
CDH1 (E-cadherin) Gene Methylation in Human Breast Cancer: Critical Appraisal of a Long and Twisted Story


Abstract
:Simple Summary
Abstract
1. Introduction
2. Genetic or Epigenetic in Activation
3. DNA Methylation
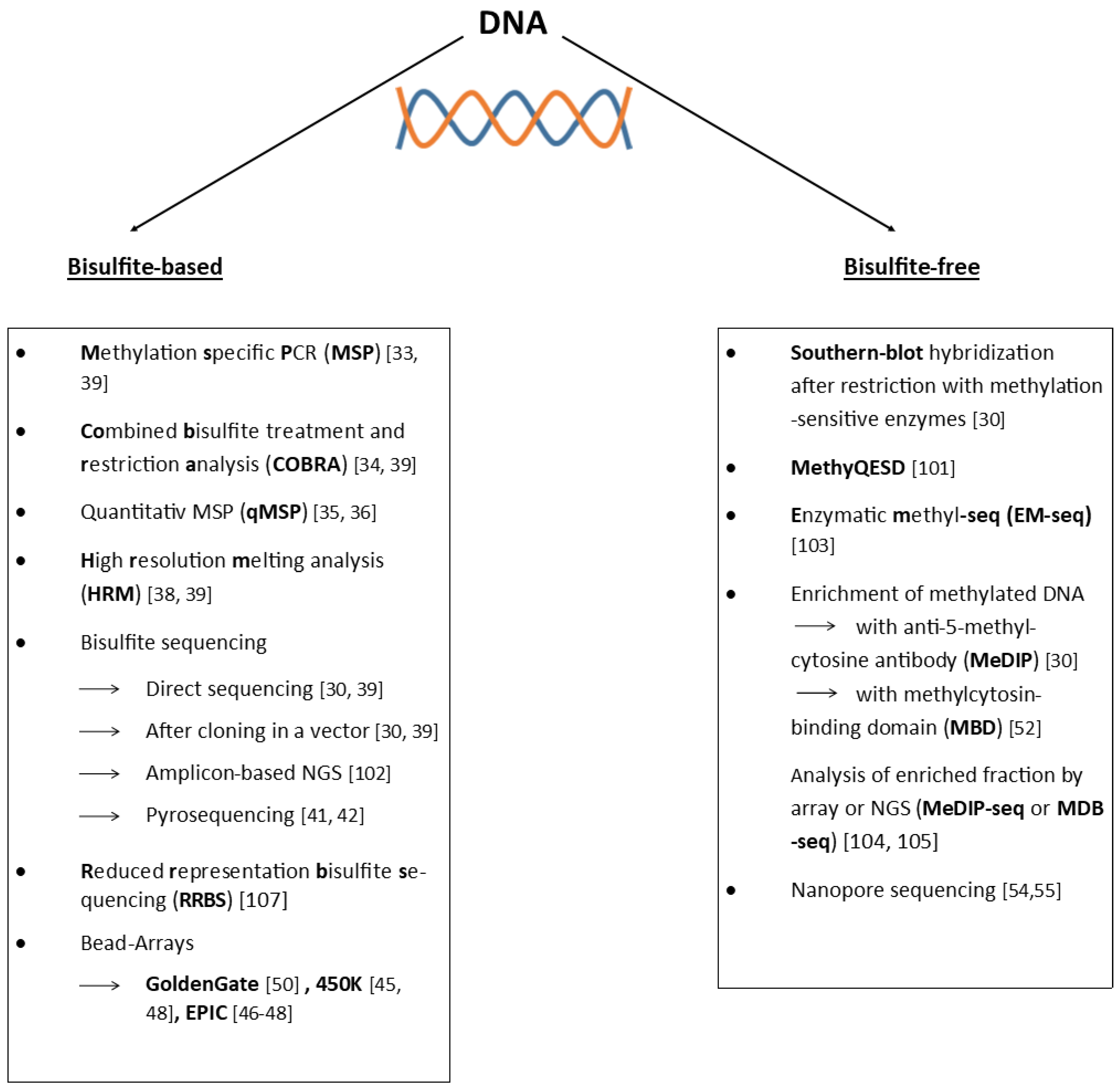
4. Detection of DNA Methylation
4.1. Bisulfite-Based
4.2. Bisulfite-Free
5. CDH1 Gene Methylation
6. In Breast Cancer
7. CDH1 Gene Methylation in Lobular Breast Cancer
8. DNA Methylation Is Cell-Type Specific
9. The Problem with Reviews
10. Wider Implications
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van Roy, F.; Berx, G. The cell-cell adhesion molecule E-cadherin. Cell Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Bruner, H.C.; Derksen, P.W.B. Loss of E-cadherin-dependent cell-cell adhesion and the development and progression of cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a029330. [Google Scholar] [CrossRef]
- Daulagala, A.C.; Bridges, M.C.; Kourtidis, A. E-cadherin beyond structure: A signaling hub in colon homeostasis and disease. Int. J. Mol. Sci. 2019, 20, 2756. [Google Scholar] [CrossRef]
- Takeichi, M. Functional correlation between cell adhesive properties and some cell surface proteins. J. Cell Biol. 1977, 75, 464–474. [Google Scholar] [CrossRef]
- Leckband, D.E.; de Rooij, J. Cadherin adhesion and mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin switch in tumor progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef]
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet Oncol. 2020, 21, e386–e397. [Google Scholar] [CrossRef]
- Corso, G.; Montagna, G.; Figueiredo, J.; La Vecchia, C.; Romario, U.F.; Fernandes, M.S.; Seixas, S.; Roviello, F.; Trovato, C.; Guerini-Rocco, E.; et al. Hereditary gastric and breast cancer syndromes related to cdh1 germline mutation: A multidisciplinary clinical review. Cancers 2020, 12, 1598. [Google Scholar] [CrossRef]
- Weinberg, R.A. The Biology of Cancer, 2nd ed.; Garland Science: New York, NY, USA, 2014; p. 876. [Google Scholar]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Gheldof, A.; Berx, G. Cadherins and epithelial-to-mesenchymal transition. Prog. Mol. Biol. Transl. Sci. 2013, 116, 317–336. [Google Scholar] [PubMed]
- Sarkies, P. Encyclopaedia of eukaryotic DNA methylation: From patterns to mechanisms and functions. Biochem. Soc. Trans. 2022, 50, 1179–1190. [Google Scholar] [CrossRef]
- Doerfler, W. DNA methylation—A regulatory signal in eukaryotic gene expression. J. Gen. Virol. 1981, 57, 1–20. [Google Scholar] [CrossRef]
- Deaton, A.M.; Bird, A. Cpg islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Smith, J.; Sen, S.; Weeks, R.J.; Eccles, M.R.; Chatterjee, A. Promoter DNA hypermethylation and paradoxical gene activation. Trends Cancer 2020, 6, 392–406. [Google Scholar] [CrossRef]
- Jones, P.A. DNA methylation and cancer. Cancer Res. 1986, 46, 461–466. [Google Scholar]
- Toyota, M.; Issa, J.P. Epigenetic changes in solid and hematopoietic tumors. Semin. Oncol. 2005, 32, 521–530. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Brandes, J.C.; Vertino, P.M. Cancer DNA methylation: Molecular mechanisms and clinical implications. Clin. Cancer Res. 2009, 15, 3927–3937. [Google Scholar] [CrossRef] [PubMed]
- Witte, T.; Plass, C.; Gerhauser, C. Pan-cancer patterns of DNA methylation. Genome Med. 2014, 6, 66. [Google Scholar] [CrossRef]
- Greger, V.; Passarge, E.; Hopping, W.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum. Genet. 1989, 83, 155–158. [Google Scholar] [CrossRef]
- Buiting, K.; Kanber, D.; Horsthemke, B.; Lohmann, D. Imprinting of rb1 (the new kid on the block). Brief. Funct. Genom. 2010, 9, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Clark, S.J.; Harrison, J.; Paul, C.L.; Frommer, M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994, 22, 2990–2997. [Google Scholar]
- Ushijima, T. Detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer 2005, 5, 223–231. [Google Scholar] [CrossRef]
- Ammerpohl, O.; Martin-Subero, J.I.; Richter, J.; Vater, I.; Siebert, R. Hunting for the 5th base: Techniques for analyzing DNA methylation. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 847–862. [Google Scholar] [CrossRef]
- Grunau, C.; Clark, S.J.; Rosenthal, A. Bisulfite genomic sequencing: Systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001, 29, e65. [Google Scholar] [CrossRef]
- Warnecke, P.M.; Stirzaker, C.; Song, J.; Grunau, C.; Melki, J.R.; Clark, S.J. Identification and resolution of artifacts in bisulfite sequencing. Methods 2002, 27, 101–107. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific pcr: A novel pcr assay for methylation status of cpg islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Laird, P.W. Cobra: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. Methylight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, E32. [Google Scholar] [CrossRef]
- Campan, M.; Weisenberger, D.J.; Trinh, B.; Laird, P.W. Methylight and digital methylight. Methods Mol. Biol. 2018, 1708, 497–513. [Google Scholar]
- Warnecke, P.M.; Stirzaker, C.; Melki, J.R.; Millar, D.S.; Paul, C.L.; Clark, S.J. Detection and measurement of pcr bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res. 1997, 25, 4422–4426. [Google Scholar] [CrossRef]
- Wojdacz, T.K.; Dobrovic, A.; Hansen, L.L. Methylation-sensitive high-resolution melting. Nat. Protoc. 2008, 3, 1903–1908. [Google Scholar] [CrossRef]
- Mikeska, T.; Candiloro, I.L.; Dobrovic, A. The implications of heterogeneous DNA methylation for the accurate quantification of methylation. Epigenomics 2010, 2, 561–573. [Google Scholar] [CrossRef]
- Barros-Silva, D.; Marques, C.J.; Henrique, R.; Jeronimo, C. Profiling DNA methylation based on next-generation sequencing approaches: New insights and clinical applications. Genes 2018, 9, 429. [Google Scholar] [CrossRef]
- Tost, J.; Dunker, J.; Gut, I.G. Analysis and quantification of multiple methylation variable positions in cpg islands by pyrosequencing. Biotechniques 2003, 35, 152–156. [Google Scholar] [CrossRef]
- Roessler, J.; Lehmann, U. Quantitative DNA methylation analysis by pyrosequencing (r). Methods Mol. Biol. 2015, 1315, 175–188. [Google Scholar] [PubMed]
- Wojdacz, T.K.; Hansen, L.L.; Dobrovic, A. A new approach to primer design for the control of pcr bias in methylation studies. BMC Res. Notes 2008, 1, 54. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, E.A.; Zavgorodnij, M.G.; Majorova, S.P.; Vorobjev, I.A.; Jandaghi, P.; Bure, I.V.; Hoheisel, J.D. Correction of pcr-bias in quantitative DNA methylation studies by means of cubic polynomial regression. Nucleic Acids Res. 2011, 39, e77. [Google Scholar] [CrossRef] [Green Version]
- Dedeurwaerder, S.; Defrance, M.; Calonne, E.; Denis, H.; Sotiriou, C.; Fuks, F. Evaluation of the infinium methylation 450k technology. Epigenomics 2011, 3, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the illumina methylationepic beadchip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef]
- Kling, T.; Wenger, A.; Beck, S.; Caren, H. Validation of the methylationepic beadchip for fresh-frozen and formalin-fixed paraffin-embedded tumours. Clin. Epigenetics 2017, 9, 33. [Google Scholar] [CrossRef]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of infinium DNA methylation beadchip probes. Nucleic Acids Res. 2017, 45, e22. [Google Scholar] [CrossRef]
- Dedeurwaerder, S.; Defrance, M.; Bizet, M.; Calonne, E.; Bontempi, G.; Fuks, F. A comprehensive overview of infinium humanmethylation450 data processing. Brief. Bioinform. 2014, 15, 929–941. [Google Scholar] [CrossRef]
- Bibikova, M.; Fan, J.B. Goldengate assay for DNA methylation profiling. Methods Mol. Biol. 2009, 507, 149–163. [Google Scholar]
- Rauch, T.A.; Pfeifer, G.P. DNA methylation profiling using the methylated-cpg island recovery assay (mira). Methods 2010, 52, 213–217. [Google Scholar] [CrossRef]
- Nair, S.S.; Coolen, M.W.; Stirzaker, C.; Song, J.Z.; Statham, A.L.; Strbenac, D.; Robinson, M.D.; Clark, S.J. Comparison of methyl-DNA immunoprecipitation (medip) and methyl-cpg binding domain (mbd) protein capture for genome-wide DNA methylation analysis reveal cpg sequence coverage bias. Epigenetics 2011, 6, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.F.; Shabalin, A.A.; Xie, L.Y.; Adkins, D.E.; Zhao, M.; Turecki, G.; Clark, S.L.; Aberg, K.A.; van den Oord, E. Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome. Nucleic Acids Res. 2017, 45, e97. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Gouil, Q.; Keniry, A. Latest techniques to study DNA methylation. Essays Biochem. 2019, 63, 639–648. [Google Scholar] [PubMed]
- Bettstetter, M.; Dechant, S.; Ruemmele, P.; Vogel, C.; Kurz, K.; Morak, M.; Keller, G.; Holinski-Feder, E.; Hofstaedter, F.; Dietmaier, W. MethyQESD, a robust and fast method for quantitative methylation analyses in HNPCC diagnostics using formalin-fixed and paraffin-embedded tissue samples. Lab. Investig. 2008, 88, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Potapova, A.; Albat, C.; Hasemeier, B.; Haeussler, K.; Lamprecht, S.; Suerbaum, S.; Kreipe, H.; Lehmann, U. Systematic cross-validation of 454 sequencing and pyrosequencing for the exact quantification of DNA methylation patterns with single cpg resolution. BMC Biotechnol. 2011, 11, 6. [Google Scholar] [CrossRef]
- Vaisvila, R.; Ponnaluri, V.K.C.; Sun, Z.; Langhorst, B.W.; Saleh, L.; Guan, S.; Dai, N.; Campbell, M.A.; Sexton, B.S.; Marks, K.; et al. Enzymatic methyl sequencing detects DNA methylation at single-base resolution from pictograms of DNA. Genome Res. 2021, 31, 1280–1289. [Google Scholar] [CrossRef]
- Ben Maamar, M.; Riggleman, I.S.; Beck, D.; Skinner, M.K. Genome-Wide Mapping of DNA Methylation 5mC by Methylated DNA Immunoprecipitation (MeDIP)-Sequencing. Methods Mol. Biol. 2021, 2198, 301–310. [Google Scholar]
- Brinkmann, A.B.; Simmer, F.; Ma, K.; Kaan, A.; Zhu, J.; Stunnenberg, H.G. Whole-genome DNA methylation profiling using MethylCapseq. Methods 2010, 52, 232–236. [Google Scholar] [CrossRef]
- Gu, H.; Smith, Z.D.; Boch, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of reduced representation bisulfite sequencing libraries for genomic-scale DNA methylation profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef]
- Sharma, M.; Verma, R.K.; Kumar, S.; Kumar, V. Computational challenges in detection of cancer using cell-free DNA methylation. Comput. Struct. Biotechnol. J. 2021, 20, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Yoshiura, K.; Kanai, Y.; Ochiai, A.; Shimoyama, Y.; Sugimura, T.; Hirohashi, S. Silencing of the E-cadherin invasion-suppressor gene by cpg methylation in human carcinomas. Proc. Natl. Acad. Sci. USA 1995, 92, 7416–7419. [Google Scholar] [CrossRef] [PubMed]
- Das, P.M.; Singal, R. DNA methylation and cancer. J. Clin. Oncol. 2004, 22, 4632–4642. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Antequera, F.; Boyes, J.; Bird, A. High levels of de novo methylation and altered chromatin structure at cpg islands in cell lines. Cell 1990, 62, 503–514. [Google Scholar] [CrossRef]
- Smiraglia, D.J.; Rush, L.J.; Fruhwald, M.C.; Dai, Z.; Held, W.A.; Costello, J.F.; Lang, J.C.; Eng, C.; Li, B.; Wright, F.A.; et al. Excessive cpg island hypermethylation in cancer cell lines versus primary human malignancies. Hum. Mol. Genet. 2001, 10, 1413–1419. [Google Scholar] [CrossRef]
- Hennessey, P.T.; Ochs, M.F.; Mydlarz, W.W.; Hsueh, W.; Cope, L.; Yu, W.; Califano, J.A. Promoter methylation in head and neck squamous cell carcinoma cell lines is significantly different than methylation in primary tumors and xenografts. PLoS ONE 2011, 6, e20584. [Google Scholar] [CrossRef]
- Roessler, J.; Ammerpohl, O.; Gutwein, J.; Steinemann, D.; Schlegelberger, B.; Weyer, V.; Sariyar, M.; Geffers, R.; Arnold, N.; Schmutzler, R.; et al. The cpg island methylator phenotype in breast cancer is associated with the lobular subtype. Epigenomics 2015, 7, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar]
- Graff, J.R.; Herman, J.G.; Myohanen, S.; Baylin, S.B.; Vertino, P.M. Mapping patterns of cpg island methylation in normal and neoplastic cells implicates both upstream and downstream regions in de novo methylation. J. Biol. Chem. 1997, 272, 22322–22329. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Shinozaki, M.; Hoon, D.S.; Giuliano, A.E.; Hansen, N.M.; Wang, H.J.; Turner, R.; Taback, B. Distinct hypermethylation profile of primary breast cancer is associated with sentinel lymph node metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, J.R.; Prando, E.C.; Quevedo, F.C.; Neto, F.A.; Rainho, C.A.; Rogatto, S.R. Cdh1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer 2006, 6, 48. [Google Scholar] [CrossRef] [PubMed]
- Droufakou, S.; Deshmane, V.; Roylance, R.; Hanby, A.; Tomlinson, I.; Hart, I.R. Multiple ways of silencing E-cadherin gene expression in lobular carcinoma of the breast. Int. J. Cancer 2001, 92, 404–408. [Google Scholar] [CrossRef]
- Toyooka, K.O.; Toyooka, S.; Maitra, A.; Feng, Q.; Kiviat, N.C.; Smith, A.; Minna, J.D.; Ashfaq, R.; Gazdar, A.F. Establishment and validation of real-time polymerase chain reaction method for cdh1 promoter methylation. Am. J. Pathol. 2002, 161, 629–634. [Google Scholar] [CrossRef]
- Sebova, K.; Zmetakova, I.; Bella, V.; Kajo, K.; Stankovicova, I.; Kajabova, V.; Krivulcik, T.; Lasabova, Z.; Tomka, M.; Galbavy, S.; et al. Rassf1a and cdh1 hypermethylation as potential epimarkers in breast cancer. Cancer Biomark. 2011, 10, 13–26. [Google Scholar] [CrossRef]
- Swift-Scanlan, T.; Blackford, A.; Argani, P.; Sukumar, S.; Fackler, M.J. Two-color quantitative multiplex methylation-specific pcr. Biotechniques 2006, 40, 210–219. [Google Scholar] [CrossRef]
- Swift-Scanlan, T.; Vang, R.; Blackford, A.; Fackler, M.J.; Sukumar, S. Methylated genes in breast cancer: Associations with clinical and histopathological features in a familial breast cancer cohort. Cancer Biol. Ther. 2011, 11, 853–865. [Google Scholar] [CrossRef]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between cpg methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, R57. [Google Scholar] [CrossRef]
- Liu, J.; Sun, X.; Qin, S.; Wang, H.; Du, N.; Li, Y.; Pang, Y.; Wang, C.; Xu, C.; Ren, H. Cdh1 promoter methylation correlates with decreased gene expression and poor prognosis in patients with breast cancer. Oncol. Lett. 2016, 11, 2635–2643. [Google Scholar] [CrossRef]
- Naghitorabi, M.; Mohammadi-Asl, J.; Sadeghi, H.M.; Rabbani, M.; Jafarian-Dehkordi, A.; Javanmard, S.H. Quantitation of cdh1 promoter methylation in formalin-fixed paraffin-embedded tissues of breast cancer patients using differential high resolution melting analysis. Adv. Biomed. Res. 2016, 5, 91. [Google Scholar] [PubMed]
- Pixberg, C.F.; Raba, K.; Muller, F.; Behrens, B.; Honisch, E.; Niederacher, D.; Neubauer, H.; Fehm, T.; Goering, W.; Schulz, W.A.; et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene 2017, 36, 3223–3231. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Gorringe, K.L.; Pang, J.B.; Byrne, D.J.; Takano, E.A.; kConFab, I.; Dobrovic, A.; Fox, S.B. Brca2 carriers with male breast cancer show elevated tumour methylation. BMC Cancer 2017, 17, 641. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Leone, J.P. Male breast cancer: An updated review of epidemiology, clinicopathology, and treatment. J. Oncol. 2022, 2022, 1734049. [Google Scholar] [CrossRef] [PubMed]
- McCullough, L.E.; Collin, L.J.; Conway, K.; White, A.J.; Cho, Y.H.; Shantakumar, S.; Terry, M.B.; Teitelbaum, S.L.; Neugut, A.I.; Santella, R.M.; et al. Reproductive characteristics are associated with gene-specific promoter methylation status in breast cancer. BMC Cancer 2019, 19, 926. [Google Scholar] [CrossRef]
- Sivadas, A.; Kok, V.C.; Ng, K.L. Multi-omics analyses provide novel biological insights to distinguish lobular ductal types of invasive breast cancers. Breast Cancer Res. Treat. 2022, 193, 361–379. [Google Scholar] [CrossRef]
- Christgen, M.; Cserni, G.; Floris, G.; Marchio, C.; Djerroudi, L.; Kreipe, H.; Derksen, P.W.B.; Vincent-Salomon, A. Lobular breast cancer: Histomorphology and different concepts of a special spectrum of tumors. Cancers 2021, 13, 3695. [Google Scholar] [CrossRef]
- Sarrio, D.; Moreno-Bueno, G.; Hardisson, D.; Sanchez-Estevez, C.; Guo, M.; Herman, J.G.; Gamallo, C.; Esteller, M.; Palacios, J. Epigenetic and genetic alterations of apc and cdh1 genes in lobular breast cancer: Relationships with abnormal E-cadherin and catenin expression and microsatellite instability. Int. J. Cancer 2003, 106, 208–215. [Google Scholar] [CrossRef]
- Lombaerts, M.; Middeldorp, J.W.; van der Weide, E.; Philippo, K.; van Wezel, T.; Smit, V.T.; Cornelisse, C.J.; Cleton-Jansen, A.M. Infiltrating leukocytes confound the detection of E-cadherin promoter methylation in tumors. Biochem. Biophys. Res. Commun. 2004, 319, 697–704. [Google Scholar] [CrossRef]
- Zou, D.; Yoon, H.S.; Perez, D.; Weeks, R.J.; Guilford, P.; Humar, B. Epigenetic silencing in non-neoplastic epithelia identifies E-cadherin (cdh1) as a target for chemoprevention of lobular neoplasia. J. Pathol. 2009, 218, 265–272. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Mariani, O.; Meaudre, C.; Fuhrmann, L.; Xiao, H.; Naidoo, K.; Gillespie, A.; Roxanis, I.; Vincent-Salomon, A.; Haider, S.; et al. Assessment of the molecular heterogeneity of E-cadherin expression in invasive lobular breast cancer. Cancers 2022, 14, 295. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, U.; Langer, F.; Feist, H.; Glockner, S.; Hasemeier, B.; Kreipe, H. Quantitative assessment of promoter hypermethylation during breast cancer development. Am. J. Pathol. 2002, 160, 605–612. [Google Scholar] [CrossRef]
- Strathdee, G.; Sim, A.; Soutar, R.; Holyoake, T.L.; Brown, R. Hoxa5 is targeted by cell-type-specific cpg island methylation in normal cells and during the development of acute myeloid leukaemia. Carcinogenesis 2007, 28, 299–309. [Google Scholar] [CrossRef]
- Christgen, M.; Steinemann, D.; Kuhnle, E.; Langer, F.; Gluz, O.; Harbeck, N.; Kreipe, H. Lobular breast cancer: Clinical, molecular and morphological characteristics. Pathol. Res. Pract. 2016, 212, 583–597. [Google Scholar] [CrossRef]
- Kristiansen, S.; Jorgensen, L.M.; Guldberg, P.; Soletormos, G. Aberrantly methylated DNA as a biomarker in breast cancer. Int. J. Biol. Markers 2013, 28, 141–150. [Google Scholar] [CrossRef]
- Huang, R.; Ding, P.; Yang, F. Clinicopathological significance and potential drug target of cdh1 in breast cancer: A meta-analysis and literature review. Drug Des. Dev. Ther. 2015, 9, 5277–5285. [Google Scholar]
- Davalos, V.; Martinez-Cardus, A.; Esteller, M. The epigenomic revolution in breast cancer: From single-gene to genome-wide next-generation approaches. Am. J. Pathol. 2017, 187, 2163–2174. [Google Scholar] [CrossRef]
- de Ruijter, T.C.; van der Heide, F.; Smits, K.M.; Aarts, M.J.; van Engeland, M.; Heijnen, V.C.G. Prognostic DNA methylation markers for hormone receptor breast cancer: A systematic review. Breast Cancer Res. 2020, 22, 13. [Google Scholar] [CrossRef]
- Mouabbi, J.A.; Hassan, A.; Lim, B.; Hortobagyi, G.N.; Tripathy, D.; Layman, R.M. Invasive lobular carcinoma: An understudied emergent subtype of breast cancer. Breast Cancer Res. Treat. 2022, 193, 253–264. [Google Scholar] [CrossRef]
- Lehmann, U. Lobular breast cancer–the most common special subtype or a most special common subtype? Breast Cancer Res. 2015, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Kalachand, R.D.; Stordal, B.; Madden, S.; Chandler, B.; Cunningham, J.; Goode, E.L.; Braicu, E.I.; Sehouli, J.; Ignatow, A.; Yu, H.; et al. BRCA1 Promotor Methylation and Clinical Outcomes in Ovarian Cancer: An Individual patient Data Meta-Analysis. J. Natl. Cancer Inst. 2020, 11, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Sahnane, N.; Carnevali, I.; Formenti, G.; Casarin, J.; Facchi, S.; Bombelli, R.; Di Lauro, E.; Memoli, D.; Salvati, A.; Rizzo, F.; et al. Brca methylation testing identifies a subset of ovarian carcinomas without germline variants that can benefit from parp inhibitor. Int. J. Mol. Sci. 2020, 21, 9708. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef]
- Menghi, F.; Banda, K.; Kumar, P.; Straub, R.; Dobrolecki, L.; Rodriguez, I.V.; Yost, S.E.; Chandok, H.; Radke, M.R.; Somlo, G.; et al. Genomic and Epigenomic BRCA alterations predict adaptive resistance and response to platinum-based therapy in aptients with triple-negative breast and ovarian carcinomas. Sci. Trans. Med. 2022, 14, eabn1926. [Google Scholar] [CrossRef]
- van Vlodrop, I.J.; Niessen, H.E.; Derks, S.; Baldewijns, M.M.; van Criekinge, W.; Herman, J.G.; van Engeland, M. Analysis of promoter cpg island hypermethylation in cancer: Location, location, location! Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Study | n = | Methylated Cases | Method | Reference |
|---|---|---|---|---|
| Droufakou et al. Int. J. Cancer 2001 | 22 | 17 (77%) | MSP | [75] |
| Sarrio et al. Int. J. Cancer | 46 | 19 (41%) | MSP | [89] |
| Lombaerts et al. BBRC | 11 | 8 (73%) | MSP | [90] |
| Shinozaki et al. Clin. Cancer Res. 2005 | 33 | 12 (36%) | MSP | [73] |
| Zou et al. J. Pathol. 2009 | 14 | 13 (93%) | MSP | [91] |
| Ciriello et al. Cell 2015 | 127 | 0 | 450 k array + NGS | [92] |
| Liu et al. Oncol. Lett. 2016 | 31 | 8 (26%) | MSP | [81] |
| Alexander et al. Cancers 2022 | 18 | 0 | EPIC array | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bücker, L.; Lehmann, U. CDH1 (E-cadherin) Gene Methylation in Human Breast Cancer: Critical Appraisal of a Long and Twisted Story. Cancers 2022, 14, 4377. https://doi.org/10.3390/cancers14184377
Bücker L, Lehmann U. CDH1 (E-cadherin) Gene Methylation in Human Breast Cancer: Critical Appraisal of a Long and Twisted Story. Cancers. 2022; 14(18):4377. https://doi.org/10.3390/cancers14184377
Chicago/Turabian StyleBücker, Lara, and Ulrich Lehmann. 2022. "CDH1 (E-cadherin) Gene Methylation in Human Breast Cancer: Critical Appraisal of a Long and Twisted Story" Cancers 14, no. 18: 4377. https://doi.org/10.3390/cancers14184377
APA StyleBücker, L., & Lehmann, U. (2022). CDH1 (E-cadherin) Gene Methylation in Human Breast Cancer: Critical Appraisal of a Long and Twisted Story. Cancers, 14(18), 4377. https://doi.org/10.3390/cancers14184377