabian StylePang, Zhaojun, Zhongyi Wang, Fengqi Li, Chunjing Feng, and Xin Mu. 2022. "Current Progress of CAR-NK Therapy in Cancer Treatment" Cancers 14, no. 17: 4318. https://doi.org/10.3390/cancers14174318
APA StylePang, Z., Wang, Z., Li, F., Feng, C., & Mu, X. (2022). Current Progress of CAR-NK Therapy in Cancer Treatment. Cancers, 14(17), 4318. https://doi.org/10.3390/cancers14174318