
Knockdown of NCOR2 Inhibits Cell Proliferation via BDNF/TrkB/ERK in NF1-Derived MPNSTs

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Patients and Specimens
2.3. Quantitative Real-Time Polymerase Chain Reaction (qRT–PCR)
2.4. Western Blotting (WB)
2.5. Cell Transfection
2.6. Cell Viability Assay
2.7. Colony Formation Assay
2.8. Cell Invasion and Migration Assays
2.9. Immunohistochemistry (IHC)
2.10. TUNEL Assays
2.11. Flow Cytometry (FCM) Experiments
2.12. RNA Sequencing (RNA-Seq)
2.13. Enzyme-Linked Immunosorbent Assay (ELISA)
2.14. Xenograft Tumour Models
2.15. Statistical Analysis
3. Results
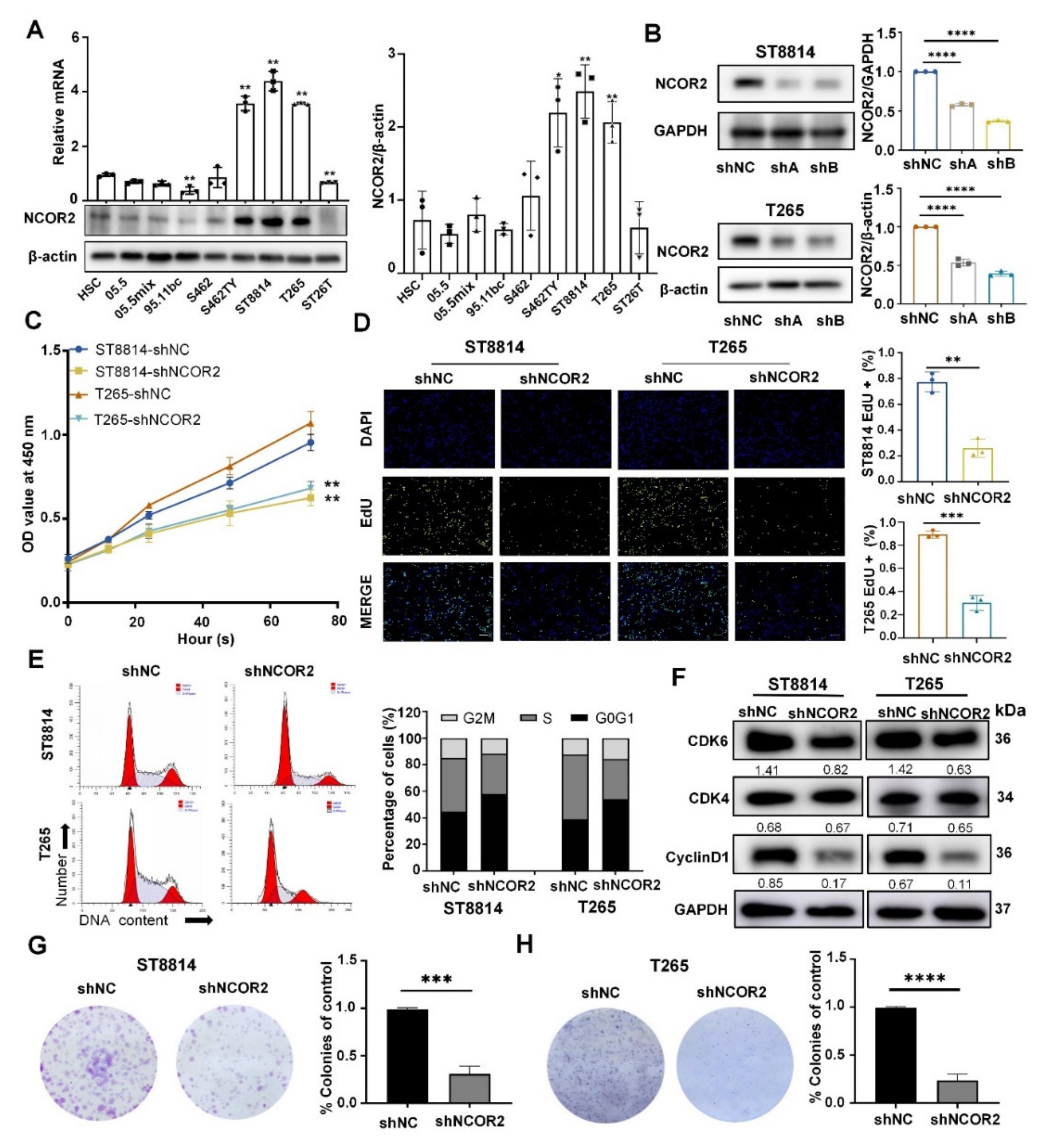
3.1. NCOR2 Expression Is Significantly Elevated in NF1-Derived MPNSTs and Is Associated with Patient 10-Year Survival Time
3.2. NCOR2 Knockdown Leads to Inhibition of NF1-Derived MPNST Cell Proliferation In Vitro
3.3. NCOR2 Knockdown Reduces the Tumourigenicity of NF1-Derived MPNST Cells In Vivo
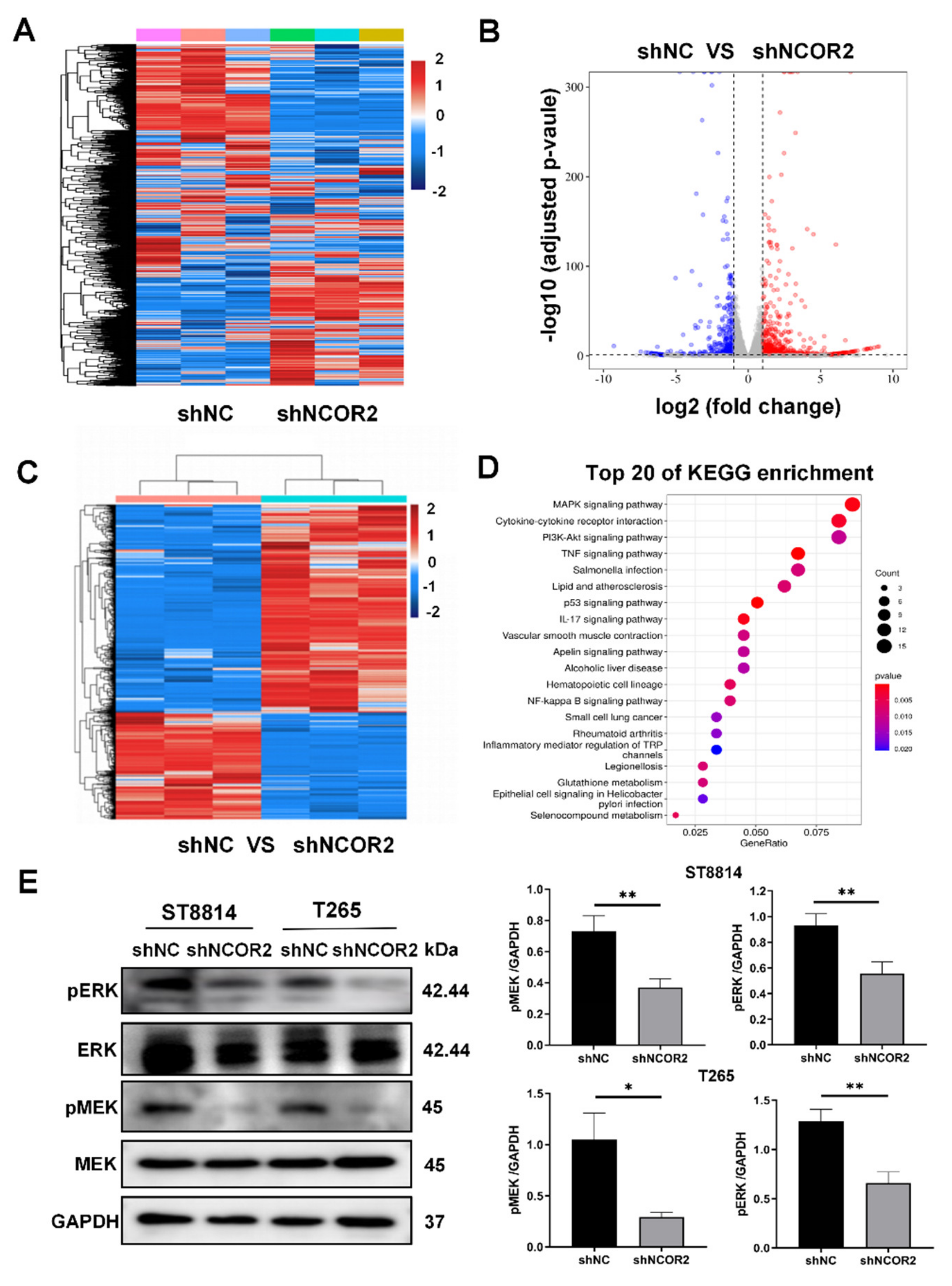
3.4. NCOR2 Regulates the MAPK Signalling Activation in NF1-Derived MPNST Cells
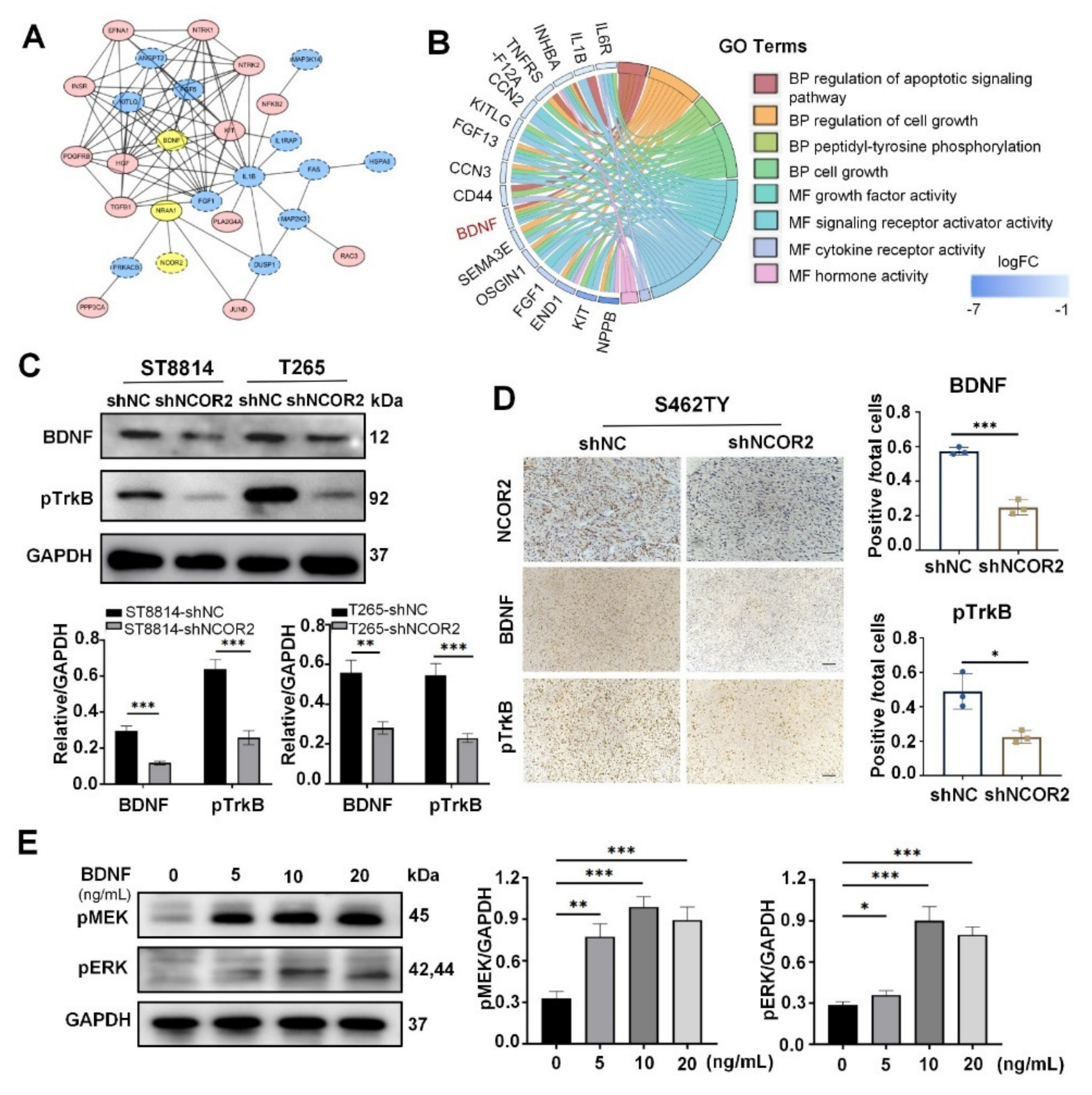
3.5. BDNF Plays a Key Role in ERK Signal in NF1-Derived MPNST Cells
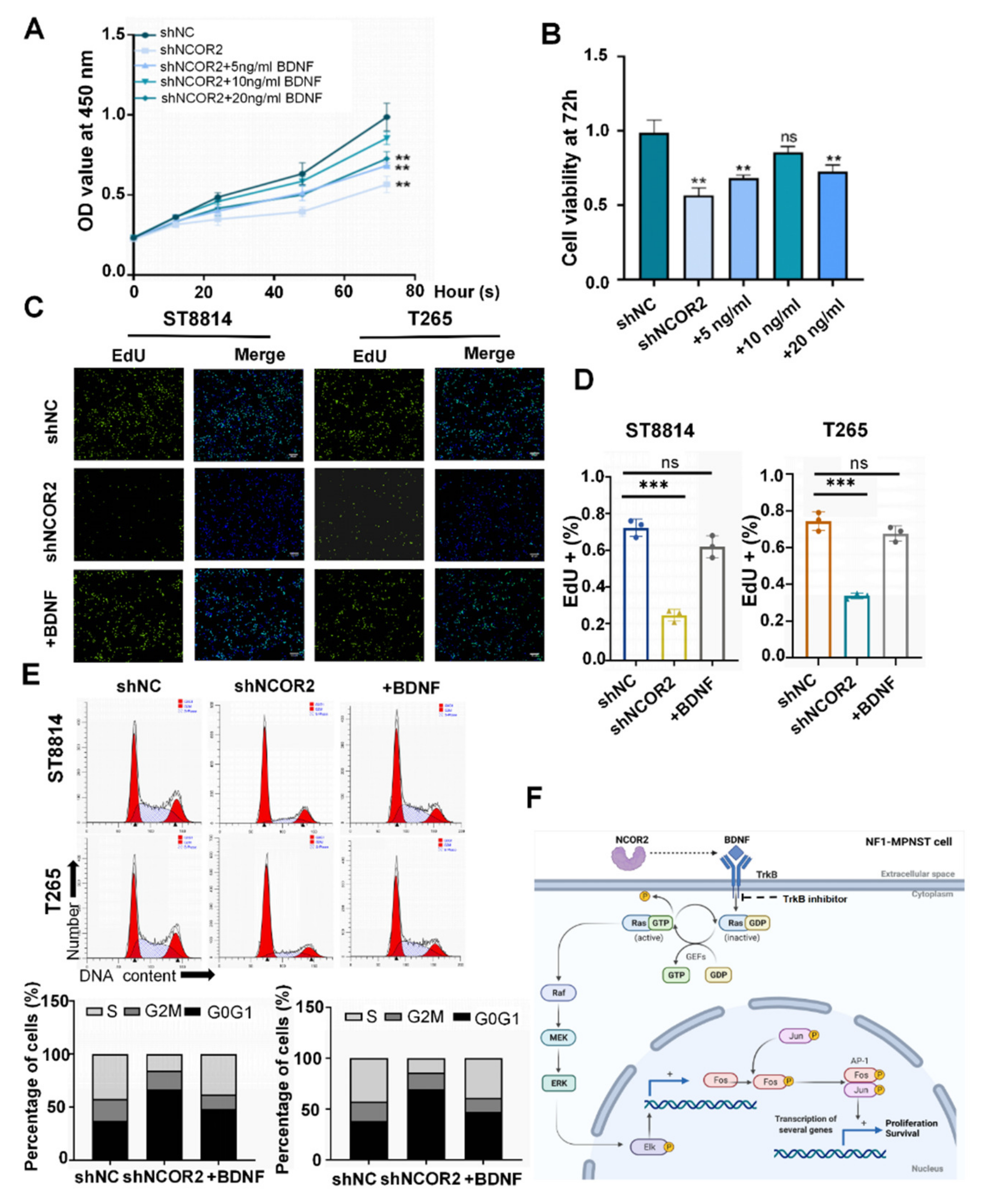
3.6. Human Recombinant BDNF Can Reverse the Effects of NCOR2 Knockdown on NF1-Derived MPNST Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [Green Version]
- Ducatman, B.S.; Scheithauer, B.W.; Piepgras, D.G.; Reiman, H.M.; Ilstrup, D.M. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer 1986, 57, 2006–2021. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, E.; Leppävirta, J.; Koffert, A.; Suominen, S.B.; Vahtera, J.; Vahlberg, T.; Pöyhönen, M.; Peltonen, J.; Peltonen, S. Incidence and Mortality of Neurofibromatosis: A Total Population Study in Finland. J. Investig. Dermatol. 2015, 135, 904–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, 17004. [Google Scholar] [CrossRef] [PubMed]
- Ferner, R.E. Neurofibromatosis 1. Eur. J. Hum. Genet. 2007, 15, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Shurell, E.; Tran, L.M.; Nakashima, J.; Smith, K.B.; Tam, B.M.; Li, Y.; Dry, S.M.; Federman, N.; Tap, W.D.; Wu, H.; et al. Gender dimorphism and age of onset in malignant peripheral nerve sheath tumor preclinical models and human patients. BMC Cancer 2014, 14, 827. [Google Scholar] [CrossRef] [Green Version]
- Watson, K.L.; Al Sannaa, G.A.; Kivlin, C.M.; Ingram, D.R.; Landers, S.M.; Roland, C.L.; Cormier, J.N.; Hunt, K.K.; Feig, B.W.; Guadagnolo, B.A.; et al. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1–associated, and radiation-associated malignant peripheral nerve sheath tumors. J. Neurosurg. 2017, 126, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Sohier, P.; Luscan, A.; Lloyd, A.; Ashelford, K.; Laurendeau, I.; Briand-Suleau, A.; Vidaud, D.; Ortonne, N.; Pasmant, E.; Upadhyaya, M. Confirmation of mutation landscape of NF1-associated malignant peripheral nerve sheath tumors. Genes Chromosom. Cancer 2017, 56, 421–426. [Google Scholar] [CrossRef]
- Mantripragada, K.K.; de Ståhl, T.D.; Patridge, C.; Menzel, U.; Andersson, R.; Chuzhanova, N.; Kluwe, L.; Guha, A.; Mautner, V.; Dumanski, J.P.; et al. Genome-wide high-resolution analysis of DNA copy number alterations in NF1-associated malignant peripheral nerve sheath tumors using 32K BAC array. Genes Chromosom. Cancer 2009, 48, 897–907. [Google Scholar] [CrossRef]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef]
- Gu, Y.; Wei, C.; Chung, M.; Li, H.; Guo, Z.; Long, M.; Li, Y.; Wang, W.; Aimaier, R.; Li, Q.; et al. Concurrent inhibition of FAK/SRC and MEK overcomes MEK inhibitor resistance in Neurofibromatosis Type I related malignant peripheral nerve sheath tumors. Front. Oncol. 2022, 12, 910505. [Google Scholar] [CrossRef]
- Wang, J.; Pollard, K.; Calizo, A.; Pratilas, C.A. Activation of Receptor Tyrosine Kinases Mediates Acquired Resistance to MEK Inhibition in Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2021, 81, 747–762. [Google Scholar] [CrossRef]
- Jiang, Q.; Galiègue-Zouitina, S.; Roumier, C.; Hildebrand, M.P.; Thomas, S.; Coignet, L.J. Genomic organization and refined mapping of the human nuclear corepressor 2 (NCOR2)/silencing mediator of retinoid and thyroid hormone receptor (SMRT) gene on chromosome 12q24.3. Cytogenet. Cell Genet. 2001, 92, 217–220. [Google Scholar] [CrossRef]
- Codina, A.; Love, J.D.; Li, Y.; Lazar, M.A.; Neuhaus, D.; Schwabe, J.W.R. Structural insights into the interaction and activation of histone deacetylase 3 by nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2005, 102, 6009–6014. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, K.; Hermanson, O.; Onami, T.M.; Gleiberman, A.S.; Lunyak, V.; McEvilly, R.J.; Kurokawa, R.; Kumar, V.; Liu, F.; Seto, E.; et al. Combinatorial Roles of the Nuclear Receptor Corepressor in Transcription and Development. Cell 2000, 102, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Jepsen, K.; Gleiberman, A.S.; Shi, C.; Simon, D.I.; Rosenfeld, M.G. Cooperative regulation in development by SMRT and FOXP1. Genes Dev. 2008, 22, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, S.; Gao, T.; Pace, M.C.; Oesterreich, S.; Smith, C.L. Cooperative Activation of Cyclin D1 and Progesterone Receptor Gene Expression by the SRC-3 Coactivator and SMRT Corepressor. Mol. Endocrinol. 2010, 24, 1187–1202. [Google Scholar] [CrossRef] [Green Version]
- Green, A.R.; Burney, C.; Granger, C.J.; Paish, E.C.; El-Sheikh, S.; Rakha, E.A.; Powe, D.G.; Macmillan, R.D.; Ellis, I.O.; Stylianou, E. The prognostic significance of steroid receptor co-regulators in breast cancer: Co-repressor NCOR2/SMRT is an independent indicator of poor outcome. Breast Cancer Res. Treat. 2008, 110, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Gong, C.; Lau, S.L.; Yang, N.; Wong, O.G.; Cheung, A.N.; Tsang, J.W.; Chan, K.Y.; Khoo, U.S. SpliceArray profiling of breast cancer reveals a novel variant of NCOR2/SMRT that is associated with tamoxifen resistance and control of ERα transcriptional activity. Cancer Res. 2013, 73, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Migliaccio, I.; Chaubal, V.; Wu, M.-F.; Pace, M.C.; Hartmaier, R.; Jiang, S.; Edwards, D.P.; Gutiérrez, M.C.; Hilsenbeck, S.G.; et al. Elevated nuclear expression of the SMRT corepressor in breast cancer is associated with earlier tumor recurrence. Breast Cancer Res. Treat. 2012, 136, 253–265. [Google Scholar] [CrossRef]
- Khanim, F.L.; Gommersall, L.M.; Wood, V.H.; Smith, K.L.; Montalvo, L.; O’Neill, L.P.; Xu, Y.; Peehl, D.M.; Stewart, P.M.; Turner, B.M.; et al. Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene 2004, 23, 6712–6725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedin, S.A.; Banwell, C.M.; Colston, K.W.; Carlberg, C.; Campbell, M.J. Epigenetic corruption of VDR signalling in malignancy. Anticancer Res. 2006, 26, 2557–2566. [Google Scholar]
- Long, M.D.; Jacobi, J.J.; Singh, P.K.; Llimos, G.; Wani, S.A.; Rowsam, A.M.; Rosario, S.R.; Hoogstraat, M.; Linder, S.; Kirk, J.; et al. Reduced NCOR2 expression accelerates androgen deprivation therapy failure in prostate cancer. Cell Rep. 2021, 37, 110109. [Google Scholar] [CrossRef]
- Mori, T.; Verma, R.; Nakamoto-Matsubara, R.; Siu, K.T.; Panaroni, C.; Fulzele, K.S.; Mukaihara, K.; Onyewadume, C.; Maebius, A.; Kato, H.; et al. Low NCOR2 levels in multiple myeloma patients drive multidrug resistance via MYC upregulation. Blood Cancer J. 2021, 11, 194. [Google Scholar] [CrossRef]
- Campos, B.; Bermejo, J.L.; Han, L.; Felsberg, J.; Ahmadi, R.; Grabe, N.; Reifenberger, G.; Unterberg, A.; Herold-Mende, C. Expression of nuclear receptor corepressors and class I histone deacetylases in astrocytic gliomas. Cancer Sci. 2011, 102, 387–392. [Google Scholar] [CrossRef]
- Li, X.X.; Zhang, S.J.; Chiu, A.P.; Lo, L.H.; Huang, J.; Rowlands, D.K.; Wang, J.; Keng, V.W. Targeting of AKT/ERK/CTNNB1 by DAW22 as a potential therapeutic compound for malignant peripheral nerve sheath tumor. Cancer Med. 2018, 7, 4791–4800. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Yang, J.; Ylipää, A.; Zhu, Z. RETRACTED ARTICLE: Genomic amplification and high expression of EGFR are key targetable oncogenic events in malignant peripheral nerve sheath tumor. J. Hematol. Oncol. 2013, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.-Y.; Gu, Y.-H.; Cui, X.-W.; Long, M.-M.; Wang, W.; Wei, C.-J.; Gu, B.; Zhang, H.-B.; Li, Q.-F.; Wang, Z.-C. Protein Tyrosine Phosphatase Receptor S Acts as a Metastatic Suppressor in Malignant Peripheral Nerve Sheath Tumor via Profilin 1-Induced Epithelial-Mesenchymal Transition. Front. Cell Dev. Biol. 2020, 8, 582220. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.J.; Jessen, W.J.; Mehta, T.; Hardiman, A.; Sites, E.; Kaiser, S.; Jegga, A.G.; Li, H.; Upadhyaya, M.; Giovannini, M.; et al. Integrative genomic analyses of neurofibromatosis tumours identify SOX9 as a biomarker and survival gene. EMBO Mol. Med. 2009, 1, 236–248. [Google Scholar] [CrossRef]
- Jessen, W.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.-O.; Dombi, E.; et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Investig. 2013, 123, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Guo, W.; Ren, T.; Huang, Y.; Han, Y.; Zhang, H.; Zhang, J. Knockdown of HMGA2 regulates the level of autophagy via interactions between MSI2 and Beclin1 to inhibit NF1-associated malignant peripheral nerve sheath tumour growth. J. Exp. Clin. Cancer Res. 2019, 38, 185. [Google Scholar] [CrossRef] [Green Version]
- STRING Database—Functional Protein Association Networks. Available online: https://string-db.org/ (accessed on 15 February 2022).
- Li, J.; Wang, X.; Wang, H.; Wang, R.; Guo, Y.; Xu, L.; Zhang, G.; Wu, J.; Wang, G. The BDNF-TrkB signaling pathway in the rostral anterior cingulate cortex is involved in the development of pain aversion in rats with bone cancer via NR2B and ERK-CREB signaling. Brain. Res. Bull. 2022, 185, 18–27. [Google Scholar] [CrossRef]
- Hua, Z.; Gu, X.; Dong, Y.; Tan, F.; Liu, Z.; Thiele, C.J.; Li, Z. PI3K and MAPK pathways mediate the BDNF/TrkB-increased metastasis in neuroblastoma. Tumour Biol 2016, 37, 16227–16236. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Yang, J.; Cao, Z.; Zhao, J.; Zhang, J.; Lu, Y.; Chu, L.; Zhang, S.; Chen, Y.; Pei, L. Baicalin ameliorates chronic unpredictable mild stress-induced depression through the BDNF/ERK/CREB signaling pathway. Behav. Brain Res. 2021, 414, 113463. [Google Scholar] [CrossRef]
- Hutchison, M.R. BDNF Alters ERK/p38 MAPK Activity Ratios to Promote Differentiation in Growth Plate Chondrocytes. Mol. Endocrinol. 2012, 26, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Anghileri, M.; Miceli, R.; Fiore, M.; Mariani, L.; Ferrari, A.; Mussi, C.; Lozza, L.; Collini, P.; Olmi, P.; Casali, P.G.; et al. Malignant peripheral nerve sheath tumors: Prognostic factors and survival in a series of patients treated at a single institution. Cancer 2006, 107, 1065–1074. [Google Scholar] [CrossRef]
- Zou, C.; Smith, K.D.; Liu, J.; Lahat, G.; Myers, S.; Wang, W.-L.; Zhang, W.; McCutcheon, I.E.; Slopis, J.M.; Lazar, A.J.; et al. Clinical, Pathological, and Molecular Variables Predictive of Malignant Peripheral Nerve Sheath Tumor Outcome. Ann. Surg. 2009, 249, 1014–1022. [Google Scholar] [CrossRef]
- Chang, F.; Steelman, L.S.; Lee, J.T.; Shelton, J.G.; Navolanic, P.M.; Blalock, W.L.; Franklin, R.A.; McCubrey, J. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: Potential targeting for therapeutic intervention. Leukemia 2003, 17, 1263–1293. [Google Scholar] [CrossRef]
- Johansson, G.; Mahller, Y.Y.; Collins, M.H.; Kim, M.-O.; Nobukuni, T.; Perentesis, J.; Cripe, T.P.; Lane, H.A.; Kozma, S.C.; Thomas, G.; et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol. Cancer Ther. 2008, 7, 1237–1245. [Google Scholar] [CrossRef]
- Holtkamp, N.; Malzer, E.; Zietsch, J.; Okuducu, A.F.; Mucha, J.; Mawrin, C.; Mautner, V.-F.; Schildhaus, H.-U.; von Deimling, A. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro-Oncology 2008, 10, 946–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, E.; Faivre, S.; Armand, J.P. Epidermal Growth Factor Receptor Tyrosine Kinase as a Target for Anticancer Therapy. Drugs 2000, 60 (Suppl. 1), 15–23, discussion 41–42. [Google Scholar] [CrossRef]
- D’Adamo, D.R.; Anderson, S.E.; Albritton, K.; Yamada, J.; Riedel, E.; Scheu, K.; Schwartz, G.K.; Chen, H.; Maki, R.G. Phase II Study of Doxorubicin and Bevacizumab for Patients With Metastatic Soft-Tissue Sarcomas. J. Clin. Oncol. 2005, 23, 7135–7142. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updat. 2008, 11, 32–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Müller, R.; Pouysségur, J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.; Tubiana-Hulin, M.; Lidereau, R.; Bieche, I. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: Evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res. 2003, 9, 1259–1266. [Google Scholar]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Hempstead, B.L. Dissecting the Diverse Actions of Pro- and Mature Neurotrophins. Curr. Alzheimer Res. 2006, 3, 19–24. [Google Scholar] [CrossRef]
- Bibel, M.; Barde, Y.-A. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [Green Version]
- Tolwani, R.J.; Varma, S.; Jacob, R.; Kuo, L.E.; Shooter, E.M. BDNF overexpression produces a long-term increase in myelin formation in the peripheral nervous system. J. Neurosci. Res. 2004, 77, 662–669. [Google Scholar] [CrossRef]
- Gagliano, T.; Shah, K.; Gargani, S.; Lao, L.; Alsaleem, M.; Chen, J.; Ntafis, V.; Huang, P.; Ditsiou, A.; Vella, V.; et al. PIK3Cδ expression by fibroblasts promotes triple-negative breast cancer progression. J. Clin. Investig. 2020, 130, 3188–3204. [Google Scholar] [CrossRef]
- Al-Qudah, M.A.; Al-Dwairi, A. Mechanisms and regulation of neurotrophin synthesis and secretion. Neurosciences 2016, 21, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Jiang, L. Advances in Regulating Tumorigenicity and Metastasis of Cancer through TrkB Signaling. Curr. Cancer Drug Targets 2020, 20, 779–788. [Google Scholar] [CrossRef]
- Carter, B.D.; Zirrgiebel, U.; Barde, Y. Differential Regulation of p21ras Activation in Neurons by Nerve Growth Factor and Brain-derived Neurotrophic Factor. J. Biol. Chem. 1995, 270, 21751–21757. [Google Scholar] [CrossRef] [Green Version]
- Nakagawara, A.; Azar, C.G.; Scavarda, N.J.; Brodeur, G.M. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol. Cell Biol. 1994, 14, 759–767. [Google Scholar] [CrossRef]
- Xiong, J.; Zhou, L.; Lim, Y.; Yang, M.; Zhu, Y.-H.; Li, Z.-W.; Fu, D.-L.; Zhou, X.-F. Mature brain-derived neurotrophic factor and its receptor TrkB are upregulated in human glioma tissues. Oncol. Lett. 2015, 10, 223–227. [Google Scholar] [CrossRef] [Green Version]
- Thomaz, A.; Jaeger, M.; Brunetto, A.L.; Brunetto, A.T.; Gregianin, L.; De Farias, C.B.; Ramaswamy, V.; Nör, C.; Taylor, M.D.; Roesler, R. Neurotrophin Signaling in Medulloblastoma. Cancers 2020, 12, 2542. [Google Scholar] [CrossRef]
- Iyer, R.; Varela, C.R.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Light, J.E.; Evans, A.E.; Zhao, H.; Thress, K.; Brown, J.L.; et al. AZ64 inhibits TrkB and enhances the efficacy of chemotherapy and local radiation in neuroblastoma xenografts. Cancer Chemother. Pharmacol. 2012, 70, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Jaboin, J.; Kim, C.J.; Kaplan, D.R.; Thiele, C.J. Brain-derived neurotrophic factor activation of TrkB protects neuroblastoma cells from chemotherapy-induced apoptosis via phosphatidylinositol 3’-kinase pathway. Cancer Res. 2002, 62, 6756–6763. [Google Scholar]
- Li, Z.; Jaboin, J.; Dennis, P.A.; Thiele, C.J. Genetic and Pharmacologic Identification of Akt as a Mediator of Brain-Derived Neurotrophic Factor/TrkB Rescue of Neuroblastoma Cells from Chemotherapy-Induced Cell Death. Cancer Res. 2005, 65, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deland, L.; Keane, S.; Olsson Bontell, T.; Sjögren, H.; Fagman, H.; Øra, I.; De La Cuesta, E.; Tisell, M.; Nilsson, J.A.; Ejeskär, K.; et al. Discovery of a rare GKAP1-NTRK2 fusion in a pediatric low-grade glioma, leading to targeted treatment with TRK-inhibitor larotrectinib. Cancer Biol. Ther. 2021, 22, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.E.; Kisselbach, K.D.; Liu, X.; Eggert, A.; Ikegaki, N.; Camoratto, A.M.; Dionne, C.; Brodeur, G. Effect of CEP-751 (KT-6587) on neuroblastoma xenografts expressing TrkB. Med. Pediatr. Oncol. 2001, 36, 181–184. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCOR2 Expression | p-Value | ||
| high | low | ||
| Gender | |||
| Male | 15 | 11 | 0.449 |
| Female | 19 | 9 | |
| Age | |||
| <45 | 18 | 10 | 0.838 |
| >45 | 16 | 10 | |
| Tumor size | |||
| T1 (<5) | 9 | 8 | 0.447 |
| T2 (5–10) | 10 | 5 | |
| T3 (10–15) | 6 | 4 | |
| T4 (>15) | 5 | 2 | |
| Tumor site | |||
| Head and neck | 11 | 4 | 0.292 |
| Trunk | 9 | 6 | |
| Limbs | 11 | 9 | |
| NF1 | |||
| With | 14 | 6 | 0.427 |
| Without | 17 | 12 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Chung, M.; Aimaier, R.; Wei, C.; Wang, W.; Ge, L.; Zhu, B.; Guo, Z.; Wang, M.; Gu, Y.; et al. Knockdown of NCOR2 Inhibits Cell Proliferation via BDNF/TrkB/ERK in NF1-Derived MPNSTs. Cancers 2022, 14, 5798. https://doi.org/10.3390/cancers14235798
Li Y, Chung M, Aimaier R, Wei C, Wang W, Ge L, Zhu B, Guo Z, Wang M, Gu Y, et al. Knockdown of NCOR2 Inhibits Cell Proliferation via BDNF/TrkB/ERK in NF1-Derived MPNSTs. Cancers. 2022; 14(23):5798. https://doi.org/10.3390/cancers14235798
Chicago/Turabian StyleLi, Yuehua, Manhon Chung, Rehanguli Aimaier, Chengjiang Wei, Wei Wang, Lingling Ge, Beiyao Zhu, Zizhen Guo, Mingyang Wang, Yihui Gu, and et al. 2022. "Knockdown of NCOR2 Inhibits Cell Proliferation via BDNF/TrkB/ERK in NF1-Derived MPNSTs" Cancers 14, no. 23: 5798. https://doi.org/10.3390/cancers14235798
APA StyleLi, Y., Chung, M., Aimaier, R., Wei, C., Wang, W., Ge, L., Zhu, B., Guo, Z., Wang, M., Gu, Y., Zhang, H., Li, Q., & Wang, Z. (2022). Knockdown of NCOR2 Inhibits Cell Proliferation via BDNF/TrkB/ERK in NF1-Derived MPNSTs. Cancers, 14(23), 5798. https://doi.org/10.3390/cancers14235798