
CRISPR-Cas9 Screen Identifies DYRK1A as a Target for Radiotherapy Sensitization in Pancreatic Cancer

 ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. The sgRNA Library and Lentivirus Production
2.3. CRISPR/Cas9 Knockout Screening
2.4. Genomic DNA Sequencing and Data Analysis
2.5. CRISPR-Cas9 Gene Editing
2.6. Clonogenic Assay
2.7. Alkaline Comet Assay
2.8. Western Blot
2.9. Immunofluorescence
2.10. Apoptosis Assay
2.11. Statistical Analysis
3. Results
3.1. CRISPR-Cas9 Loss-of-Function Screen Identifies DYRK1A as Candidate for Radiotherapy Resistance
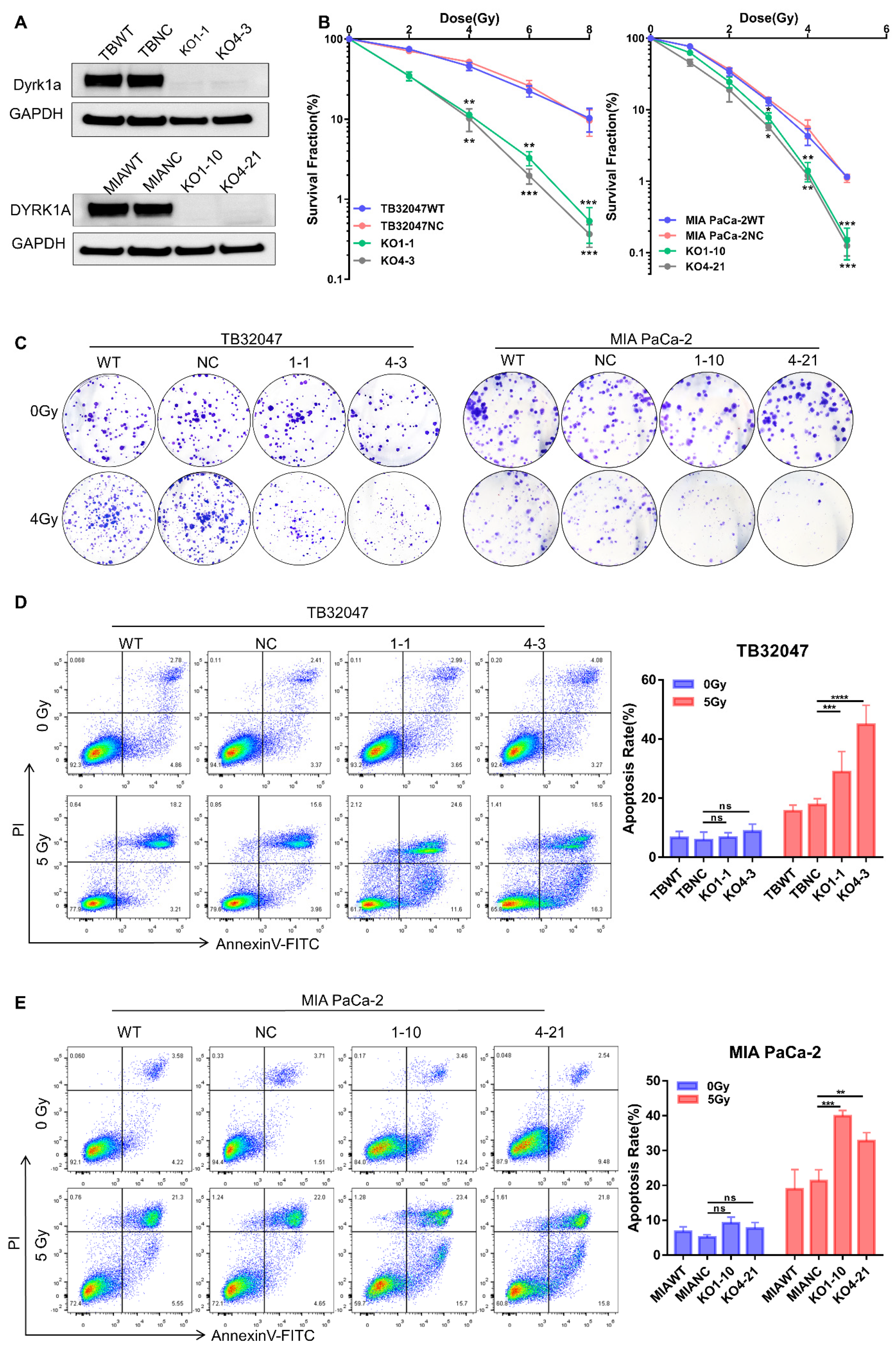
3.2. DYRK1A Knockout Enhances Radiotherapy Efficacy in Pancreatic Cancer Cells
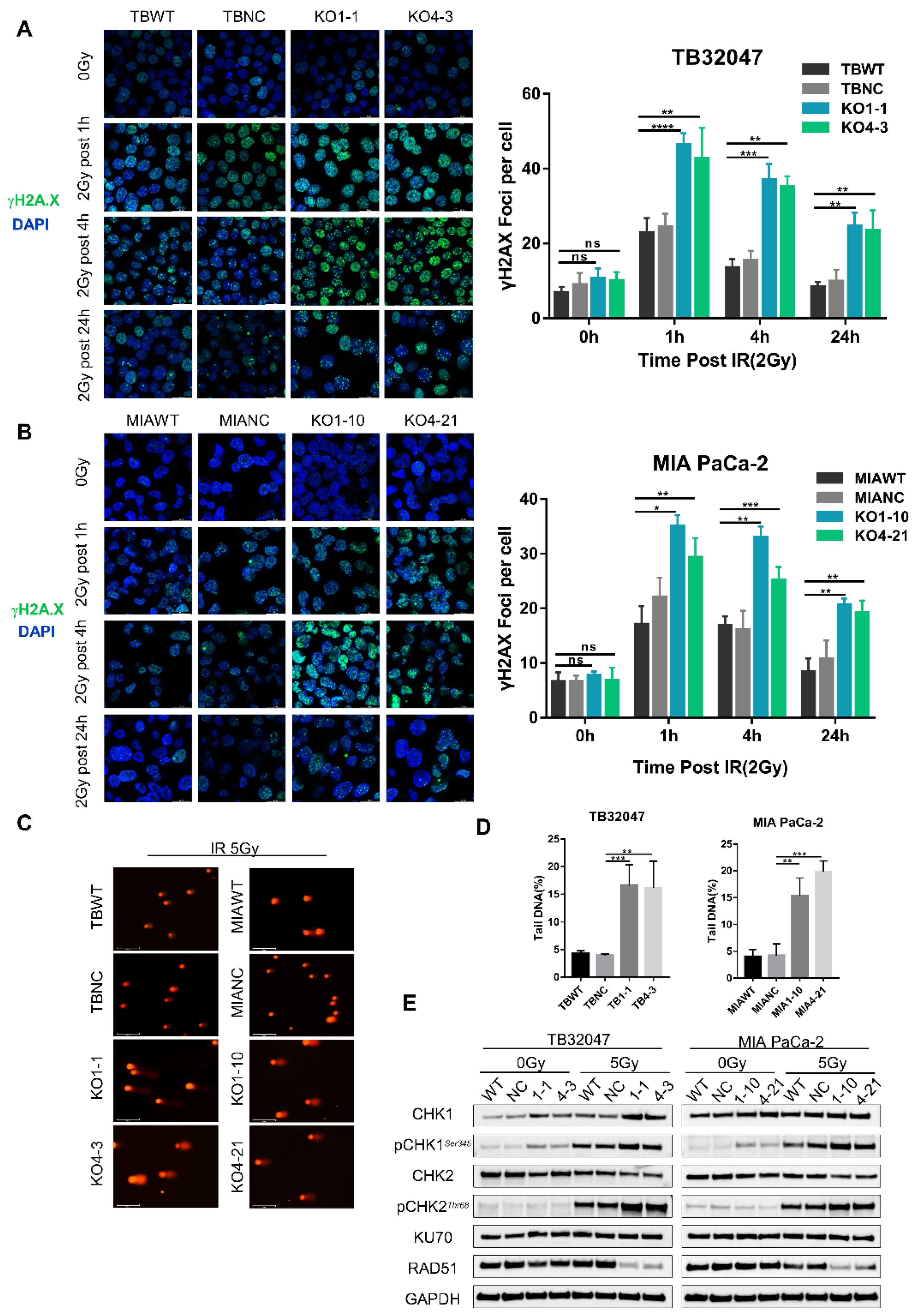
3.3. DYRK1A Knockout Increases DNA Damage and DNA Double-Strand Breaks in Pancreatic Cancer Cells after Radiotherapy
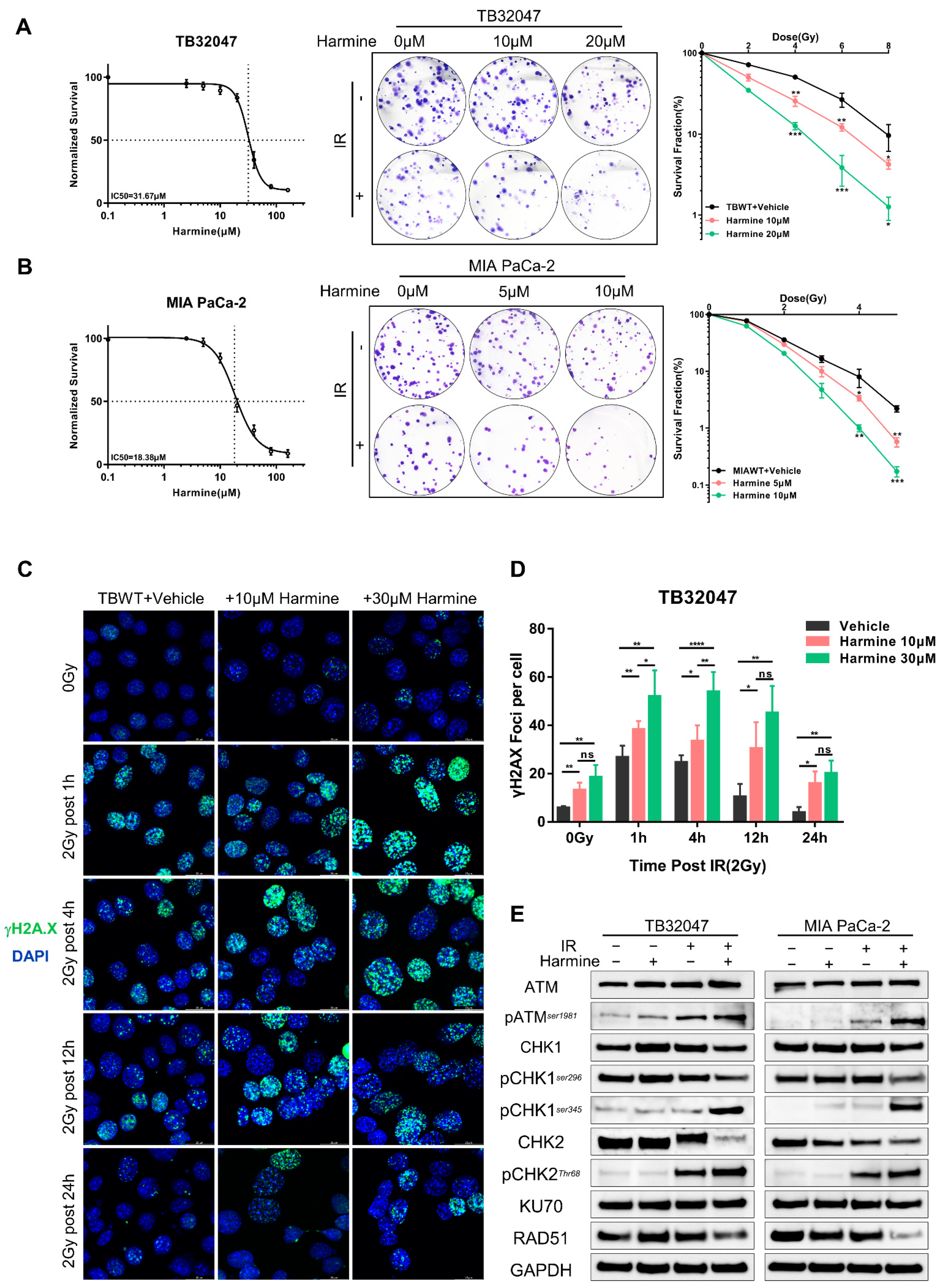
3.4. Targeted Inhibition of DYRK1A Promotes Radiosensitivity in Pancreatic Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Baine, M.J.; Souchek, J.J.; Menning, M.; Kaur, S.; Yan, Y.; Ouellette, M.M.; Jain, M.; Lin, C.; Batra, S.K. Biological determinants of radioresistance and their remediation in pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 69–92. [Google Scholar] [CrossRef]
- Hu, J.X.; Zhao, C.F.; Chen, W.B.; Liu, Q.C.; Li, Q.W.; Lin, Y.Y.; Gao, F. Pancreatic cancer: A review of epidemiology, trend, and risk factors. World J. Gastroenterol. 2021, 27, 4298–4321. [Google Scholar] [CrossRef]
- Fietkau, R.; Grutzmann, R.; Wittel, U.A.; Croner, R.S.; Jacobasch, L.; Neumann, U.P.; Reinacher-Schick, A.; Imhoff, D.; Boeck, S.; Keilholz, L.; et al. R0 resection following chemo (radio)therapy improves survival of primary inoperable pancreatic cancer patients. Interim results of the German randomized CONKO-007± trial. Strahlenther. Onkol. 2021, 197, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Versteijne, E.; Suker, M.; Groothuis, K.; Akkermans-Vogelaar, J.M.; Besselink, M.G.; Bonsing, B.A.; Buijsen, J.; Busch, O.R.; Creemers, G.M.; van Dam, R.M.; et al. Preoperative Chemoradiotherapy Versus Immediate Surgery for Resectable and Borderline Resectable Pancreatic Cancer: Results of the Dutch Randomized Phase III PREOPANC Trial. J. Clin. Oncol. 2020, 38, 1763–1773. [Google Scholar] [CrossRef]
- Machairas, N.; Raptis, D.A.; Velazquez, P.S.; Sauvanet, A.; Rueda de Leon, A.; Oba, A.; Koerkamp, B.G.; Lovasik, B.; Chan, C.; Yeo, C.J.; et al. The Impact of Neoadjuvant Treatment on Survival in Patients Undergoing Pancreatoduodenectomy with Concomitant Portomesenteric Venous Resection: An International Multicenter Analysis. Ann. Surg. 2021, 274, 721–728. [Google Scholar] [CrossRef]
- Cloyd, J.M.; Chen, H.C.; Wang, X.; Tzeng, C.D.; Kim, M.P.; Aloia, T.A.; Vauthey, J.N.; Lee, J.E.; Katz, M.H.G. Chemotherapy Versus Chemoradiation as Preoperative Therapy for Resectable Pancreatic Ductal Adenocarcinoma: A Propensity Score Adjusted Analysis. Pancreas 2019, 48, 216–222. [Google Scholar] [CrossRef]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, I.; Manic, G.; De Maria, R.; Kroemer, G.; Galluzzi, L. DNA Damage in Stem Cells. Mol. Cell 2017, 66, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Cordes, N. ATM controls DNA repair and mitochondria transfer between neighboring cells. Cell Commun. Signal 2019, 17, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waissi, W.; Paix, A.; Nicol, A.; Noel, G.; Burckel, H. Targeting DNA repair in combination with radiotherapy in pancreatic cancer: A systematic review of preclinical studies. Crit. Rev. Oncol. Hematol. 2020, 153, 103060. [Google Scholar] [CrossRef] [PubMed]
- Parsels, L.A.; Engelke, C.G.; Parsels, J.; Flanagan, S.A.; Zhang, Q.; Tanska, D.; Wahl, D.R.; Canman, C.E.; Lawrence, T.S.; Morgan, M.A. Combinatorial Efficacy of Olaparib with Radiation and ATR Inhibitor Requires PARP1 Protein in Homologous Recombination-Proficient Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Gorte, J.; Danen, E.; Cordes, N. Therapy-Naive and Radioresistant 3-Dimensional Pancreatic Cancer Cell Cultures Are Effectively Radiosensitized by β1 Integrin Targeting. Int. J. Radiat. Oncol. Biol. Phys. 2021. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Koster, J.; Xu, H.; Chen, C.H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef] [Green Version]
- Lentsch, E.; Li, L.; Pfeffer, S.; Ekici, A.B.; Taher, L.; Pilarsky, C.; Grutzmann, R. CRISPR/Cas9-Mediated Knock-Out of Kras(G12D) Mutated Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konca, K.; Lankoff, A.; Banasik, A.; Lisowska, H.; Kuszewski, T.; Gozdz, S.; Koza, Z.; Wojcik, A. A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res. 2003, 534, 15–20. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Y.; Xiong, J.; Lan, B.; Wang, X.; Liu, J.; Lin, J.; Fei, Z.; Zheng, X.; Chen, C. Role of PRKDC in cancer initiation, progression, and treatment. Cancer Cell Int. 2021, 21, 563. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Purman, C.; Porter, S.I.; Nganga, V.; Saini, A.; Hayer, K.E.; Gurewitz, G.L.; Sleckman, B.P.; Bednarski, J.J.; Bassing, C.H.; et al. DNA double-strand breaks induce H2Ax phosphorylation domains in a contact-dependent manner. Nat. Commun. 2020, 11, 3158. [Google Scholar] [CrossRef]
- Barroso, S.I.; Aguilera, A. Detection of DNA Double-Strand Breaks by gamma-H2AX Immunodetection. Methods Mol. Biol. 2021, 2153, 1–8. [Google Scholar] [CrossRef]
- Wassing, I.E.; Esashi, F. RAD51: Beyond the break. Semin. Cell Dev. Biol. 2021, 113, 38–46. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Liang, S.Q.; Yang, H.; Xu, D.; Bruggmann, R.; Gao, Y.; Deng, H.; Berezowska, S.; Hall, S.R.R.; Marti, T.M.; et al. CRISPR-Mediated Kinome Editing Prioritizes a Synergistic Combination Therapy for FGFR1-Amplified Lung Cancer. Cancer Res. 2021, 81, 3121–3133. [Google Scholar] [CrossRef]
- Deville, S.S.; Luft, S.; Kaufmann, M.; Cordes, N. Keap1 inhibition sensitizes head and neck squamous cell carcinoma cells to ionizing radiation via impaired non-homologous end joining and induced autophagy. Cell Death Dis. 2020, 11, 887. [Google Scholar] [CrossRef] [PubMed]
- Vellayappan, B.A.; Soon, Y.Y.; Ku, G.Y.; Leong, C.N.; Lu, J.J.; Tey, J.C. Chemoradiotherapy versus chemoradiotherapy plus surgery for esophageal cancer. Cochrane Database Syst. Rev. 2017, 8, CD010511. [Google Scholar] [CrossRef]
- Kunigenas, L.; Stankevicius, V.; Dulskas, A.; Budginaite, E.; Alzbutas, G.; Stratilatovas, E.; Cordes, N.; Suziedelis, K. 3D Cell Culture-Based Global miRNA Expression Analysis Reveals miR-142-5p as a Theranostic Biomarker of Rectal Cancer Following Neoadjuvant Long-Course Treatment. Biomolecules 2020, 10, 613. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Lee, W.C.; Aust, D.; Pilarsky, C.; Cordes, N. β8 Integrin Mediates Pancreatic Cancer Cell Radiochemoresistance. Mol. Cancer Res. 2019, 17, 2126–2138. [Google Scholar] [CrossRef]
- Laham, A.J.; Saber-Ayad, M.; El-Awady, R. DYRK1A: A down syndrome-related dual protein kinase with a versatile role in tumorigenesis. Cell. Mol. Life Sci. 2021, 78, 603–619. [Google Scholar] [CrossRef]
- Luna, J.; Boni, J.; Cuatrecasas, M.; Bofill-De Ros, X.; Nunez-Manchon, E.; Gironella, M.; Vaquero, E.C.; Arbones, M.L.; de la Luna, S.; Fillat, C. DYRK1A modulates c-MET in pancreatic ductal adenocarcinoma to drive tumour growth. Gut 2019, 68, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Clarke, P.R. p38α- and DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site in response to hyperosmotic stress. Cell. Signal. 2009, 21, 1626–1633. [Google Scholar] [CrossRef]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef] [Green Version]
- Roewenstrunk, J.; Di Vona, C.; Chen, J.; Borras, E.; Dong, C.; Arato, K.; Sabido, E.; Huen, M.S.Y.; de la Luna, S. A comprehensive proteomics-based interaction screen that links DYRK1A to RNF169 and to the DNA damage response. Sci. Rep. 2019, 9, 6014. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Olaparib: First global approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef]
- Huang, R.X.; Zhou, P.K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, F.; Zhang, W.; Chen, L.; Gao, N.; Men, Y.; Xu, X.; Jiang, Y. Harmine suppresses homologous recombination repair and inhibits proliferation of hepatoma cells. Cancer Biol. Ther. 2015, 16, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.W.; Zhang, J.K.; Rao, M.; Zhang, Z.Y.; Zhu, H.J.; Zhang, C. Harmine suppresses the proliferation of pancreatic cancer cells and sensitizes pancreatic cancer to gemcitabine treatment. OncoTargets Ther. 2019, 12, 4585–4593. [Google Scholar] [CrossRef] [Green Version]
- Recasens, A.; Humphrey, S.J.; Ellis, M.; Hoque, M.; Abbassi, R.H.; Chen, B.; Longworth, M.; Needham, E.J.; James, D.E.; Johns, T.G.; et al. Global phosphoproteomics reveals DYRK1A regulates CDK1 activity in glioblastoma cells. Cell Death Discov. 2021, 7, 81. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, B.; Zeng, S.; Zhang, S.; Ren, X.; Xing, Y.; Kutschick, I.; Pfeffer, S.; Frey, B.; Britzen-Laurent, N.; Grützmann, R.; et al. CRISPR-Cas9 Screen Identifies DYRK1A as a Target for Radiotherapy Sensitization in Pancreatic Cancer. Cancers 2022, 14, 326. https://doi.org/10.3390/cancers14020326
Lan B, Zeng S, Zhang S, Ren X, Xing Y, Kutschick I, Pfeffer S, Frey B, Britzen-Laurent N, Grützmann R, et al. CRISPR-Cas9 Screen Identifies DYRK1A as a Target for Radiotherapy Sensitization in Pancreatic Cancer. Cancers. 2022; 14(2):326. https://doi.org/10.3390/cancers14020326
Chicago/Turabian StyleLan, Bin, Siyuan Zeng, Shuman Zhang, Xiaofan Ren, Yuming Xing, Isabella Kutschick, Susanne Pfeffer, Benjamin Frey, Nathalie Britzen-Laurent, Robert Grützmann, and et al. 2022. "CRISPR-Cas9 Screen Identifies DYRK1A as a Target for Radiotherapy Sensitization in Pancreatic Cancer" Cancers 14, no. 2: 326. https://doi.org/10.3390/cancers14020326
APA StyleLan, B., Zeng, S., Zhang, S., Ren, X., Xing, Y., Kutschick, I., Pfeffer, S., Frey, B., Britzen-Laurent, N., Grützmann, R., Cordes, N., & Pilarsky, C. (2022). CRISPR-Cas9 Screen Identifies DYRK1A as a Target for Radiotherapy Sensitization in Pancreatic Cancer. Cancers, 14(2), 326. https://doi.org/10.3390/cancers14020326