
Identifying Tissue- and Cohort-Specific RNA Regulatory Modules in Cancer Cells Using Multitask Learning


Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Previous Studies
1.2. Our Contributions
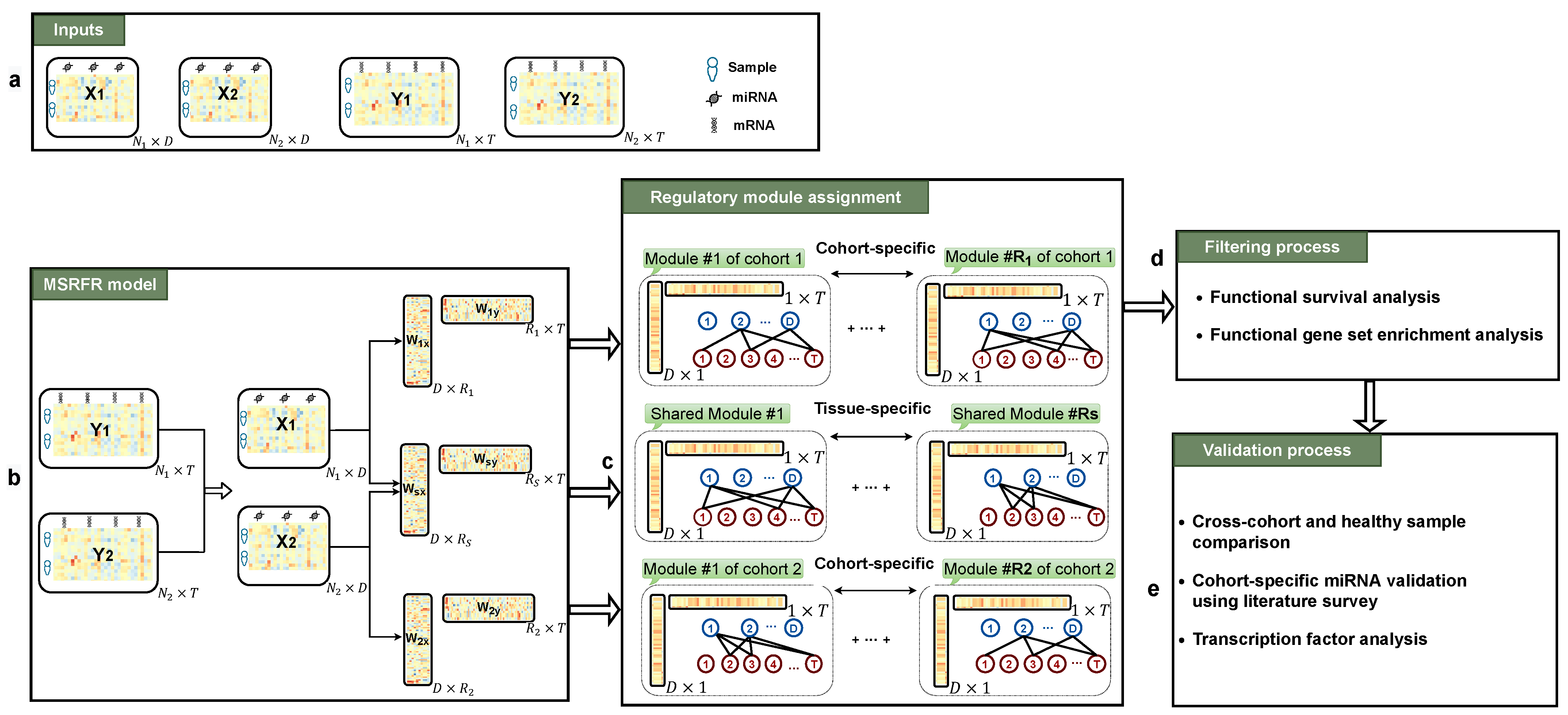
2. Materials and Methods
2.1. Datasets
2.2. Problem Definition
2.3. Method
2.4. Experimental Setting
- Blood ,
- Kidney ,
- Lung .
3. Results
3.1. Predictive Performance Comparison
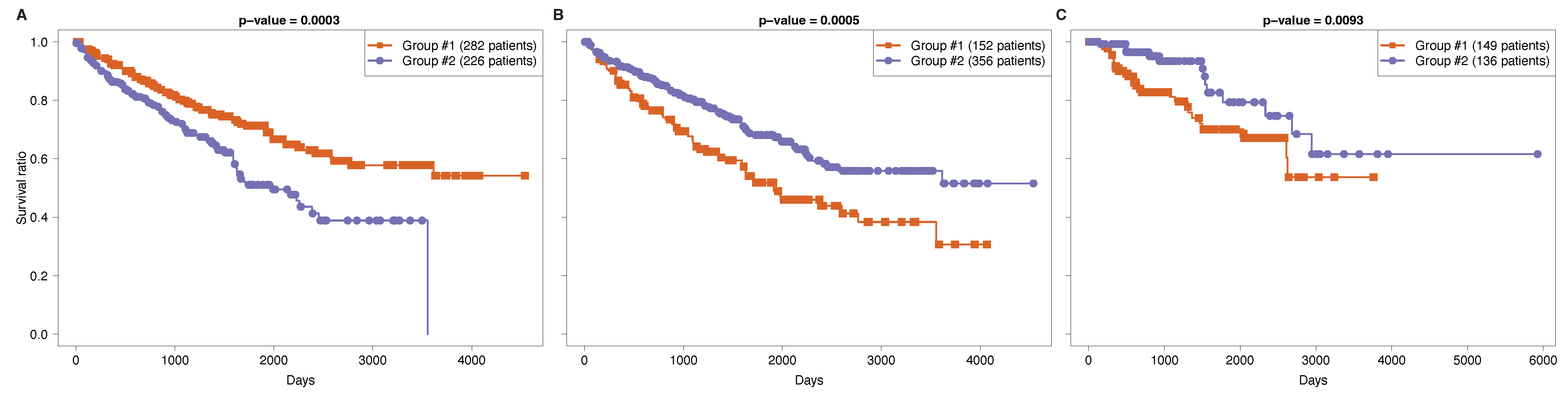
3.2. Functional Survival Analysis of Identified Regulatory Modules
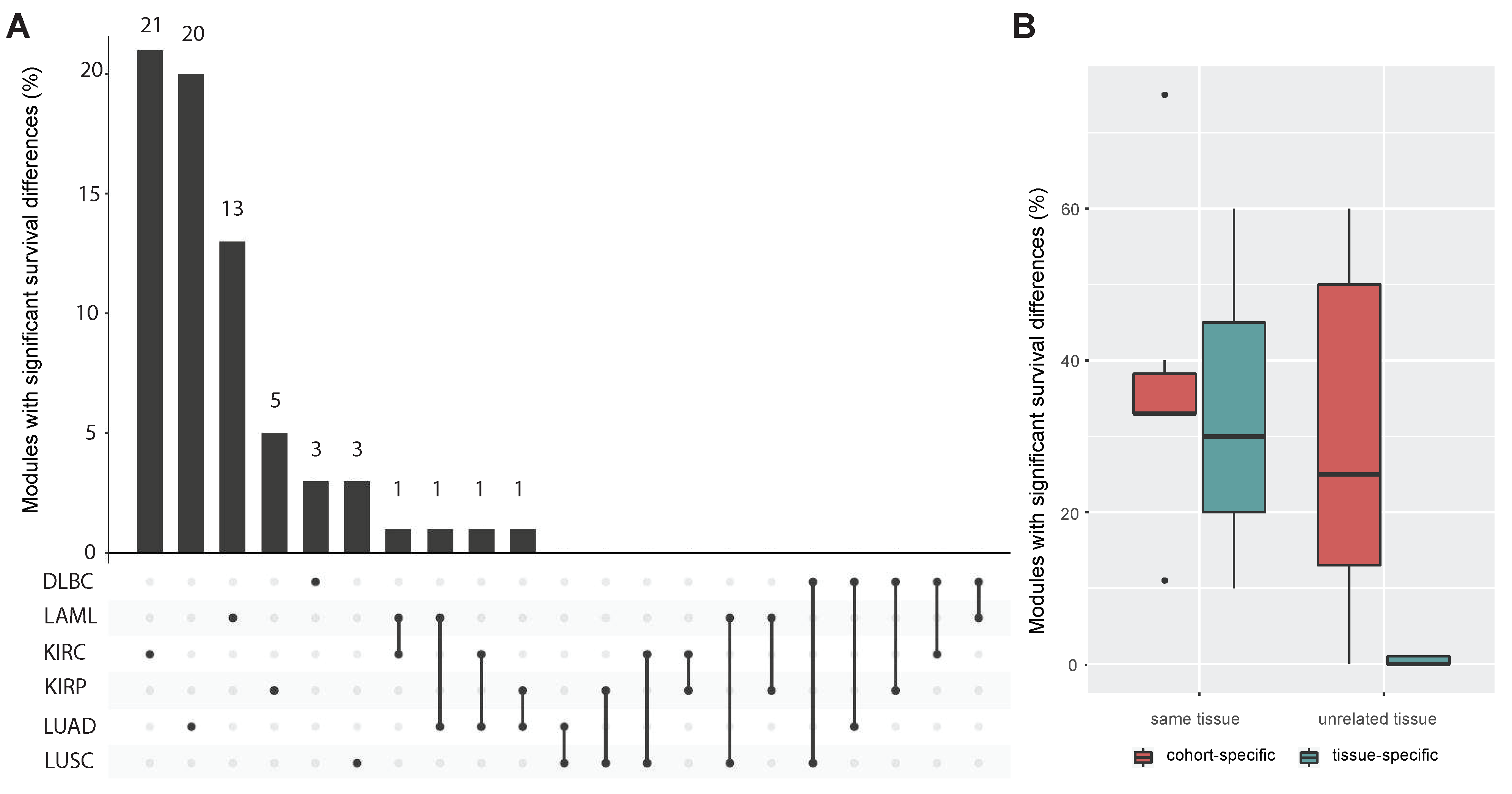
3.3. Tissue-Specificity and Disease Association of Key Regulatory Modules
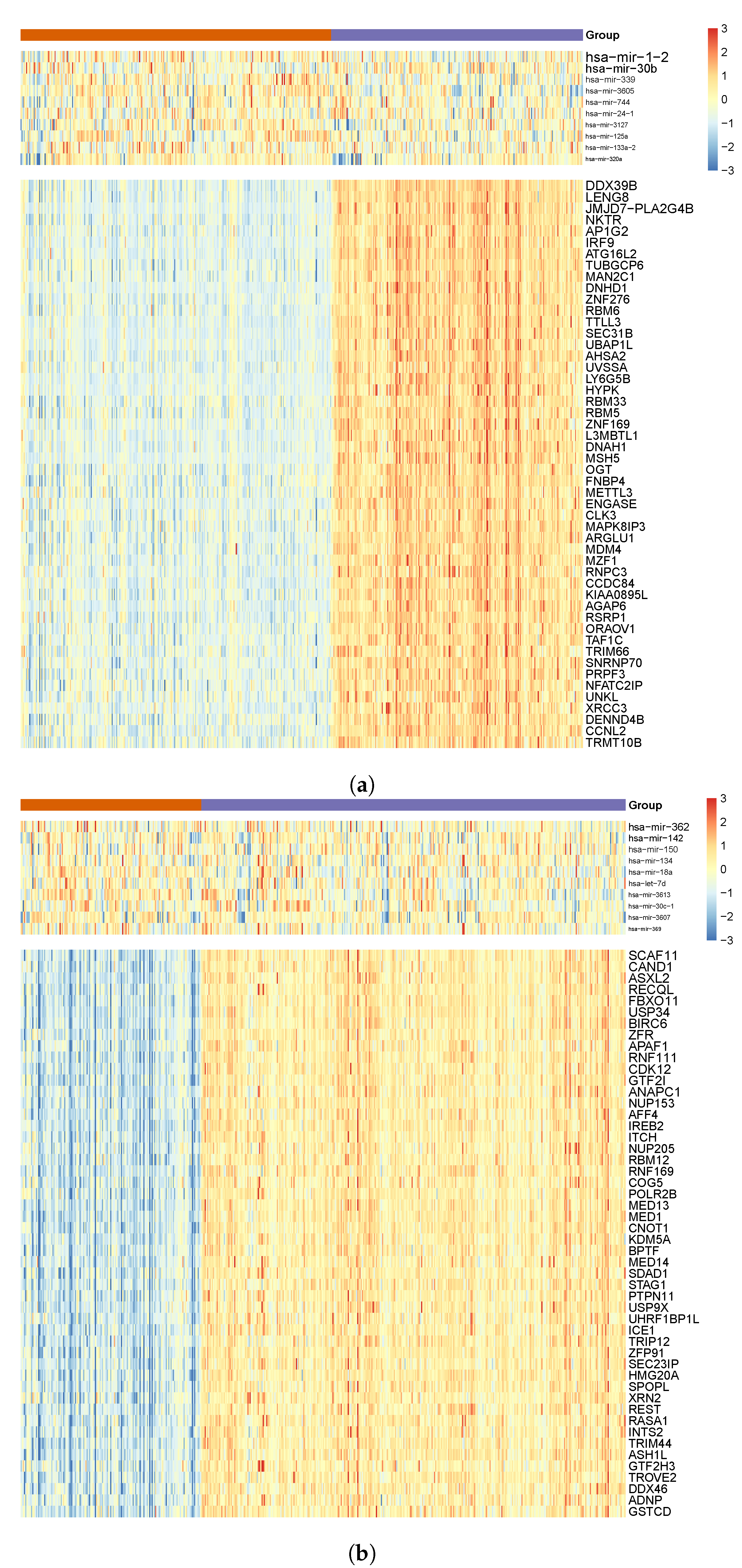
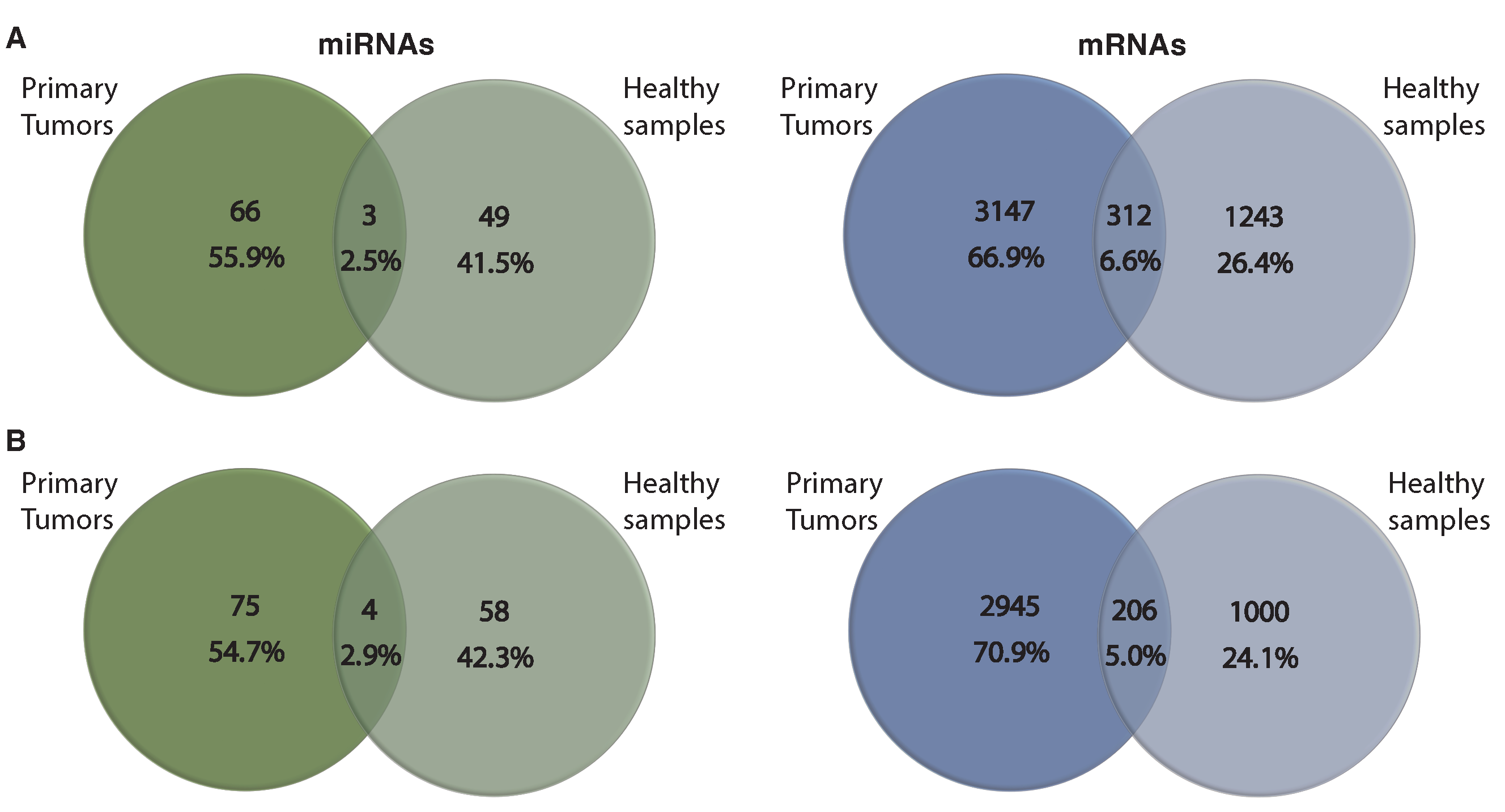
3.4. Comparing miRNA–mRNA Signatures in Cross-Cohort Combinations and with Healthy Samples
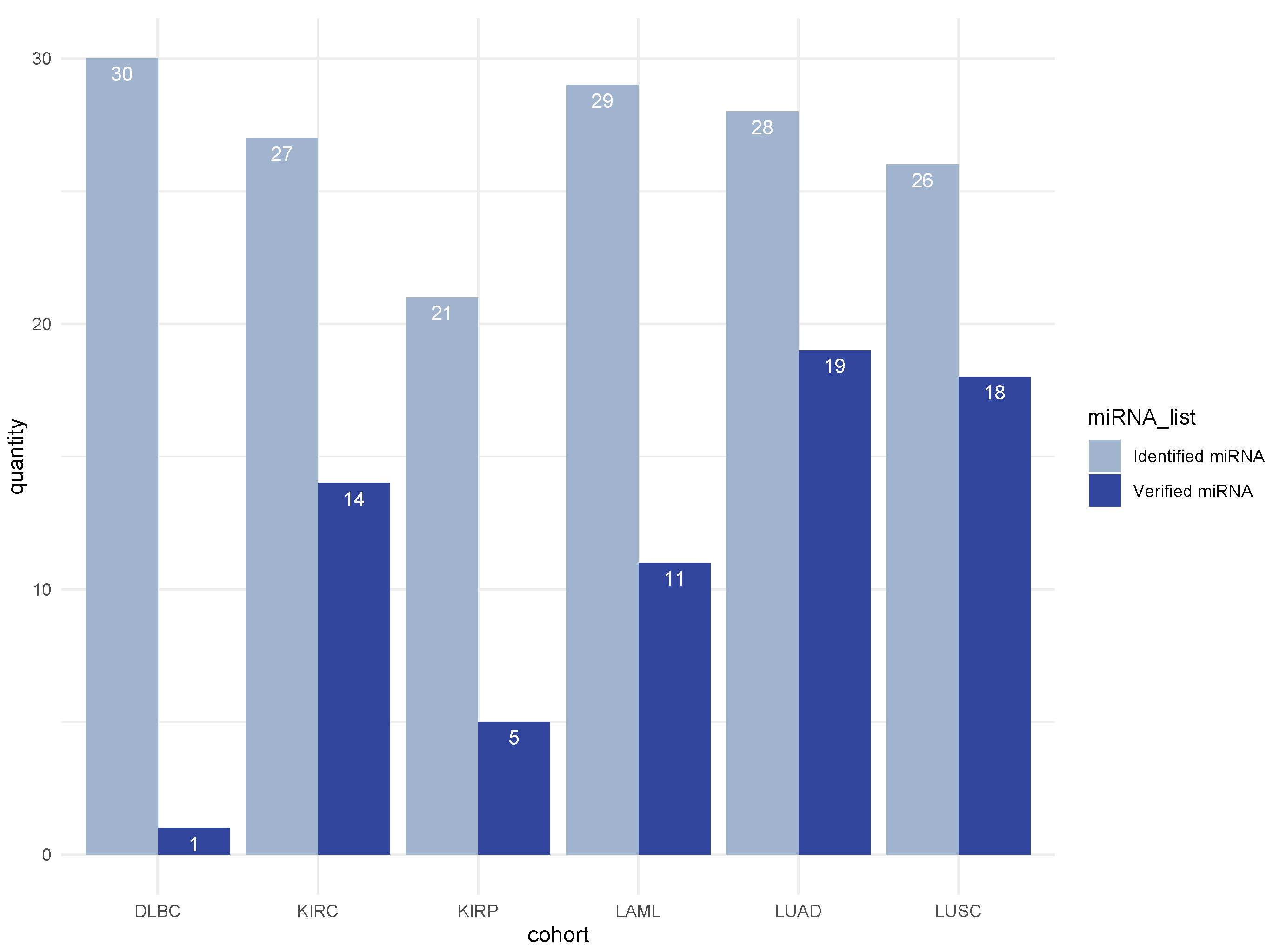
3.5. Literature Validation of Identified miRNA–mRNA Signatures
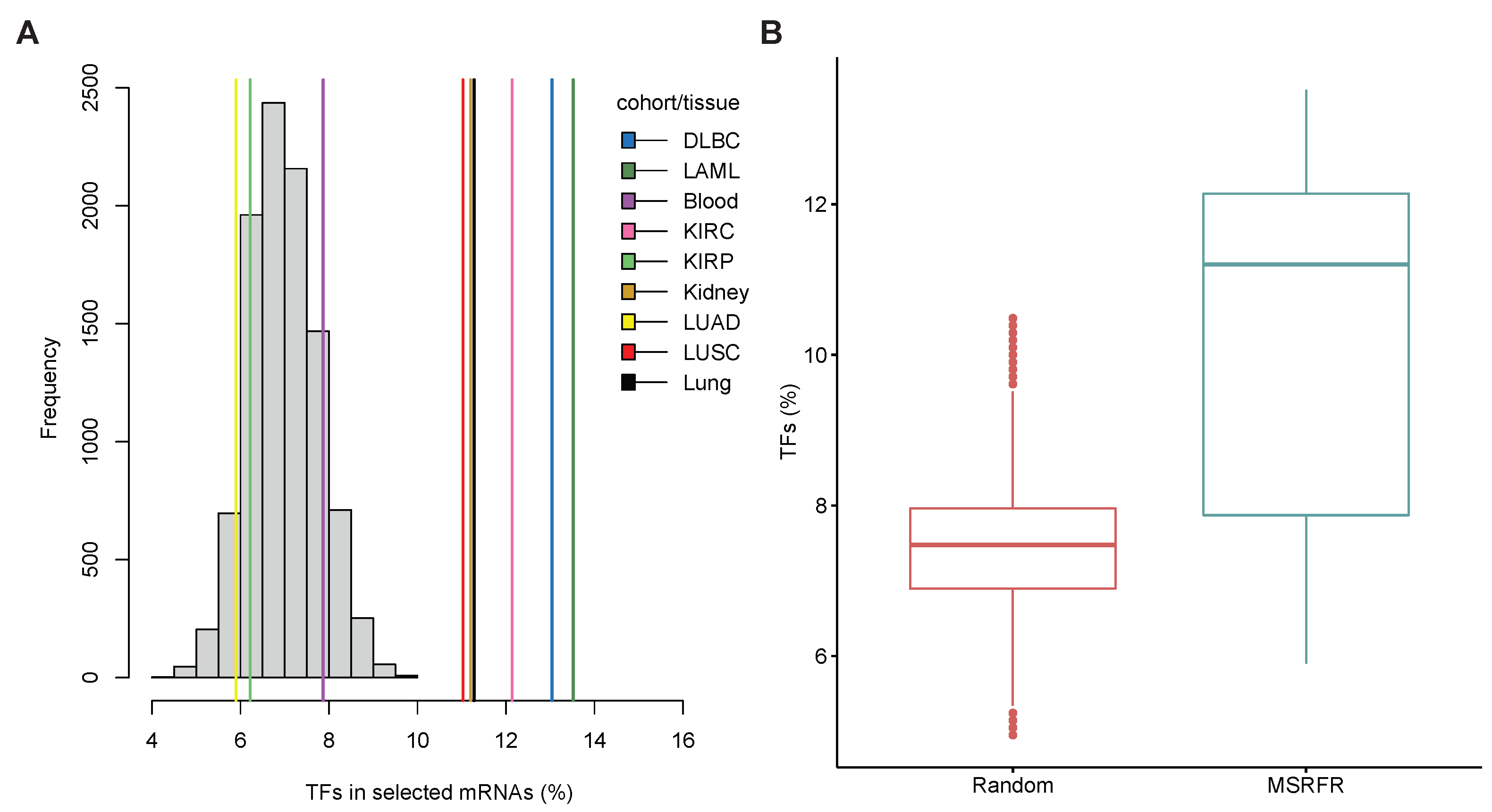
3.6. Abundance of Transcription Factor mRNAs in miRNA–mRNA Signatures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| miRNA | MicroRNA |
| mRNA | Messenger RNA |
| MSRFR | Multitask Learning Sparse Regularized Factor Regression |
| NRMSE | Normalized Root Mean Squared Error |
| TCGA | The Cancer Genome Atlas |
| DLBC | Lymphoid neoplasm diffuse large B-cell lymphoma |
| LAML | Acute myeloid leukemia |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| TF | Transcription Factor |
Appendix A
Appendix B
Appendix C
| Algorithm A1 Optimization algorithm |
|
References
- Nikolaou, M.; Pavlopoulou, A.; Georgakilas, A.G.; Kyrodimos, E. The challenge of drug resistance in cancer treatment: A current overview. Clin. Exp. Metastasis 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Lung cancers: Molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers 2018, 10, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Lee, Y.; Lee, J.S. RNA-binding proteins in cancer: Functional and therapeutic perspectives. Cancers 2020, 12, 2699. [Google Scholar] [CrossRef] [PubMed]
- Drula, R.; Ott, L.F.; Berindan-Neagoe, I.; Pantel, K.; Calin, G.A. MicroRNAs from liquid biopsy derived extracellular vesicles: Recent advances in detection and characterization methods. Cancers 2020, 12, 2009. [Google Scholar] [CrossRef] [PubMed]
- Arif, K.; Elliott, E.K.; Haupt, L.M.; Griffiths, L.R. Regulatory mechanisms of epigenetic miRNA relationships in human cancer and potential as therapeutic targets. Cancers 2020, 12, 2922. [Google Scholar] [CrossRef] [PubMed]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; de Stricker, K.; Grauslund, M.; Sørensen, J.B. Intrinsic resistance to EGFR-tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: Differences and similarities with acquired resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.; Zhang, J.; Chaterji, S. Tiresias: Context-sensitive approach to decipher the presence and strength of MicroRNA regulatory interactions. Theranostics 2018, 8, 277. [Google Scholar] [CrossRef]
- Xiao, Q.; Luo, J.; Liang, C.; Cai, J.; Li, G.; Cao, B. CeModule: An integrative framework for discovering regulatory patterns from genomic data in cancer. BMC Bioinform. 2019, 20, 67. [Google Scholar] [CrossRef] [Green Version]
- Mokhtaridoost, M.; Gönen, M. Identifying key miRNA–mRNA regulatory modules in cancer using sparse multivariate factor regression. In Proceedings of the International Conference on Machine Learning, Optimization, and Data Science, Siena, Italy, 19–23 July 2020; pp. 422–433. [Google Scholar]
- Bonnet, E.; Tatari, M.; Joshi, A.; Michoel, T.; Marchal, K.; Berx, G.; Van de Peer, Y. Module network inference from a cancer gene expression data set identifies microRNA regulated modules. PLoS ONE 2010, 5, e10162. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Goldenberg, A.; Wong, K.C.; Zhang, Z. A probabilistic approach to explore human miRNA targetome by integrating miRNA-overexpression data and sequence information. Bioinformatics 2014, 30, 621–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, R.; Pandey, P. Surveying computational algorithms for identification of miRNA–mRNA regulatory modules. Nucleus 2017, 60, 165–174. [Google Scholar] [CrossRef]
- Zhang, J.; Le, T.D.; Liu, L.; Liu, B.; He, J.; Goodall, G.J.; Li, J. Identifying direct miRNA–mRNA causal regulatory relationships in heterogeneous data. J. Biomed. Inform. 2014, 52, 438–447. [Google Scholar] [CrossRef]
- Pham, V.V.; Zhang, J.; Liu, L.; Truong, B.; Xu, T.; Nguyen, T.T.; Li, J.; Le, T.D. Identifying miRNA-mRNA regulatory relationships in breast cancer with invariant causal prediction. BMC Bioinform. 2019, 20, 143. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Tang, Y.; Maquat, L.E. mRNA–mRNA duplexes that autoelicit Staufen1-mediated mRNA decay. Nat. Struct. Mol. Biol. 2013, 20, 1214–1220. [Google Scholar] [CrossRef] [Green Version]
- Mokhtaridoost, M.; Gönen, M. An efficient framework to identify key miRNA–mRNA regulatory modules in cancer. Bioinformatics 2020, 36, i592–i600. [Google Scholar] [CrossRef]
- Caruana, R. Multitask learning. Mach. Learn. 1997, 28, 41–75. [Google Scholar] [CrossRef]
- Xu, Q.; Yang, Q. A survey of transfer and multitask learning in bioinformatics. J. Comput. Sci. Eng. 2011, 5, 257–268. [Google Scholar] [CrossRef]
- Gönen, M.; Margolin, A.A. Drug susceptibility prediction against a panel of drugs using kernelized Bayesian multitask learning. Bioinformatics 2014, 30, i556–i563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dereli, O.; Oğuz, C.; Gönen, M. A multitask multiple kernel learning algorithm for survival analysis with application to cancer biology. In Proceedings of the International Conference on Machine Learning, Long Beach, CA, USA, 10–15 June 2019; pp. 1576–1585. [Google Scholar]
- Rahimi, A.; Gönen, M. A multitask multiple kernel learning formulation for discriminating early-and late-stage cancers. Bioinformatics 2020, 36, 3766–3772. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Liu, F.; Liu, K.; Yao, X.; Risacher, S.L.; Han, J.; Guo, L.; Saykin, A.J.; Shen, L.; Initiative, A.D.N. Identifying diagnosis-specific genotype–phenotype associations via joint multitask sparse canonical correlation analysis and classification. Bioinformatics 2020, 36, i371–i379. [Google Scholar] [CrossRef]
- Zhang, X.; Xing, Y.; Sun, K.; Guo, Y. OmiEmbed: A unified multi-task deep learning framework for multi-omics data. Cancers 2021, 13, 3047. [Google Scholar] [CrossRef]
- Nikolova, O.; Moser, R.; Kemp, C.; Gönen, M.; Margolin, A.A. Modeling gene-wise dependencies improves the identification of drug response biomarkers in cancer studies. Bioinformatics 2017, 33, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- List, M.; Dehghani Amirabad, A.; Kostka, D.; Schulz, M.H. Large-scale inference of competing endogenous RNA networks with sparse partial correlation. Bioinformatics 2019, 35, i596–i604. [Google Scholar] [CrossRef] [PubMed]
- Kharratzadeh, M.; Coates, M. Sparse multivariate factor regression. In Proceedings of the IEEE Statistical Signal Processing Workshop, Palma de Mallorca, Spain, 26–29 June 2016; pp. 1–5. [Google Scholar]
- Parikh, N.; Boyd, S. Proximal algorithms. Found. Trends Opt. 2014, 1, 127–239. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The human transcription factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Pi nero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Oto, J.; Herranz, R.; Plana, E.; Sánchez-González, J.V.; Pérez-Ardavín, J.; Hervás, D.; Fernández-Pardo, Á.; Cana, F.; Vera-Donoso, C.D.; Martínez-Sarmiento, M.; et al. Identification of miR-20a-5p as robust normalizer for urine microRNAstudies in Renal Cell Carcinoma and a profile of dysregulated microRNAs. Int. J. Mol. Sci. 2021, 22, 7913. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Ghosh, A.; Datta, S.; Dasgupta, D.; Das, S.; Ray, S.; Gupta, S.; Datta, S.; Chowdhury, A.; Chatterjee, R.; et al. Hepatic miR-126 is a potential plasma biomarker for detection of hepatitis B virus infected hepatocellular carcinoma. Int. J. Cancer 2016, 138, 2732–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M. Functional proteomics of the epigenetic regulators ASXL1, ASXL2 and ASXL3: A convergence of proteomics and epigenetics for translational medicine. Expert Rev. Proteom. 2015, 12, 317–328. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Definition |
|---|---|
| mRNA expression profile matrix | |
| miRNA expression profile matrix | |
| Weight matrix of regression model | |
| Error term matrix | |
| Weight matrix to project miRNA profiles into low-dimensional space | |
| Weight matrix of linear regression in projected space | |
| N | Number of tumors |
| D | Number of miRNAs |
| T | Number of mRNAs |
| R | Dimensionality of projected space |
| k | Index of cohorts () |
| S | Index of tissue (shared between cohorts) |
| Frobenius norm | |
| norm | |
| Regularization parameters |
| MSRFR | Single-Task | |||
|---|---|---|---|---|
| Cohort | Rank | NRMSE | Rank | NRMSE |
| DLBC | 13 | 0.7664 | 19 | 0.5803 |
| LAML | 19 | 0.6708 | 20 | 0.7479 |
| KIRC | 18 | 0.7412 | 20 | 0.9796 |
| KIRP | 16 | 0.7626 | 20 | 0.7838 |
| LUAD | 20 | 0.7840 | 20 | 0.8832 |
| LUSC | 19 | 0.8039 | 20 | 0.8870 |
| MSRFR | Single-Task | |||
|---|---|---|---|---|
| Cohort or Tissue | All | Survival | All | Survival |
| DLBC | 3 | 1 | 19 | 0 |
| LAML | 9 | 3 | 20 | 4 |
| Blood | 10 | 3 | - | - |
| KIRC | 8 | 6 | 20 | 8 |
| KIRP | 6 | 2 | 20 | 0 |
| Kidney | 10 | 6 | - | - |
| LUAD | 10 | 4 | 20 | 14 |
| LUSC | 9 | 1 | 20 | 2 |
| Lung | 10 | 1 | - | - |
| Total (%) | 75 | 27 (36) | 119 | 28 (23.53) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokhtaridoost, M.; Maass, P.G.; Gönen, M. Identifying Tissue- and Cohort-Specific RNA Regulatory Modules in Cancer Cells Using Multitask Learning. Cancers 2022, 14, 4939. https://doi.org/10.3390/cancers14194939
Mokhtaridoost M, Maass PG, Gönen M. Identifying Tissue- and Cohort-Specific RNA Regulatory Modules in Cancer Cells Using Multitask Learning. Cancers. 2022; 14(19):4939. https://doi.org/10.3390/cancers14194939
Chicago/Turabian StyleMokhtaridoost, Milad, Philipp G. Maass, and Mehmet Gönen. 2022. "Identifying Tissue- and Cohort-Specific RNA Regulatory Modules in Cancer Cells Using Multitask Learning" Cancers 14, no. 19: 4939. https://doi.org/10.3390/cancers14194939
APA StyleMokhtaridoost, M., Maass, P. G., & Gönen, M. (2022). Identifying Tissue- and Cohort-Specific RNA Regulatory Modules in Cancer Cells Using Multitask Learning. Cancers, 14(19), 4939. https://doi.org/10.3390/cancers14194939