

Neurofibroma Development in Neurofibromatosis Type 1: Insights from Cellular Origin and Schwann Cell Lineage Development


Abstract
:Simple Summary
Abstract
1. Introduction
2. Neurofibroma Formation
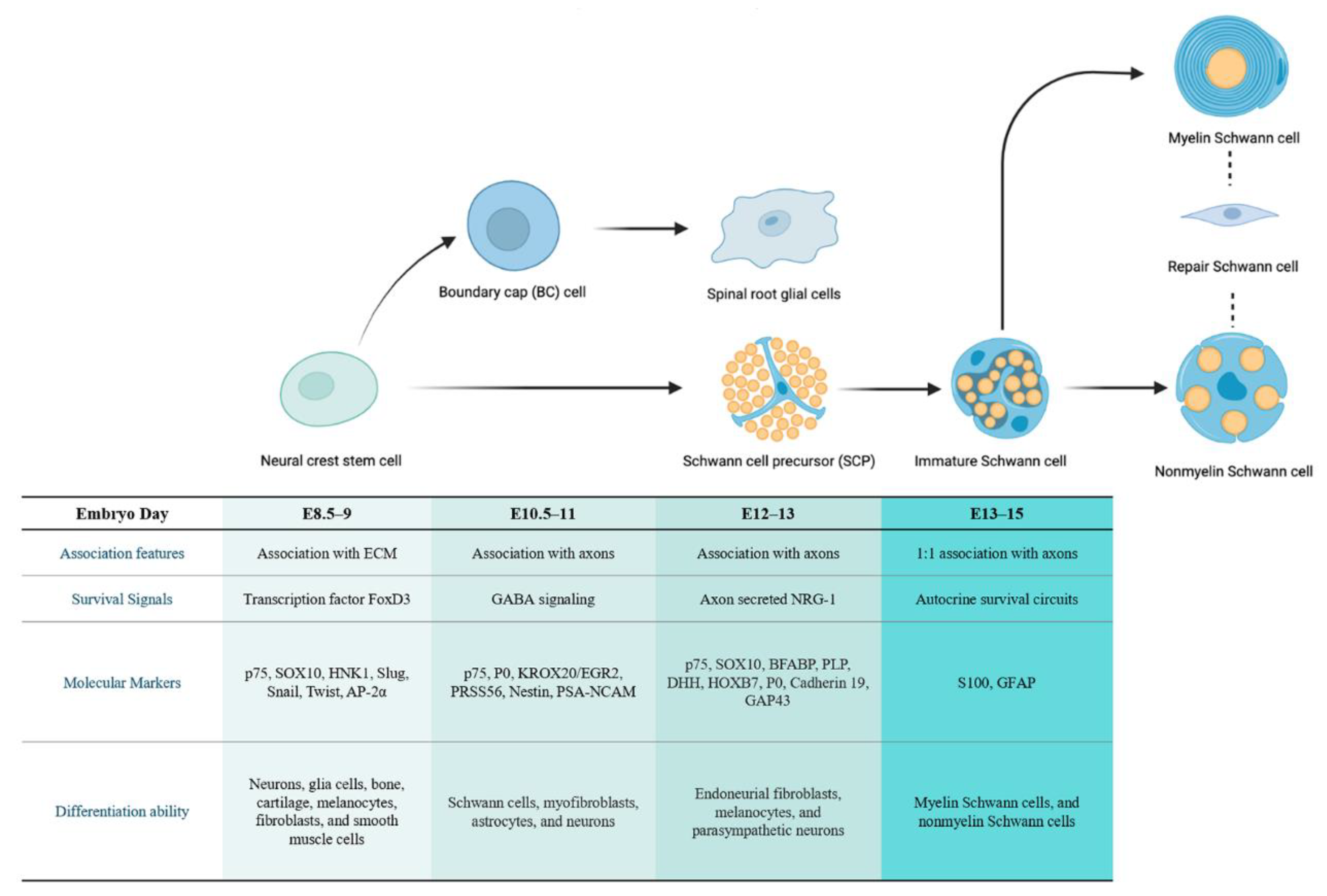
2.1. The Developmental Origin of SC Lineages
2.1.1. The Cellular Origin of Neurofibroma
2.1.2. The Cellular Origin of pNF
2.1.3. The Cellular Origin of cNF
2.1.4. Associate pNF and cNF with a Common Stage of Origin
2.2. Alterations in SCs in the Early Stage of Tumorigenesis
3. Neurofibroma Progression
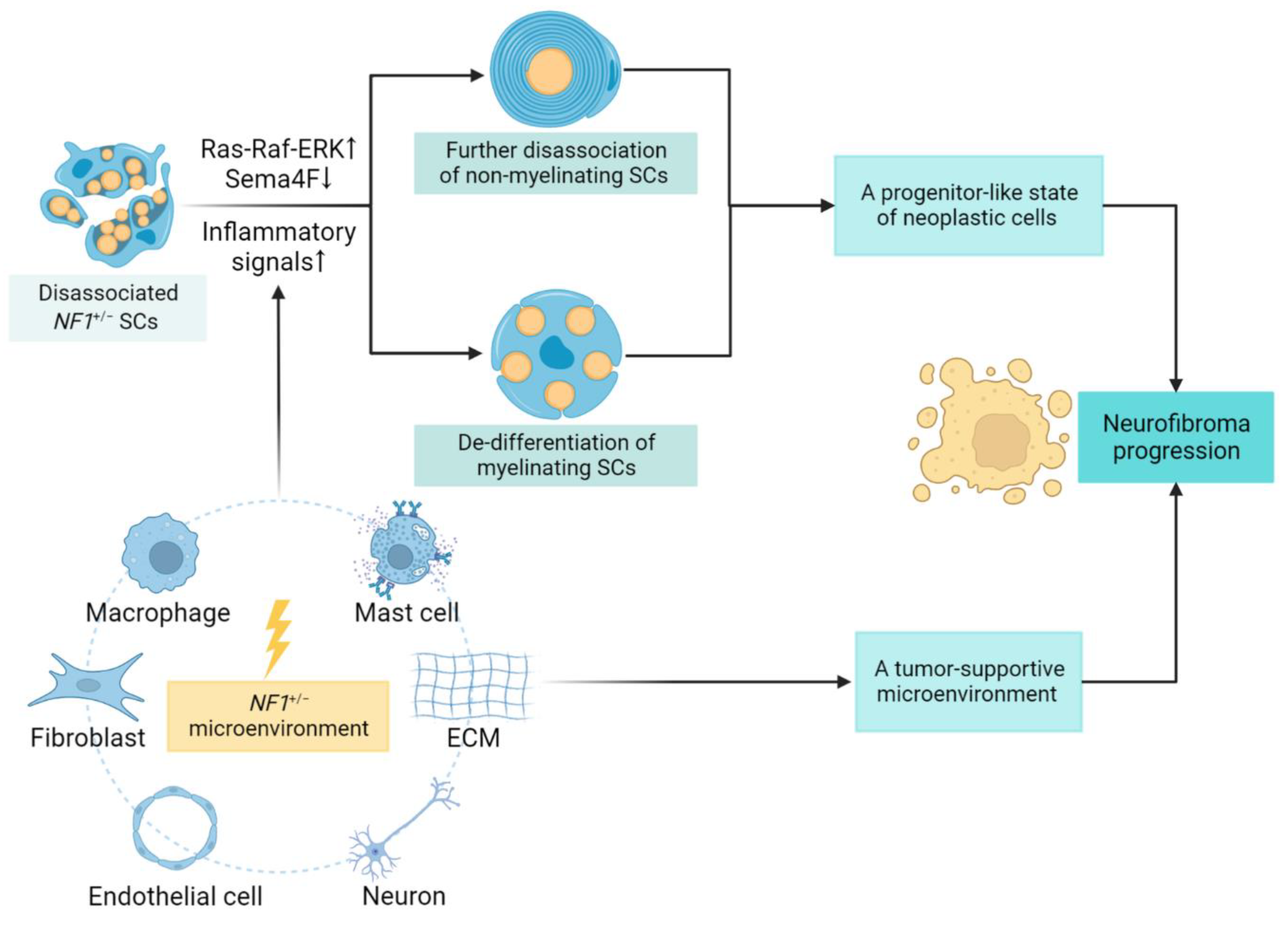
3.1. SCs Contribution and Lineage Shift
3.2. Role of the Tumor Microenvironment
4. Malignant Transformation of Neurofibroma
4.1. SCs Transition and Microenvironment Alteration
4.2. Accumulation of Additional Gene Mutations
4.3. Dysregulated Signaling Pathways
5. Discussion and Future Directions
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xu, G.F.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R.; et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- Friedman, J.M. Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 1–6. [Google Scholar] [CrossRef]
- Scala, M.; Schiavetti, I.; Madia, F.; Chelleri, C.; Piccolo, G.; Accogli, A.; Riva, A.; Salpietro, V.; Bocciardi, R.; Morcaldi, G.; et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: A Single-Center Cohort Study. Cancers 2021, 13, 1879. [Google Scholar] [CrossRef] [PubMed]
- Ferner, R.E. Neurofibromatosis 1. Eur. J. Hum. Genet. 2007, 15, 131–138. [Google Scholar] [CrossRef]
- Dugoff, L.; Sujansky, E. Neurofibromatosis type 1 and pregnancy. Am. J. Med. Genet. 1996, 66, 7–10. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Pichard, D.C.; Legius, E.H.; Wolkenstein, P.; Lavker, R.M.; Blakeley, J.O.; Riccardi, V.M.; Verma, S.K.; Brownell, I.; Le, L.Q. The biology of cutaneous neurofibromas: Consensus recommendations for setting research priorities. Neurology 2018, 91, S14–S20. [Google Scholar] [CrossRef]
- Akshintala, S.; Baldwin, A.; Liewehr, D.J.; Goodwin, A.; Blakeley, J.O.; Gross, A.M.; Steinberg, S.M.; Dombi, E.; Widemann, B.C. Longitudinal evaluation of peripheral nerve sheath tumors in neurofibromatosis type 1: Growth analysis of plexiform neurofibromas and distinct nodular lesions. Neuro-Oncol. 2020, 22, 1368–1378. [Google Scholar] [CrossRef]
- Woodruff, J.M. Pathology of tumors of the peripheral nerve sheath in type 1 neurofibromatosis. Am. J. Med. Genet. 1999, 89, 23–30. [Google Scholar] [CrossRef]
- Sørensen, S.A.; Mulvihill, J.J.; Nielsen, A. Long-term follow-up of von Recklinghausen neurofibromatosis. Survival and malignant neoplasms. N. Engl. J. Med. 1986, 314, 1010–1015. [Google Scholar] [CrossRef]
- Evans, D.G.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Mohamad, T.; Plante, C.; Brosseau, J.P. Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development. Int. J. Mol. Sci. 2021, 22, 8620. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Ghosh, P.; Charnay, P.; Burns, D.K.; Parada, L.F. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 2002, 296, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Sheela, S.; Riccardi, V.M.; Ratner, N. Angiogenic and invasive properties of neurofibroma Schwann cells. J. Cell Biol. 1990, 111, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mo, J.; Brosseau, J.P.; Shipman, T.; Wang, Y.; Liao, C.P.; Cooper, J.M.; Allaway, R.J.; Gosline, S.J.C.; Guinney, J.; et al. Spatiotemporal Loss of NF1 in Schwann Cell Lineage Leads to Different Types of Cutaneous Neurofibroma Susceptible to Modification by the Hippo Pathway. Cancer Discov. 2019, 9, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Crump, T. Translation of case reports in Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen by F. v. Recklinghausen. Adv. Neurol. 1981, 29, 259–275. [Google Scholar] [PubMed]
- Kamata, Y. Study on the ultrastructure and acetylcholinesterase activity in von Recklinghausen’s neurofibromatosis. Acta Pathol. Jpn. 1978, 28, 393–410. [Google Scholar] [CrossRef]
- Le, L.Q.; Liu, C.; Shipman, T.; Chen, Z.; Suter, U.; Parada, L.F. Susceptible stages in Schwann cells for NF1-associated plexiform neurofibroma development. Cancer Res. 2011, 71, 4686–4695. [Google Scholar] [CrossRef]
- Mayes, D.A.; Rizvi, T.A.; Cancelas, J.A.; Kolasinski, N.T.; Ciraolo, G.M.; Stemmer-Rachamimov, A.O.; Ratner, N. Perinatal or adult Nf1 inactivation using tamoxifen-inducible PlpCre each cause neurofibroma formation. Cancer Res. 2011, 71, 4675–4685. [Google Scholar] [CrossRef]
- Keng, V.W.; Rahrmann, E.P.; Watson, A.L.; Tschida, B.R.; Moertel, C.L.; Jessen, W.J.; Rizvi, T.A.; Collins, M.H.; Ratner, N.; Largaespada, D.A. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res. 2012, 72, 3405–3413. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Patel, A.J.; Liao, C.P.; Wang, Y.; Le, L.Q. Cells of origin in the embryonic nerve roots for NF1-associated plexiform neurofibroma. Cancer Cell 2014, 26, 695–706. [Google Scholar] [CrossRef] [Green Version]
- Chaney, K.E.; Perrino, M.R.; Kershner, L.J.; Patel, A.V.; Wu, J.; Choi, K.; Rizvi, T.A.; Dombi, E.; Szabo, S.; Largaespada, D.A.; et al. Cdkn2a Loss in a Model of Neurofibroma Demonstrates Stepwise Tumor Progression to Atypical Neurofibroma and MPNST. Cancer Res. 2020, 80, 4720–4730. [Google Scholar] [CrossRef] [PubMed]
- Stemple, D.L.; Anderson, D.J. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 1992, 71, 973–985. [Google Scholar] [CrossRef]
- Achilleos, A.; Trainor, P.A. Neural crest stem cells: Discovery, properties and potential for therapy. Cell Res. 2012, 22, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Soldatov, R.; Kaucka, M.; Kastriti, M.E.; Petersen, J.; Chontorotzea, T.; Englmaier, L.; Akkuratova, N.; Yang, Y.; Häring, M.; Dyachuk, V.; et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 2019, 364, 9536. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.; Ding, X.; Wang, A.; Li, S. Neural crest-like stem cells for tissue regeneration. Stem Cells Transl. Med. 2021, 10, 681–693. [Google Scholar] [CrossRef]
- Golding, J.P.; Cohen, J. Border controls at the mammalian spinal cord: Late-surviving neural crest boundary cap cells at dorsal root entry sites may regulate sensory afferent ingrowth and entry zone morphogenesis. Mol. Cell. Neurosci. 1997, 9, 381–396. [Google Scholar] [CrossRef]
- Niederländer, C.; Lumsden, A. Late emigrating neural crest cells migrate specifically to the exit points of cranial branchiomotor nerves. Development 1996, 122, 2367–2374. [Google Scholar] [CrossRef]
- Radomska, K.J.; Topilko, P. Boundary cap cells in development and disease. Curr. Opin. Neurobiol. 2017, 47, 209–215. [Google Scholar] [CrossRef]
- Topilko, P.; Schneider-Maunoury, S.; Levi, G.; Baron-Van Evercooren, A.; Chennoufi, A.B.; Seitanidou, T.; Babinet, C.; Charnay, P. Krox-20 controls myelination in the peripheral nervous system. Nature 1994, 371, 796–799. [Google Scholar] [CrossRef]
- Zujovic, V.; Thibaud, J.; Bachelin, C.; Vidal, M.; Deboux, C.; Coulpier, F.; Stadler, N.; Charnay, P.; Topilko, P.; Baron-Van Evercooren, A. Boundary cap cells are peripheral nervous system stem cells that can be redirected into central nervous system lineages. Proc. Natl. Acad. Sci. USA 2011, 108, 10714–10719. [Google Scholar] [CrossRef] [Green Version]
- Gresset, A.; Coulpier, F.; Gerschenfeld, G.; Jourdon, A.; Matesic, G.; Richard, L.; Vallat, J.M.; Charnay, P.; Topilko, P. Boundary Caps Give Rise to Neurogenic Stem Cells and Terminal Glia in the Skin. Stem Cell Rep. 2015, 5, 278–290. [Google Scholar] [CrossRef] [PubMed]
- Coulpier, F.; Le Crom, S.; Maro, G.S.; Manent, J.; Giovannini, M.; Maciorowski, Z.; Fischer, A.; Gessler, M.; Charnay, P.; Topilko, P. Novel features of boundary cap cells revealed by the analysis of newly identified molecular markers. Glia 2009, 57, 1450–1457. [Google Scholar] [CrossRef]
- Dong, Z.; Sinanan, A.; Parkinson, D.; Parmantier, E.; Mirsky, R.; Jessen, K.R. Schwann cell development in embryonic mouse nerves. J. Neurosci. Res. 1999, 56, 334–348. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005, 6, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Maro, G.S.; Vermeren, M.; Voiculescu, O.; Melton, L.; Cohen, J.; Charnay, P.; Topilko, P. Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat. Neurosci. 2004, 7, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci. 1999, 22, 402–410. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Schwann Cell Precursors; Multipotent Glial Cells in Embryonic Nerves. Front. Mol. Neurosci. 2019, 12, 69. [Google Scholar] [CrossRef]
- Soto, J.; Monje, P.V. Axon contact-driven Schwann cell dedifferentiation. Glia 2017, 65, 864–882. [Google Scholar] [CrossRef]
- Feltri, M.L.; Poitelon, Y.; Previtali, S.C. How Schwann Cells Sort Axons: New Concepts. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2016, 22, 252–265. [Google Scholar] [CrossRef]
- Zhu, Y.; Parada, L.F. The molecular and genetic basis of neurological tumours. Nat. Rev. Cancer 2002, 2, 616–626. [Google Scholar] [CrossRef]
- Joseph, N.M.; Mosher, J.T.; Buchstaller, J.; Snider, P.; McKeever, P.E.; Lim, M.; Conway, S.J.; Parada, L.F.; Zhu, Y.; Morrison, S.J. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell 2008, 13, 129–140. [Google Scholar] [CrossRef]
- Zheng, H.; Chang, L.; Patel, N.; Yang, J.; Lowe, L.; Burns, D.K.; Zhu, Y. Induction of abnormal proliferation by nonmyelinating schwann cells triggers neurofibroma formation. Cancer Cell 2008, 13, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Mattiaccio, A.; Conti, A.; Turco, L.; Seri, M.; Piscaglia, F.; Morelli, M.C. Genetics in Familial Intrahepatic Cholestasis: Clinical Patterns and Development of Liver and Biliary Cancers: A Review of the Literature. Cancers 2022, 14, 3421. [Google Scholar] [CrossRef]
- Saito, H.; Yoshida, T.; Yamazaki, H.; Suzuki, N. Conditional N-rasG12V expression promotes manifestations of neurofibromatosis in a mouse model. Oncogene 2007, 26, 4714–4719. [Google Scholar] [CrossRef]
- Wu, J.; Williams, J.P.; Rizvi, T.A.; Kordich, J.J.; Witte, D.; Meijer, D.; Stemmer-Rachamimov, A.O.; Cancelas, J.A.; Ratner, N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 2008, 13, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Le, L.Q.; Shipman, T.; Burns, D.K.; Parada, L.F. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell 2009, 4, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Radomska, K.J.; Coulpier, F.; Gresset, A.; Schmitt, A.; Debbiche, A.; Lemoine, S.; Wolkenstein, P.; Vallat, J.M.; Charnay, P.; Topilko, P. Cellular Origin, Tumor Progression, and Pathogenic Mechanisms of Cutaneous Neurofibromas Revealed by Mice with Nf1 Knockout in Boundary Cap Cells. Cancer Discov. 2019, 9, 130–147. [Google Scholar] [CrossRef]
- Mo, J.; Anastasaki, C.; Chen, Z.; Shipman, T.; Papke, J.; Yin, K.; Gutmann, D.H.; Le, L.Q. Humanized neurofibroma model from induced pluripotent stem cells delineates tumor pathogenesis and developmental origins. J. Clin. Investig. 2021, 131, e139807. [Google Scholar] [CrossRef]
- Corfas, G.; Velardez, M.O.; Ko, C.P.; Ratner, N.; Peles, E. Mechanisms and roles of axon-Schwann cell interactions. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 9250–9260. [Google Scholar] [CrossRef]
- Parrinello, S.; Noon, L.A.; Harrisingh, M.C.; Wingfield Digby, P.; Rosenberg, L.H.; Cremona, C.A.; Echave, P.; Flanagan, A.M.; Parada, L.F.; Lloyd, A.C. NF1 loss disrupts Schwann cell-axonal interactions: A novel role for semaphorin 4F. Genes Dev. 2008, 22, 3335–3348. [Google Scholar] [CrossRef] [Green Version]
- Stonecypher, M.S.; Byer, S.J.; Grizzle, W.E.; Carroll, S.L. Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene 2005, 24, 5589–5605. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, S.; Lloyd, A.C. Neurofibroma development in NF1-insights into tumour initiation. Trends Cell Biol. 2009, 19, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef] [PubMed]
- Shamash, S.; Reichert, F.; Rotshenker, S. The cytokine network of Wallerian degeneration: Tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J. Neurosci. 2002, 22, 3052–3060. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, T.A.; Huang, Y.; Sidani, A.; Atit, R.; Largaespada, D.A.; Boissy, R.E.; Ratner, N. A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve. J. Neurosci. 2002, 22, 9831–9840. [Google Scholar] [CrossRef]
- Ribeiro, S.; Napoli, I.; White, I.J.; Parrinello, S.; Flanagan, A.M.; Suter, U.; Parada, L.F.; Lloyd, A.C. Injury signals cooperate with Nf1 loss to relieve the tumor-suppressive environment of adult peripheral nerve. Cell Rep. 2013, 5, 126–136. [Google Scholar] [CrossRef]
- Singer, A.J.; Clark, R.A. Cutaneous wound healing. N. Engl. J. Med. 1999, 341, 738–746. [Google Scholar] [CrossRef]
- Liao, C.P.; Booker, R.C.; Brosseau, J.P.; Chen, Z.; Mo, J.; Tchegnon, E.; Wang, Y.; Clapp, D.W.; Le, L.Q. Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. J. Clin. Investig. 2018, 128, 2848–2861. [Google Scholar] [CrossRef]
- Yang, F.C.; Ingram, D.A.; Chen, S.; Zhu, Y.; Yuan, J.; Li, X.; Yang, X.; Knowles, S.; Horn, W.; Li, Y.; et al. Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell 2008, 135, 437–448. [Google Scholar] [CrossRef]
- Li, F.; Munchhof, A.M.; White, H.A.; Mead, L.E.; Krier, T.R.; Fenoglio, A.; Chen, S.; Wu, X.; Cai, S.; Yang, F.C.; et al. Neurofibromin is a novel regulator of RAS-induced signals in primary vascular smooth muscle cells. Hum. Mol. Genet. 2006, 15, 1921–1930. [Google Scholar] [CrossRef] [Green Version]
- Staser, K.; Yang, F.C.; Clapp, D.W. Pathogenesis of plexiform neurofibroma: Tumor-stromal/hematopoietic interactions in tumor progression. Annu. Rev. Pathol. 2012, 7, 469–495. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; McKay, R.M.; Le, L.Q. Tumorigenesis in neurofibromatosis type 1: Role of the microenvironment. Oncogene 2021, 40, 5781–5787. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, E.; Rantanen, M.; Kallionpää, R.A.; Pöyhönen, M.; Leppävirta, J.; Ylä-Outinen, H.; Riccardi, V.M.; Pukkala, E.; Pitkäniemi, J.; Peltonen, S.; et al. Distinctive Cancer Associations in Patients With Neurofibromatosis Type 1. J. Clin. Oncol. 2016, 34, 1978–1986. [Google Scholar] [CrossRef]
- Carroll, S.L. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012, 123, 321–348. [Google Scholar] [CrossRef]
- Lee, P.R.; Cohen, J.E.; Tendi, E.A.; Farrer, R.; GH, D.E.V.; Becker, K.G.; Fields, R.D. Transcriptional profiling in an MPNST-derived cell line and normal human Schwann cells. Neuron. Glia Biol. 2004, 1, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef]
- Mo, W.; Chen, J.; Patel, A.; Zhang, L.; Chau, V.; Li, Y.; Cho, W.; Lim, K.; Xu, J.; Lazar, A.J.; et al. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell 2013, 152, 1077–1090. [Google Scholar] [CrossRef]
- Hirose, T.; Tani, T.; Shimada, T.; Ishizawa, K.; Shimada, S.; Sano, T. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod. Pathol. 2003, 16, 293–298. [Google Scholar] [CrossRef]
- Jouhilahti, E.M.; Peltonen, S.; Heape, A.M.; Peltonen, J. The pathoetiology of neurofibromatosis 1. Am. J. Pathol. 2011, 178, 1932–1939. [Google Scholar] [CrossRef]
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.C.; et al. Mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro-Oncol. 2019, 21, 981–992. [Google Scholar] [CrossRef]
- Menon, A.G.; Anderson, K.M.; Riccardi, V.M.; Chung, R.Y.; Whaley, J.M.; Yandell, D.W.; Farmer, G.E.; Freiman, R.N.; Lee, J.K.; Li, F.P.; et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc. Natl. Acad. Sci. USA 1990, 87, 5435–5439. [Google Scholar] [CrossRef] [PubMed]
- Verdijk, R.M.; den Bakker, M.A.; Dubbink, H.J.; Hop, W.C.; Dinjens, W.N.; Kros, J.M. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J. Neuropathol. Exp. Neurol. 2010, 69, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Hirbe, A.C.; Dahiya, S.; Friedmann-Morvinski, D.; Verma, I.M.; Clapp, D.W.; Gutmann, D.H. Spatially- and temporally-controlled postnatal p53 knockdown cooperates with embryonic Schwann cell precursor Nf1 gene loss to promote malignant peripheral nerve sheath tumor formation. Oncotarget 2016, 7, 7403–7414. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- Skotheim, R.I.; Kallioniemi, A.; Bjerkhagen, B.; Mertens, F.; Brekke, H.R.; Monni, O.; Mousses, S.; Mandahl, N.; Soeter, G.; Nesland, J.M.; et al. Topoisomerase-II alpha is upregulated in malignant peripheral nerve sheath tumors and associated with clinical outcome. J. Clin. Oncol. 2003, 21, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Thayanithy, V.; West, R.B.; Lee, C.H.; Beck, A.H.; Zhu, S.; Downs-Kelly, E.; Montgomery, K.; Goldblum, J.R.; Hogendoorn, P.C.; et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J. Pathol. 2010, 220, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.G.; Rostad, S.; Ross, J.S.; Ali, S.M.; Millis, S.Z. Genomic Profiling in Patients With Malignant Peripheral Nerve Sheath Tumors Reveals Multiple Pathways With Targetable Mutations. J. Natl. Compr. Cancer Netw. 2018, 16, 967–974. [Google Scholar] [CrossRef]
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of human malignant peripheral nerve sheath tumors. Neuro-Oncol. Adv. 2020, 2, i50–i61. [Google Scholar] [CrossRef]
- Miller, S.J.; Jessen, W.J.; Mehta, T.; Hardiman, A.; Sites, E.; Kaiser, S.; Jegga, A.G.; Li, H.; Upadhyaya, M.; Giovannini, M.; et al. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol. Med. 2009, 1, 236–248. [Google Scholar] [CrossRef]
- Watson, M.A.; Perry, A.; Tihan, T.; Prayson, R.A.; Guha, A.; Bridge, J.; Ferner, R.; Gutmann, D.H. Gene expression profiling reveals unique molecular subtypes of Neurofibromatosis Type I-associated and sporadic malignant peripheral nerve sheath tumors. Brain Pathol. 2004, 14, 297–303. [Google Scholar] [CrossRef]
- Holtkamp, N.; Okuducu, A.F.; Mucha, J.; Afanasieva, A.; Hartmann, C.; Atallah, I.; Estevez-Schwarz, L.; Mawrin, C.; Friedrich, R.E.; Mautner, V.F.; et al. Mutation and expression of PDGFRA and KIT in malignant peripheral nerve sheath tumors, and its implications for imatinib sensitivity. Carcinogenesis 2006, 27, 664–671. [Google Scholar] [CrossRef]
- Carroll, S.L.; Stonecypher, M.S. Tumor suppressor mutations and growth factor signaling in the pathogenesis of NF1-associated peripheral nerve sheath tumors: II. The role of dysregulated growth factor signaling. J. Neuropathol. Exp. Neurol. 2005, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Eckert, J.M.; Byer, S.J.; Clodfelder-Miller, B.J.; Carroll, S.L. Neuregulin-1 beta and neuregulin-1 alpha differentially affect the migration and invasion of malignant peripheral nerve sheath tumor cells. Glia 2009, 57, 1501–1520. [Google Scholar] [CrossRef] [PubMed]
- Rao, U.N.; Sonmez-Alpan, E.; Michalopoulos, G.K. Hepatocyte growth factor and c-MET in benign and malignant peripheral nerve sheath tumors. Hum. Pathol. 1997, 28, 1066–1070. [Google Scholar] [CrossRef]
- Su, W.; Gutmann, D.H.; Perry, A.; Abounader, R.; Laterra, J.; Sherman, L.S. CD44-independent hepatocyte growth factor/c-Met autocrine loop promotes malignant peripheral nerve sheath tumor cell invasion in vitro. Glia 2004, 45, 297–306. [Google Scholar] [CrossRef]
- Kadono, T.; Soma, Y.; Takehara, K.; Nakagawa, H.; Ishibashi, Y.; Kikuchi, K. The growth regulation of neurofibroma cells in neurofibromatosis type-1: Increased responses to PDGF-BB and TGF-beta 1. Biochem. Biophys. Res. Commun. 1994, 198, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, D.B.; Dong, Z.; Bunting, H.; Whitfield, J.; Meier, C.; Marie, H.; Mirsky, R.; Jessen, K.R. Transforming growth factor beta (TGFbeta) mediates Schwann cell death in vitro and in vivo: Examination of c-Jun activation, interactions with survival signals, and the relationship of TGFbeta-mediated death to Schwann cell differentiation. J. Neurosci. 2001, 21, 8572–8585. [Google Scholar] [CrossRef]
- Yang, J.; Ylipää, A.; Sun, Y.; Zheng, H.; Chen, K.; Nykter, M.; Trent, J.; Ratner, N.; Lev, D.C.; Zhang, W. Genomic and molecular characterization of malignant peripheral nerve sheath tumor identifies the IGF1R pathway as a primary target for treatment. Clin. Cancer Res. 2011, 17, 7563–7573. [Google Scholar] [CrossRef]
- Mashour, G.A.; Ratner, N.; Khan, G.A.; Wang, H.L.; Martuza, R.L.; Kurtz, A. The angiogenic factor midkine is aberrantly expressed in NF1-deficient Schwann cells and is a mitogen for neurofibroma-derived cells. Oncogene 2001, 20, 97–105. [Google Scholar] [CrossRef]
- Nebesio, T.D.; Ming, W.; Chen, S.; Clegg, T.; Yuan, J.; Yang, Y.; Estwick, S.A.; Li, Y.; Li, X.; Hingtgen, C.M.; et al. Neurofibromin-deficient Schwann cells have increased lysophosphatidic acid dependent survival and migration-implications for increased neurofibroma formation during pregnancy. Glia 2007, 55, 527–536. [Google Scholar] [CrossRef]
- Ryan, J.J.; Klein, K.A.; Neuberger, T.J.; Leftwich, J.A.; Westin, E.H.; Kauma, S.; Fletcher, J.A.; DeVries, G.H.; Huff, T.F. Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. J. Neurosci. Res. 1994, 37, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Yamamoto, H.; Setsu, N.; Kohashi, K.; Takahashi, Y.; Ishii, T.; Iida, K.; Matsumoto, Y.; Hakozaki, M.; Aoki, M.; et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2013, 19, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Luscan, A.; Shackleford, G.; Masliah-Planchon, J.; Laurendeau, I.; Ortonne, N.; Varin, J.; Lallemand, F.; Leroy, K.; Dumaine, V.; Hivelin, M.; et al. The activation of the WNT signaling pathway is a Hallmark in neurofibromatosis type 1 tumorigenesis. Clin. Cancer Res. 2014, 20, 358–371. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.L.; Rahrmann, E.P.; Moriarity, B.S.; Choi, K.; Conboy, C.B.; Greeley, A.D.; Halfond, A.L.; Anderson, L.K.; Wahl, B.R.; Keng, V.W.; et al. Canonical Wnt/β-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013, 3, 674–689. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Sathe, A.A.; Wang, Y.; Nguyen, T.; Glass, D.A., 2nd; Xing, C.; Le, L.Q. Human cutaneous neurofibroma matrisome revealed by single-cell RNA sequencing. Acta Neuropathol. Commun. 2021, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Reavis, H.D.; Chen, H.I.; Drapkin, R. Tumor Innervation: Cancer Has Some Nerve. Trends Cancer 2020, 6, 1059–1067. [Google Scholar] [CrossRef]
- Liao, C.P.; Pradhan, S.; Chen, Z.; Patel, A.J.; Booker, R.C.; Le, L.Q. The role of nerve microenvironment for neurofibroma development. Oncotarget 2016, 7, 61500–61508. [Google Scholar] [CrossRef]
- Anastasaki, C.; Mo, J.; Chen, J.K.; Chatterjee, J.; Pan, Y.; Scheaffer, S.M.; Cobb, O.; Monje, M.; Le, L.Q.; Gutmann, D.H. Neuronal hyperexcitability drives central and peripheral nervous system tumor progression in models of neurofibromatosis-1. Nat. Commun. 2022, 13, 2785. [Google Scholar] [CrossRef]
- Korfhage, J.; Lombard, D.B. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol. Cancer Res. 2019, 17, 1417–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Study ID | Subtypes of NF | GEM Model | Cell of Origin | Supported Points | Unsupported/Unknown Points |
|---|---|---|---|---|---|
| Zhu et al., 2002 [12] | pNF | Krox20-Cre | SC lineage | Use of Krox20-Cre to ablate Nf1 function within the SC lineage led to pNF. | The exact cellular origin remained unknown due to the extensive expression of Krox20 in NCSCs, SCPs, and SCs. |
| Joseph et al., 2008 [41] | pNF | P0a-Cre | Later NCSC derivatives | Loss of Nf1 function in NCSCs resulted in transient hyperproliferation instead of tumorigenesis. Neurofibromas may arise from differentiated cell types but not NCSCs. | The authors failed to detect the cellular origin of cNF, with no typical cutaneous lesions generated in any of the mouse models. |
| Zheng et al., 2008 [42] | pNF | P0a-Cre | nmSCs | The molecular signatures of the proliferating neoplastic cells were similar to nmSCs but not NCSCs. | The specific mechanism leading to the transformation of SCs from axon-associated to axon-disassociated cells in pNF remained unclear. |
| Le et al., 2011 [17] | pNF | Plp-CreERT2 | SCPs and immature SCs | The embryonic stage showed enhanced susceptibility to pNF formation compared with the adult stage. | Another study showed that loss of Nf1 at either embryonic or adult SC stages could lead to neurofibroma formation [18]. |
| Maye et al., 2011 [18] | pNF | Plp-Cre | Embryonic/adult SCs | Loss of Nf1 in either embryonic or adult SCs caused neurofibroma formation. | The capability of mature SCs to generate pNF was less supported by its clinical manifestation as a congenital lesion. |
| Keng et al., 2012 [19] | pNF | Dhh-Cre | SCs and SCPs | Loss of Pten and Nf1 was sufficient for progressing from pNFs to MPNSTs. | A previous study using the mGFAP-cre with conditional inactivation of both Pten and Nf1 failed to develop neurofibromas [43]. |
| Chen et al., 2014 [20] | pNF | Plp-Cre | GAP43+ PLP+ SCPs | GAP43+ PLP+ cells were detected in the embryonic nerve roots at E11.5, and acute loss of Nf1 in SCPs led to pNF formation. | The remaining SCPs may persist into the adult stage and retain the capacity to form pNFs. However, the overlap of cell types in the transition from NCSCs to embryonic and mature SCs remained unknown. |
| Chaney et al., 2020 [21] | pNF | Dhh-Cre | Developing SCs | Loss of Ink4a/Arf in mice (CDKN2A in humans) and Nf1 generated paraspinal neurofibromas and precursor malignant lesions. | Malignant transformation only occurred after transplantation into secondary mice, indicating the necessity of an immune microenvironment for tumor progression. |
| Saito et al., 2007 [44] | cNF | Camk2-Cre | Neural crest-derived cells | Activation of the N-Ras signaling pathway expressed in neural crest-derived cells caused cNF formation. | The differences between the Ras signals leading to cNF and pNF and the specific cell type of cNF origin remained unclear. |
| Wu et al., 2008 [45] | cNF, pNF | Dhh-Cre | SCP | Loss of Nf1 in SCs at E12.5 was sufficient to give rise to both pNF and cNF in a wild-type microenvironment. | The cNFs observed in mouse models were found outside the dermis, below the panniculus carnosus, differing from the location in humans. |
| Le et al., 2009 [46] | cNF, pNF | CMV-CreERT2 | SKP | The capability of SKPs to express Dhh and generate both pNF and cNF was identified. | Since SKPs are a heterogeneous cell population, the specific subpopulation acting as the cellular origin of cNF remained unknown. In addition, it was unclear whether there was a common cellular origin for cNF and pNF. |
| Chen et al., 2019 [14] | cNF, pNF | Hoxb7-Cre | Hoxb7 lineage-derived cells | Loss of Nf1 in Hoxb7-derived cells could recapitulate both pNF and cNF. | Loss of N1 occurring before the bifurcation into distinct SC lineages and therefore giving rise to both cNF and pNF after subsequent differentiation was not definitively confirmed. |
| Radomska et al., 2019 [47] | cNF, pNF | Prss56-Cre | BC cells | BC-derived nmSCs and subepidermal SCs constitute the major population of pathogenic cells in pNF and cNF, respectively. | The differences in phenotypes between mouse models and human neurofibroma require further investigation. |
| Mo et al., 2021 [48] | cNF, pNF | SOX10+ cells | SOX10+ stem cells | Humanized models established using hiPSCs showed that inactivation of both Nf1 alleles in mouse SOX10+ cells led to cNF and pNF formation. | This study further identified the common cells of origin for cNF and pNF, but an explanation of specific spatiotemporal differences was lacking. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ge, L.-L.; Xing, M.-Y.; Zhang, H.-B.; Wang, Z.-C. Neurofibroma Development in Neurofibromatosis Type 1: Insights from Cellular Origin and Schwann Cell Lineage Development. Cancers 2022, 14, 4513. https://doi.org/10.3390/cancers14184513
Ge L-L, Xing M-Y, Zhang H-B, Wang Z-C. Neurofibroma Development in Neurofibromatosis Type 1: Insights from Cellular Origin and Schwann Cell Lineage Development. Cancers. 2022; 14(18):4513. https://doi.org/10.3390/cancers14184513
Chicago/Turabian StyleGe, Ling-Ling, Ming-Yan Xing, Hai-Bing Zhang, and Zhi-Chao Wang. 2022. "Neurofibroma Development in Neurofibromatosis Type 1: Insights from Cellular Origin and Schwann Cell Lineage Development" Cancers 14, no. 18: 4513. https://doi.org/10.3390/cancers14184513
APA StyleGe, L. -L., Xing, M. -Y., Zhang, H. -B., & Wang, Z. -C. (2022). Neurofibroma Development in Neurofibromatosis Type 1: Insights from Cellular Origin and Schwann Cell Lineage Development. Cancers, 14(18), 4513. https://doi.org/10.3390/cancers14184513