
Increased Expression and Altered Cellular Localization of Fibroblast Growth Factor Receptor-Like 1 (FGFRL1) Are Associated with Prostate Cancer Progression

 , ,
, ,  , , , , , ,
, , , , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Expression Database Mining
2.2. Patient Data and Tissue Microarray (TMA) Construction
2.3. Collection and Statistical Analysis of Clinical Data
2.4. Cell Culture and Treatments
2.5. FGFRL1 Knockdown and Overexpression in PC3M Cells
2.6. Western Blotting
2.7. RNA Isolation and Quantitative Real-Time PCR (qRT-PCR)
2.8. Cell Proliferation and Migration Assays
2.9. Organotypic 3D Culture
2.10. Mouse Xenografts
2.11. RNA Sequencing and Data Analysis
3. Results
3.1. FGFRL1 mRNA Expression Is Increased in PCa
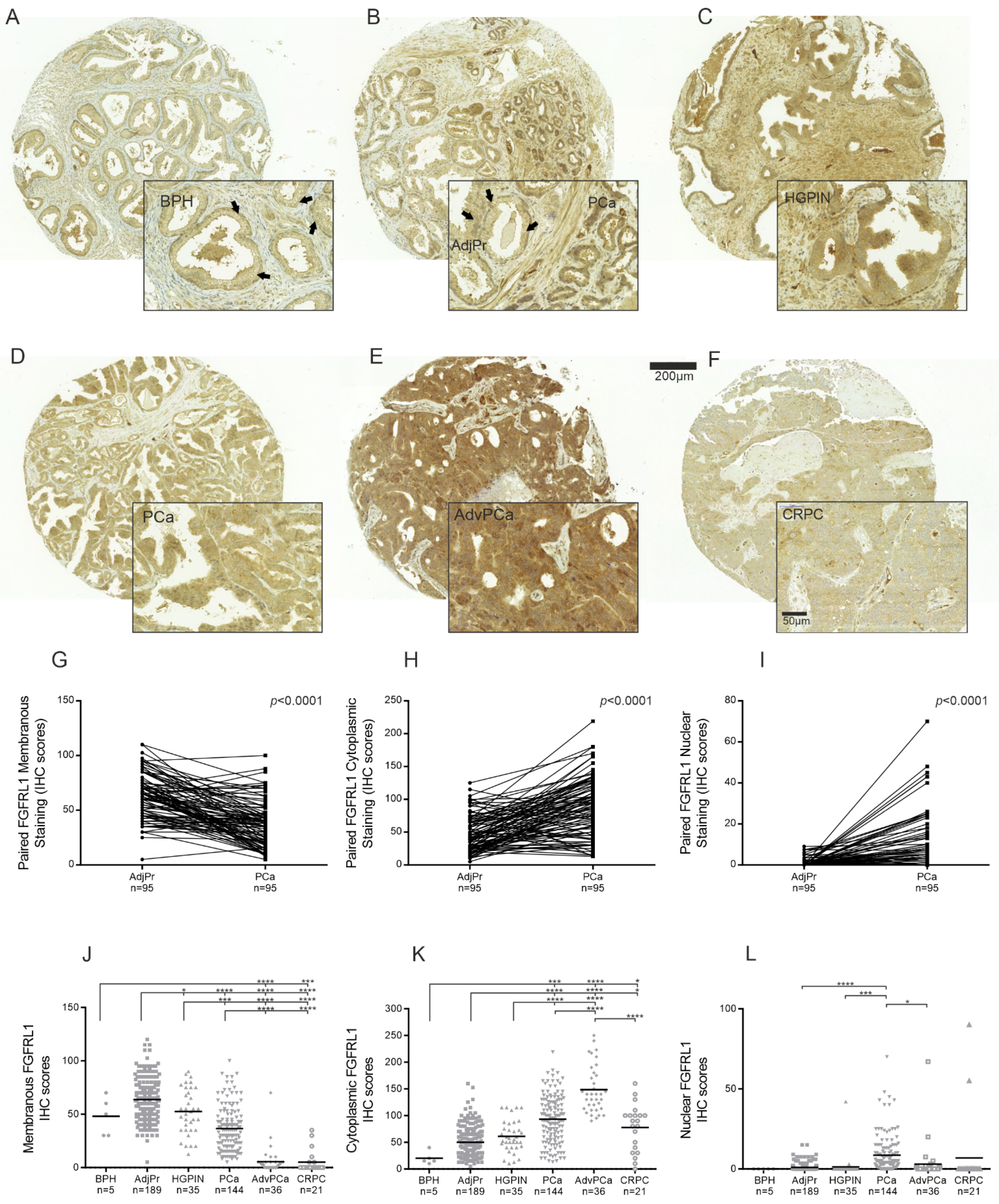
3.2. FGFRL1 Protein Shows Increased Expression and Altered Cellular Localization in HGPIN and PCa Compared to Benign Prostate
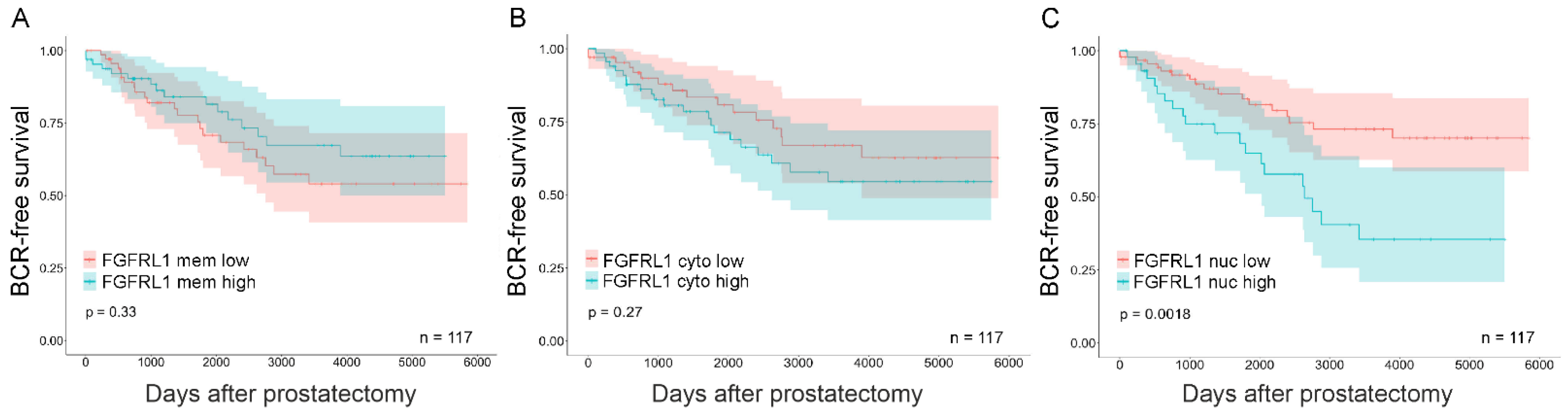
3.3. Expression of FGFRL1 Correlates with Gleason Score, Pre-Operative PSA Levels, and Ki67 Staining
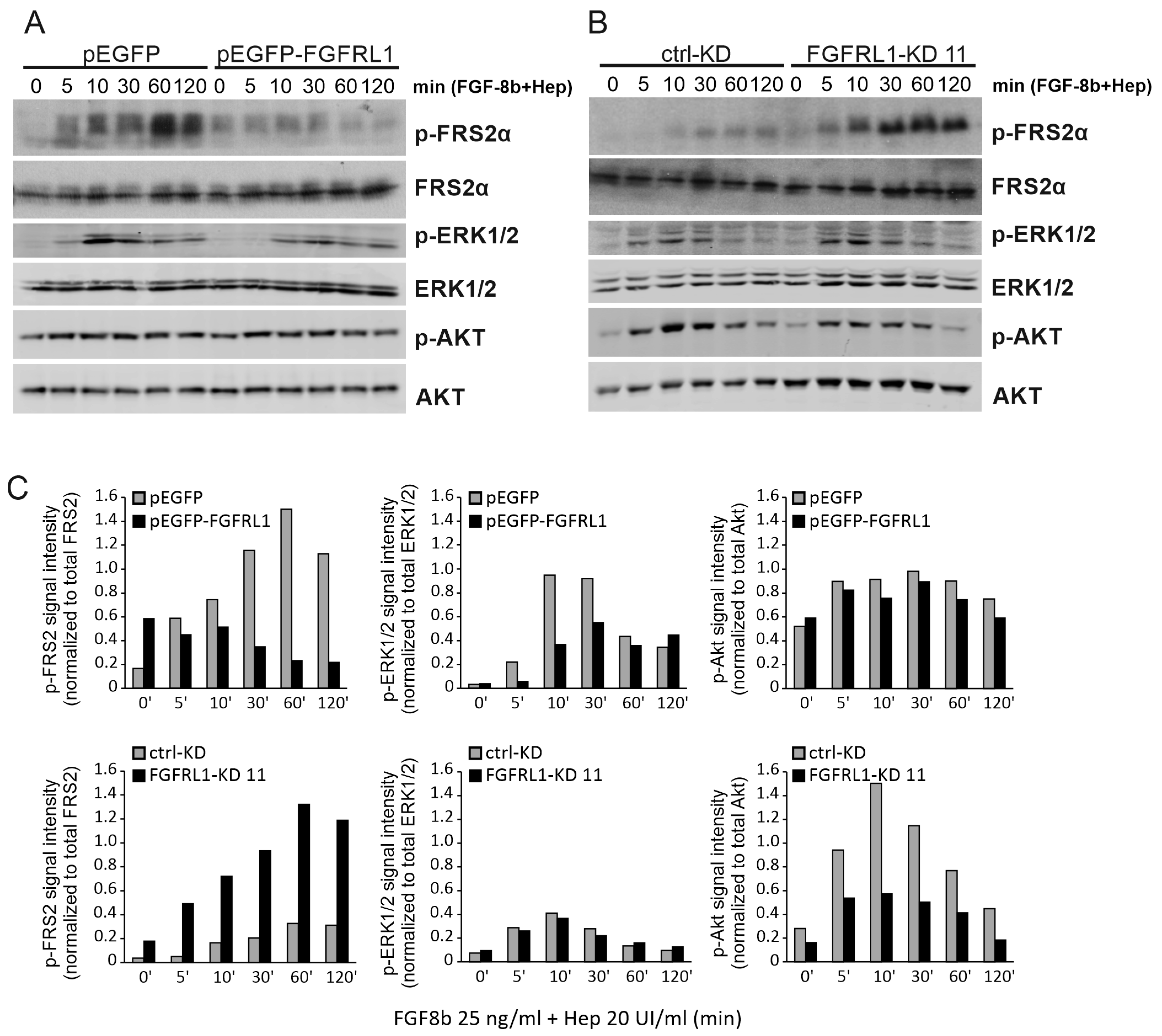
3.4. FGFRL1 Negatively Regulates FGFR Signaling in Prostate Cancer Cells
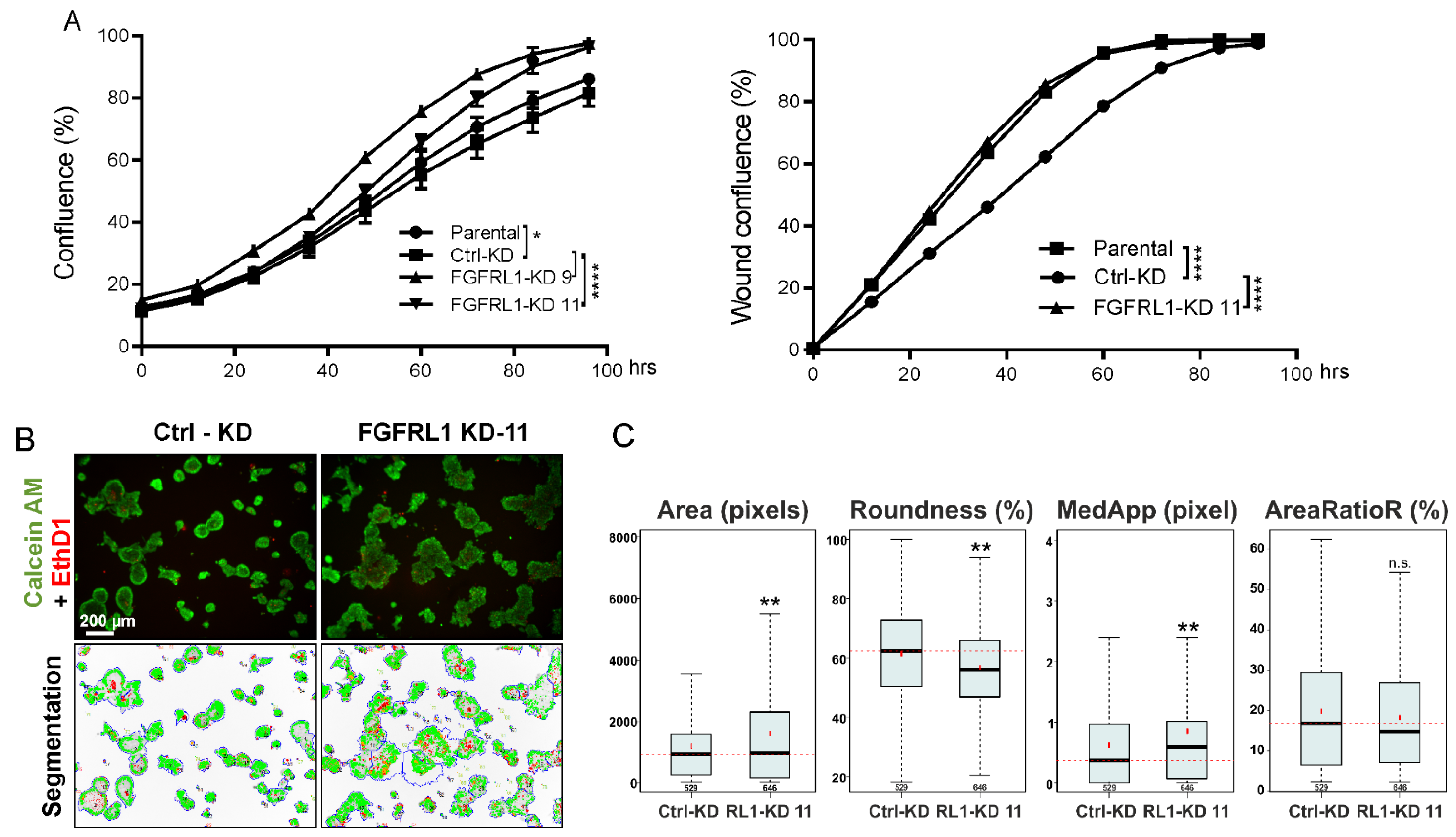
3.5. FGFRL1 Knockdown Affects PC3M Organoid Growth and Differentiation in Organotypic 3D Cultures
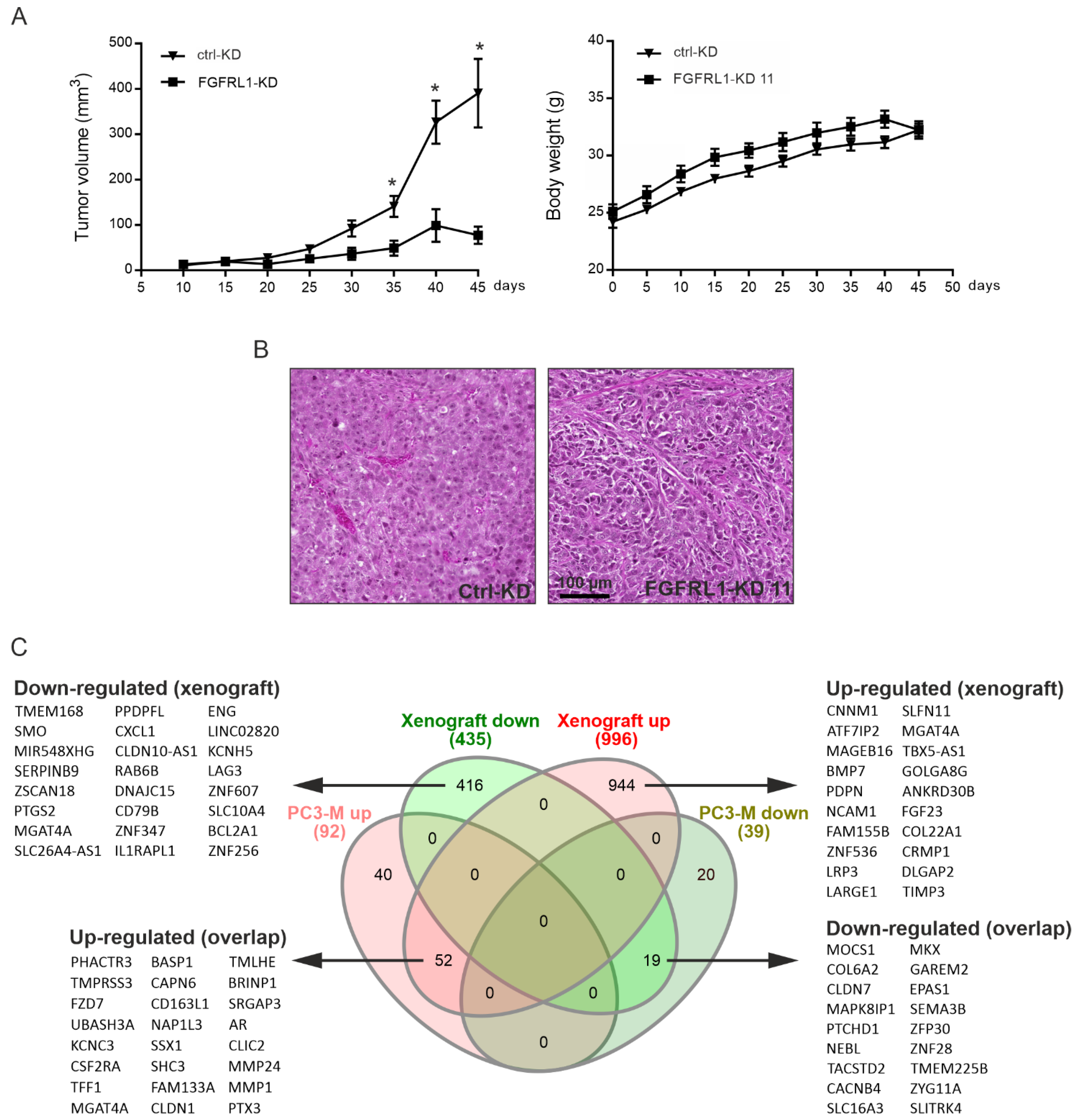
3.6. Growth of Xenografts Derived from FGFRL1-KD Cells Is Decreased
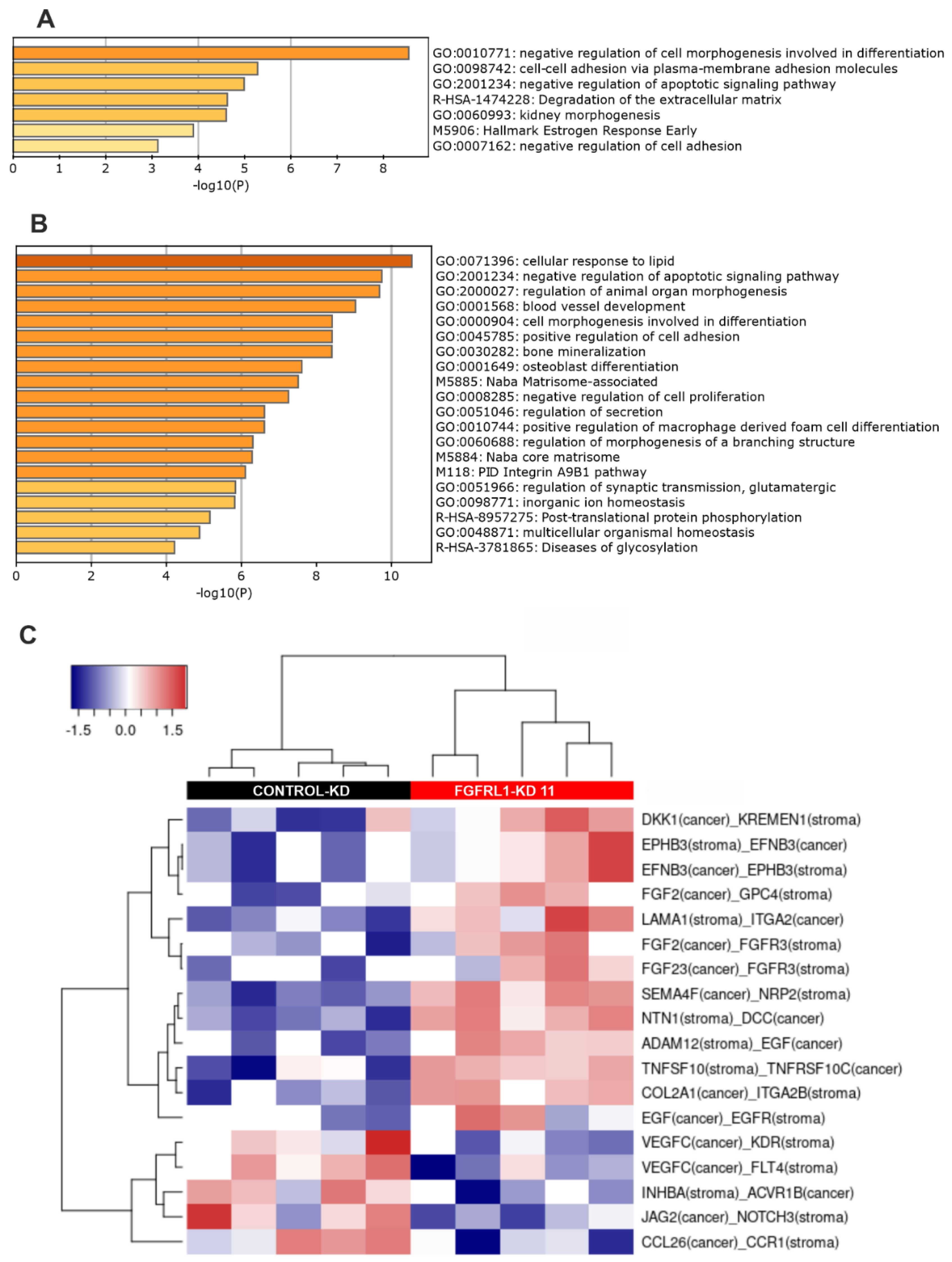
3.7. Altered Gene Expression Profiles between Control-KD and FGFRL1-KD Cells in Cultured Cells and Xenografts

3.8. Altered Ligand–Receptor Interactions in Xenografts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ingrosso, G.; Detti, B.; Scartoni, D.; Lancia, A.; Giacomelli, I.; Baki, M.; Carta, G.A.; Livi, L.; Santoni, R. Current therapeutic options in metastatic castration-resistant prostate cancer. Semin. Oncol. 2018, 45, 303–315. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Corn, P.G.; Wang, F.; McKeehan, W.L.; Navone, N. Targeting Fibroblast Growth Factor Pathways in Prostate Cancer. Clin. Cancer Res. 2013, 19, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, A.; Grillo, E.; Rezzola, S.; Ribatti, D.; Rusnati, M.; Ronca, R.; Presta, M. The FGF/FGFR system in the physiopathology of the prostate gland. Physiol. Rev. 2021, 101, 569–610. [Google Scholar] [CrossRef] [PubMed]
- Eswarakumar, J.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Iwata, T.; Leung, H.Y. Mechanisms of FGFR-mediated carcinogenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 2004, 11, 709–724. [Google Scholar] [CrossRef]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef]
- Haugsten, E.M.; Wiedlocha, A.; Olsnes, S.; Wesche, J. Roles of Fibroblast Growth Factor Receptors in Carcinogenesis. Mol. Cancer Res. 2010, 8, 1439–1452. [Google Scholar] [CrossRef]
- Elo, T.D.; Valve, E.M.; Seppänen, J.A.; Vuorikoski, H.; Mäkelä, S.; Poutanen, M.; Kujala, P.M.; Härkönen, P.L. Stromal Activation Associated with Development of Prostate Cancer in Prostate-Targeted Fibroblast Growth Factor 8b Transgenic Mice. Neoplasia 2010, 12, 915-IN19. [Google Scholar] [CrossRef]
- Valta, M.P.; Tuomela, J.; Vuorikoski, H.; Loponen, N.; Väänänen, R.-M.; Pettersson, K.; Väänänen, H.K.; Härkönen, P.L. FGF-8b induces growth and rich vascularization in an orthotopic PC-3 model of prostate cancer. J. Cell. Biochem. 2009, 107, 769–784. [Google Scholar] [CrossRef]
- Valta, M.P.; Tuomela, J.; Bjartell, A.; Valve, E.; Väänänen, H.K.; Härkönen, P. FGF-8 is involved in bone metastasis of prostate cancer. Int. J. Cancer 2008, 123, 22–31. [Google Scholar] [CrossRef]
- Trueb, B. Biology of FGFRL1, the fifth fibroblast growth factor receptor. Cell. Mol. Life Sci. 2010, 68, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.D.; Beauchamp, P.; Zhuang, L.; Villiger, P.M.; Trueb, B. Functional domains of the FgfrL1 receptor. Dev. Biol. 2020, 461, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Catela, C.; Bilbao-Cortes, D.; Slonimsky, E.; Kratsios, P.; Rosenthal, N.; Welscher, P.T. Multiple congenital malformations of Wolf-Hirschhorn syndrome are recapitulated inFgfrl1null mice. Dis. Model. Mech. 2009, 2, 283–294. [Google Scholar] [CrossRef]
- Silva, P.N.; Altamentova, S.M.; Kilkenny, D.M.; Rocheleau, J.V. Fibroblast Growth Factor Receptor Like-1 (FGFRL1) Interacts with SHP-1 Phosphatase at Insulin Secretory Granules and Induces Beta-Cell ERK1/2 Protein Activation. J. Biol. Chem. 2013, 288, 17859–17870. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Villiger, P.; Trueb, B. Interaction of the receptor FGFRL1 with the negative regulator Spred1. Cell. Signal. 2011, 23, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, F.; Zhuang, L.; Beyeler, M.; Kälin, R.E.; Mullis, P.E.; Brändli, A.W.; Trueb, B. The FGFRL1 Receptor Is Shed from Cell Membranes, Binds Fibroblast Growth Factors (FGFs), and Antagonizes FGF Signaling in Xenopus Embryos. J. Biol. Chem. 2010, 285, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Amann, R.; Trueb, B. Evidence that the novel receptor FGFRL1 signals indirectly via FGFR1. Int. J. Mol. Med. 2013, 32, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Trueb, B.; Zhuang, L.; Taeschler, S.; Wiedemann, M. Characterization of FGFRL1, a Novel Fibroblast Growth Factor (FGF) Receptor Preferentially Expressed in Skeletal Tissues. J. Biol. Chem. 2003, 278, 33857–33865. [Google Scholar] [CrossRef] [PubMed]
- Kähkönen, T.; Ivaska, K.; Jiang, M.; Büki, K.; Väänänen, H.; Härkönen, P. Role of fibroblast growth factor receptors (FGFR) and FGFR like-1 (FGFRL1) in mesenchymal stromal cell differentiation to osteoblasts and adipocytes. Mol. Cell. Endocrinol. 2018, 461, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, T.; Kotevic, I.; Trueb, B. The cell surface receptor FGFRL1 forms constitutive dimers that promote cell adhesion. Exp. Cell Res. 2008, 314, 1071–1081. [Google Scholar] [CrossRef]
- Yang, X.; Steinberg, F.; Zhuang, L.; Bessey, R.; Trueb, B. Receptor FGFRL1 does not promote cell proliferation but induces cell adhesion. Int. J. Mol. Med. 2016, 38, 30–38. [Google Scholar] [CrossRef]
- Schild, C.; Trueb, B. Aberrant expression of FGFRL1, a novel FGF receptor, in ovarian tumors. Int. J. Mol. Med. 2005, 16, 1169–1173. [Google Scholar] [CrossRef]
- Tai, H.; Wu, Z.; Sun, S.; Zhang, Z.; Xu, C. FGFRL1 Promotes Ovarian Cancer Progression by Crosstalk with Hedgehog Signaling. J. Immunol. Res. 2018, 2018, 7438608. [Google Scholar] [CrossRef]
- Takei, Y.; Matsumura, T.; Watanabe, K.; Nakamine, H.; Sudo, T.; Shimizu, K.; Shimada, Y. FGFRL1 deficiency reduces motility and tumorigenic potential of cells derived from oesophageal squamous cell carcinomas. Oncol. Lett. 2018, 16, 809–814. [Google Scholar] [CrossRef]
- Tsuchiya, S.; Fujiwara, T.; Sato, F.; Shimada, Y.; Tanaka, E.; Sakai, Y.; Shimizu, K.; Tsujimoto, G. MicroRNA-210 Regulates Cancer Cell Proliferation through Targeting Fibroblast Growth Factor Receptor-like 1 (FGFRL1). J. Biol. Chem. 2011, 286, 420–428. [Google Scholar] [CrossRef]
- Donnard, E.; Asprino, P.F.; Correa, B.R.S.; Bettoni, F.; Koyama, F.C.; Navarro, F.; Perez, R.O.; Mariadason, J.; Sieber, O.; Straussberg, R.L.; et al. Mutational analysis of genes coding for cell surface proteins in colorectal cancer cell lines reveal novel altered pathways, druggable mutations and mutated epitopes for targeted therapy. Oncotarget 2014, 5, 9199–9213. [Google Scholar] [CrossRef]
- di Martino, E.; Taylor, C.F.; Roulson, J.-A.; Knowles, M.A. An integrated genomic, transcriptional and protein investigation ofFGFRL1as a putative 4p16.3 deletion target in bladder cancer. Genes Chromosom. Cancer 2013, 52, 860–871. [Google Scholar] [CrossRef]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, S.; Autio, R.; Ojala, K.; Iljin, K.; Bucher, E.; Sara, H.; Pisto, T.; Saarela, M.; Skotheim, R.I.; Björkman, M.; et al. Systematic bioinformatic analysis of expression levels of 17,330 human genes across 9783 samples from 175 types of healthy and pathological tissues. Genome Biol. 2008, 9, R139. [Google Scholar] [CrossRef]
- Yu, L.; Toriseva, M.; Tuomala, M.; Seikkula, H.; Elo, T.; Tuomela, J.; Kallajoki, M.; Mirtti, T.; Taimen, P.; Boström, P.J.; et al. Increased expression of fibroblast growth factor 13 in prostate cancer is associated with shortened time to biochemical recurrence after radical prostatectomy. Int. J. Cancer 2016, 139, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Allsbrook, W.C., Jr.; Amin, M.B.; Egevad, L.L.; ISUP Grading Committee. The 2005 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 2005, 29, 1228–1242. [Google Scholar] [CrossRef]
- Epstein, J.I. An Update of the Gleason Grading System. J. Urol. 2010, 183, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Härmä, V.; Virtanen, J.; Mäkelä, R.; Happonen, A.; Mpindi, J.-P.; Knuuttila, M.; Kohonen, P.; Lötjönen, J.; Kallioniemi, O.; Nees, M. A Comprehensive Panel of Three-Dimensional Models for Studies of Prostate Cancer Growth, Invasion and Drug Responses. PLoS ONE 2010, 5, e10431. [Google Scholar] [CrossRef] [PubMed]
- Härmä, V.; Schukov, H.-P.; Happonen, A.; Ahonen, I.; Virtanen, J.; Siitari, H.; Åkerfelt, M.; Lötjönen, J.; Nees, M. Quantification of Dynamic Morphological Drug Responses in 3D Organotypic Cell Cultures by Automated Image Analysis. PLoS ONE 2014, 9, e96426. [Google Scholar] [CrossRef]
- Komura, D.; Isagawa, T.; Kishi, K.; Suzuki, R.; Sato, R.; Tanaka, M.; Katoh, H.; Yamamoto, S.; Tatsuno, K.; Fukayama, M.; et al. CASTIN: A system for comprehensive analysis of cancer-stromal interactome. BMC Genom. 2016, 17, 899. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, T.; Zhuang, L.; Flück, C.E.; Trueb, B. Characterization of the first FGFRL1 mutation identified in a craniosynostosis patient. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 112–121. [Google Scholar] [CrossRef]
- Porębska, N.; Latko, M.; Kucińska, M.; Zakrzewska, M.; Otlewski, J.; Opaliński, Ł. Targeting Cellular Trafficking of Fibroblast Growth Factor Receptors as a Strategy for Selective Cancer Treatment. J. Clin. Med. 2018, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.J.; Chioni, A.; Ghallab, M.; Anderson, R.K.; Lemoine, N.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR 1 and FGF 2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.; Bruce, C.; Chioni, A.; Kocher, H.; Grose, R. The Ins and Outs of Fibroblast Growth Factor Receptor Signalling. Clin. Sci. 2014, 127, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Tuzon, C.T.; Rigueur, D.; Merrill, A.E. Nuclear Fibroblast Growth Factor Receptor Signaling in Skeletal Development and Disease. Curr. Osteoporos. Rep. 2019, 17, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Polnaszek, N.; Kwabi-Addo, B.; Peterson, L.E.; Ozen, M.; Greenberg, N.M.; Ortega, S.; Basilico, C.; Ittmann, M. Fibroblast growth factor 2 promotes tumor progression in an autochthonous mouse model of prostate cancer. Cancer Res. 2003, 63, 5754–5760. [Google Scholar]
- Ronca, R.; Giacomini, A.; Di Salle, E.; Coltrini, D.; Pagano, K.; Ragona, L.; Matarazzo, S.; Rezzola, S.; Maiolo, D.; Torella, R.; et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 2015, 28, 225–239. [Google Scholar] [CrossRef]
- Ronca, R.; Tamma, R.; Coltrini, D.; Ruggieri, S.; Presta, M.; Ribatti, D. Fibroblast growth factor modulates mast cell recruitment in a murine model of prostate cancer. Oncotarget 2017, 8, 82583–82592. [Google Scholar] [CrossRef]
- Matarazzo, S.; Melocchi, L.; Rezzola, S.; Grillo, E.; Maccarinelli, F.; Giacomini, A.; Turati, M.; Taranto, S.; Zammataro, L.; Cerasuolo, M.; et al. Long Pentraxin-3 Follows and Modulates Bladder Cancer Progression. Cancers 2019, 11, 1277. [Google Scholar] [CrossRef]
- Miyazaki, H.; Watabe, T.; Kitamura, T.; Miyazono, K. BMP signals inhibit proliferation and in vivo tumor growth of androgen-insensitive prostate carcinoma cells. Oncogene 2004, 23, 9326–9335. [Google Scholar] [CrossRef]
- Jackson, H.; Defamie, V.; Waterhouse, P.; Khokha, R. TIMPs: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.R.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nat. Cell Biol. 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Dong, B.; Xu, F.; Xu, Y.; Pan, J.; Song, J.; Zhang, J.; Huang, Y.; Xue, W. CXCL1-LCN2 paracrine axis promotes progression of prostate cancer via the Src activation and epithelial-mesenchymal transition. Cell Commun. Signal. 2019, 17, 1–15. [Google Scholar] [CrossRef]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | FGFRL1 Staining IHC Scores | Spearman Rank Correlation Coefficient | p-Value |
|---|---|---|---|
| Membrane | −0.200 | 0.018 | |
| Gleason Score | Cytoplasm | 0.217 | 0.010 |
| Nucleus | 0.297 | 0.000 | |
| Membrane | −0.004 | 0.966 | |
| Pre-operative PSA | Cytoplasm | −0.141 | 0.098 |
| Nucleus | 0.199 | 0.019 | |
| Membrane | −0.269 | 0.002 | |
| Ki67 IHC scores | Cytoplasm | 0.261 | 0.002 |
| Nucleus | 0.097 | 0.261 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Toriseva, M.; Afshan, S.; Cangiano, M.; Fey, V.; Erickson, A.; Seikkula, H.; Alanen, K.; Taimen, P.; Ettala, O.; et al. Increased Expression and Altered Cellular Localization of Fibroblast Growth Factor Receptor-Like 1 (FGFRL1) Are Associated with Prostate Cancer Progression. Cancers 2022, 14, 278. https://doi.org/10.3390/cancers14020278
Yu L, Toriseva M, Afshan S, Cangiano M, Fey V, Erickson A, Seikkula H, Alanen K, Taimen P, Ettala O, et al. Increased Expression and Altered Cellular Localization of Fibroblast Growth Factor Receptor-Like 1 (FGFRL1) Are Associated with Prostate Cancer Progression. Cancers. 2022; 14(2):278. https://doi.org/10.3390/cancers14020278
Chicago/Turabian StyleYu, Lan, Mervi Toriseva, Syeda Afshan, Mario Cangiano, Vidal Fey, Andrew Erickson, Heikki Seikkula, Kalle Alanen, Pekka Taimen, Otto Ettala, and et al. 2022. "Increased Expression and Altered Cellular Localization of Fibroblast Growth Factor Receptor-Like 1 (FGFRL1) Are Associated with Prostate Cancer Progression" Cancers 14, no. 2: 278. https://doi.org/10.3390/cancers14020278
APA StyleYu, L., Toriseva, M., Afshan, S., Cangiano, M., Fey, V., Erickson, A., Seikkula, H., Alanen, K., Taimen, P., Ettala, O., Nurmi, M., Boström, P. J., Kallajoki, M., Tuomela, J., Mirtti, T., Beumer, I. J., Nees, M., & Härkönen, P. (2022). Increased Expression and Altered Cellular Localization of Fibroblast Growth Factor Receptor-Like 1 (FGFRL1) Are Associated with Prostate Cancer Progression. Cancers, 14(2), 278. https://doi.org/10.3390/cancers14020278