
Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance?

 , ,
, , 
 and
and
Abstract
:Simple Summary
Abstract
1. The Therapeutic Landscape of Metastatic Prostate Cancer
2. Immunotherapy in Metastatic Prostate Cancer Treatment: Current Status and Mechanisms of Resistance
3. Tumor-Associated Macrophages in PC
4. Involvement of TAMs in ICIs Efficacy
5. Targeting Macrophages in Cancer Therapy and Its Application in PC
5.1. Therapies Affecting TAM Precursor Recruitment
5.2. Therapies That Induce Depletion or Affect Macrophage Survival
5.3. Strategies to Reprogram Macrophage Activity
6. Discussion and Future Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Sandhu, S.; Moore, C.M.; Chiong, E.; Beltran, H.; Bristow, R.G.; Williams, S.G. Prostate cancer. Lancet 2021, 398, 1075–1090. [Google Scholar] [CrossRef]
- Davies, A.; Conteduca, V.; Zoubeidi, A.; Beltran, H. Biological Evolution of Castration-resistant Prostate Cancer. Eur. Urol. Focus 2019, 5, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.S. Metastatic Prostate Cancer. N. Engl. J. Med. 2018, 378, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Berthold, D.R.; Pond, G.R.; Soban, F.; de Wit, R.; Eisenberger, M.; Tannock, I.F. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: Updated survival in the TAX 327 study. J. Clin. Oncol. 2008, 26, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Beltran, H. The treatment landscape of metastatic prostate cancer. Cancer Lett. 2021, 519, 20–29. [Google Scholar] [CrossRef]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Violet, J.; Hicks, R.J.; Ferdinandus, J.; Thang, S.P.; Akhurst, T.; Iravani, A.; Kong, G.; Ravi Kumar, A.; Murphy, D.G.; et al. [(177)Lu]-PSMA-617 radionuclide treatment in patients with metastatic castration-resistant prostate cancer (LuPSMA trial): A single-centre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 825–833. [Google Scholar] [CrossRef]
- Corn, P.G.; Heath, E.I.; Zurita, A.; Ramesh, N.; Xiao, L.; Sei, E.; Li-Ning-Tapia, E.; Tu, S.M.; Subudhi, S.K.; Wang, J.; et al. Cabazitaxel plus carboplatin for the treatment of men with metastatic castration-resistant prostate cancers: A randomised, open-label, phase 1-2 trial. Lancet Oncol 2019, 20, 1432–1443. [Google Scholar] [CrossRef]
- Ruiz de Porras, V.; Font, A.; Aytes, A. Chemotherapy in metastatic castration-resistant prostate cancer: Current scenario and future perspectives. Cancer Lett. 2021, 523, 162–169. [Google Scholar] [CrossRef]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Ruiz de Porras, V.; Pardo, J.C.; Notario, L.; Etxaniz, O.; Font, A. Immune Checkpoint Inhibitors: A Promising Treatment Option for Metastatic Castration-Resistant Prostate Cancer? Int. J. Mol. Sci. 2021, 22, 4712. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labadie, B.W.; Balar, A.V.; Luke, J.J. Immune Checkpoint Inhibitors for Genitourinary Cancers: Treatment Indications, Investigational Approaches and Biomarkers. Cancers 2021, 13, 5415. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Callahan, M.K.; Bono, P.; Kim, J.; Spiliopoulou, P.; Calvo, E.; Pillai, R.N.; Ott, P.A.; de Braud, F.; Morse, M.; et al. Nivolumab monotherapy in recurrent metastatic urothelial carcinoma (CheckMate 032): A multicentre, open-label, two-stage, multi-arm, phase 1/2 trial. Lancet Oncol. 2016, 17, 1590–1598. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Shaukat, F.; Isaacsson Velho, P.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Blankenstein, T.; Coulie, P.G.; Gilboa, E.; Jaffee, E.M. The determinants of tumour immunogenicity. Nat. Rev. Cancer 2012, 12, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Bilusic, M.; Madan, R.A.; Gulley, J.L. Immunotherapy of Prostate Cancer: Facts and Hopes. Clin. Cancer Res. 2017, 23, 6764–6770. [Google Scholar] [CrossRef] [Green Version]
- Vitkin, N.; Nersesian, S.; Siemens, D.R.; Koti, M. The Tumor Immune Contexture of Prostate Cancer. Front. Immunol. 2019, 10, 603. [Google Scholar] [CrossRef] [Green Version]
- Sanda, M.G.; Restifo, N.P.; Walsh, J.C.; Kawakami, Y.; Nelson, W.G.; Pardoll, D.M.; Simons, J.W. Molecular characterization of defective antigen processing in human prostate cancer. J. Natl. Cancer Inst. 1995, 87, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Moreira, D.; Won, H.; White, S.W.; Duttagupta, P.; Lucia, M.; Jones, J.; Hsu, J.; Kortylewski, M. Reduced T-cell Numbers and Elevated Levels of Immunomodulatory Cytokines in Metastatic Prostate Cancer Patients De Novo Resistant to Abiraterone and/or Enzalutamide Therapy. Int. J. Mol. Sci. 2019, 20, 1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Pluddemann, A. The Mononuclear Phagocytic System. Generation of Diversity. Front. Immunol. 2019, 10, 1893. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Pluddemann, A. Tissue macrophages: Heterogeneity and functions. BMC Biol. 2017, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Sinha, M.; Datta, S.; Abas, M.; Chaffee, S.; Sen, C.K.; Roy, S. Monocyte and macrophage plasticity in tissue repair and regeneration. Am. J. Pathol. 2015, 185, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Stout, R.D.; Jiang, C.; Matta, B.; Tietzel, I.; Watkins, S.K.; Suttles, J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J. Immunol. 2005, 175, 342–349. [Google Scholar] [CrossRef]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev. Immunoll. 2012, 32, 463–488. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal. Transductl Target. Ther 2021, 6, 127. [Google Scholar] [CrossRef]
- Castells, M.; Thibault, B.; Delord, J.P.; Couderc, B. Implication of tumor microenvironment in chemoresistance: Tumor-associated stromal cells protect tumor cells from cell death. Int. J. Mol. Sci. 2012, 13, 9545–9571. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Bissell, M.J. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist. Updates 2012, 15, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Nonomura, N.; Takayama, H.; Nakayama, M.; Nakai, Y.; Kawashima, A.; Mukai, M.; Nagahara, A.; Aozasa, K.; Tsujimura, A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2011, 107, 1918–1922. [Google Scholar] [CrossRef]
- Cioni, B.; Zaalberg, A.; van Beijnum, J.R.; Melis, M.H.M.; van Burgsteden, J.; Muraro, M.J.; Hooijberg, E.; Peters, D.; Hofland, I.; Lubeck, Y.; et al. Androgen receptor signalling in macrophages promotes TREM-1-mediated prostate cancer cell line migration and invasion. Nat. Commun. 2020, 11, 4498. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson, A.; Carlsson, J.; Lundholm, M.; Falt, A.; Andersson, S.O.; Andren, O.; Davidsson, S. M2 macrophages and regulatory T cells in lethal prostate cancer. Prostate 2019, 79, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Qian, Y.; Yu, F.; Liu, W.; Wu, Y.; Fang, X.; Hao, W. Alternatively activated macrophages are associated with metastasis and poor prognosis in prostate adenocarcinoma. Oncol. Lett. 2015, 10, 1390–1396. [Google Scholar] [CrossRef] [Green Version]
- Izumi, K.; Fang, L.Y.; Mizokami, A.; Namiki, M.; Li, L.; Lin, W.J.; Chang, C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013, 5, 1383–1401. [Google Scholar] [CrossRef] [PubMed]
- Maolake, A.; Izumi, K.; Shigehara, K.; Natsagdorj, A.; Iwamoto, H.; Kadomoto, S.; Takezawa, Y.; Machioka, K.; Narimoto, K.; Namiki, M.; et al. Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 2017, 8, 9739–9751. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Wang, S.; Wang, N.; Zheng, Y.; Zhou, J.; Yang, B.; Wang, X.; Zhang, J.; Guo, L.; Wang, S.; et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating beta-catenin/STAT3 signaling. Cell Death Dis. 2020, 11, 234. [Google Scholar] [CrossRef]
- Idorn, M.; Kollgaard, T.; Kongsted, P.; Sengelov, L.; Thor Straten, P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. 2014, 63, 1177–1187. [Google Scholar] [CrossRef]
- Lopez-Bujanda, Z.; Drake, C.G. Myeloid-derived cells in prostate cancer progression: Phenotype and prospective therapies. J. Leukoc. Biol. 2017, 102, 393–406. [Google Scholar] [CrossRef] [Green Version]
- Beyzaei, Z.; Shojazadeh, A.; Geramizadeh, B. The role of regulatory T cells in liver transplantation. Transpl. Immunol. 2021, 70, 101512. [Google Scholar] [CrossRef]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karkampouna, S. The Role of Cancer-Associated Fibroblasts in Prostate Cancer Tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Gok Yavuz, B.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1(+) TAMs. Sci. Rep. 2019, 9, 3172. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundholm, M.; Hagglof, C.; Wikberg, M.L.; Stattin, P.; Egevad, L.; Bergh, A.; Wikstrom, P.; Palmqvist, R.; Edin, S. Secreted Factors from Colorectal and Prostate Cancer Cells Skew the Immune Response in Opposite Directions. Sci. Rep. 2015, 5, 15651. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, J.X.; Wang, H.; Wang, G.L.; Hu, Q.G.; Zheng, Q.C. Hepatocellular carcinoma and macrophage interaction induced tumor immunosuppression via Treg requires TLR4 signaling. World J. Gastroenterol. 2012, 18, 2938–2947. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, H.; Lu, J.; Bi, P.; Wang, F.; Liu, X.; Zhang, B.; Luo, Y.; Li, X. Tumor cells induced-M2 macrophage favors accumulation of Treg in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 2017, 10, 8389–8401. [Google Scholar]
- Gyori, D.; Lim, E.L.; Grant, F.M.; Spensberger, D.; Roychoudhuri, R.; Shuttleworth, S.J.; Okkenhaug, K.; Stephens, L.R.; Hawkins, P.T. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 2018, 3, e120631. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Wei, F.Q.; Li, W.J.; Wei, J.W.; Zhong, H.; Wen, Y.H.; Lei, W.B.; Chen, L.; Li, H.; Lin, H.Q.; et al. A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br. J. Cancer 2017, 117, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [Green Version]
- McCord, R.; Bolen, C.R.; Koeppen, H.; Kadel, E.E., 3rd; Oestergaard, M.Z.; Nielsen, T.; Sehn, L.H.; Venstrom, J.M. PD-L1 and tumor-associated macrophages in de novo DLBCL. Blood Adv. 2019, 3, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.Y.; Chen, Y.L.; Wu, W.Y.; Lin, H.W.; Chiang, Y.C.; Chang, C.F.; Tai, Y.J.; Hsu, H.C.; Chen, C.A.; Sun, W.Z.; et al. Blockade of PD-L1 Enhances Cancer Immunotherapy by Regulating Dendritic Cell Maturation and Macrophage Polarization. Cancers 2019, 11, 1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1014–1030.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liakou, C.I.; Kamat, A.; Pettaway, C.; Ward, J.F.; Tang, D.N.; Sun, J.; Jungbluth, A.A.; Troncoso, P.; Logothetis, C.; et al. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 2729–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waitz, R.; Solomon, S.B.; Petre, E.N.; Trumble, A.E.; Fasso, M.; Norton, L.; Allison, J.P. Potent induction of tumor immunity by combining tumor cryoablation with anti-CTLA-4 therapy. Cancer Res. 2012, 72, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Stanley, E.R. Role of dimerization and modification of the CSF-1 receptor in its activation and internalization during the CSF-1 response. EMBO J. 1991, 10, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, N.G.; Yu, W.; Cox, D.; Wyckoff, J.; Condeelis, J.; Stanley, E.R.; Pixley, F.J. Phosphorylation of CSF-1R Y721 mediates its association with PI3K to regulate macrophage motility and enhancement of tumor cell invasion. J. Cell Sci. 2011, 124 Pt 12, 2021–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Chen, J.; Xiong, Y.; Pixley, F.J.; Yeung, Y.G.; Stanley, E.R. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J. Biol. Chem. 2012, 287, 13694–13704. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, H.; Shen, Z.; Lin, C.; Wang, X.; Qin, J.; Qin, X.; Xu, J.; Sun, Y. Increased Expression of CSF-1 Associates With Poor Prognosis of Patients With Gastric Cancer Undergoing Gastrectomy. Medicine 2016, 95, e2675. [Google Scholar] [CrossRef]
- Richardsen, E.; Uglehus, R.D.; Johnsen, S.H.; Busund, L.T. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. 2015, 35, 865–874. [Google Scholar]
- Espinosa, I.; Edris, B.; Lee, C.H.; Cheng, H.W.; Gilks, C.B.; Wang, Y.; Montgomery, K.D.; Varma, S.; Li, R.; Marinelli, R.J.; et al. CSF1 expression in nongynecological leiomyosarcoma is associated with increased tumor angiogenesis. Am. J. Pathol. 2011, 179, 2100–2107. [Google Scholar] [CrossRef]
- Zhang, C.; Ibrahim, P.N.; Zhang, J.; Burton, E.A.; Habets, G.; Zhang, Y.; Powell, B.; West, B.L.; Matusow, B.; Tsang, G.; et al. Design and pharmacology of a highly specific dual FMS and KIT kinase inhibitor. Proc. Natl. Acad. Sci. USA 2013, 110, 5689–5694. [Google Scholar] [CrossRef] [Green Version]
- Cuccarese, M.F.; Dubach, J.M.; Pfirschke, C.; Engblom, C.; Garris, C.; Miller, M.A.; Pittet, M.J.; Weissleder, R. Heterogeneity of macrophage infiltration and therapeutic response in lung carcinoma revealed by 3D organ imaging. Nat. Commun. 2017, 8, 14293. [Google Scholar] [CrossRef]
- Tan, I.L.; Arifa, R.D.N.; Rallapalli, H.; Kana, V.; Lao, Z.; Sanghrajka, R.M.; Sumru Bayin, N.; Tanne, A.; Wojcinski, A.; Korshunov, A.; et al. CSF1R inhibition depletes tumor-associated macrophages and attenuates tumor progression in a mouse sonic Hedgehog-Medulloblastoma model. Oncogene 2021, 40, 396–407. [Google Scholar] [CrossRef]
- West, R.B.; Rubin, B.P.; Miller, M.A.; Subramanian, S.; Kaygusuz, G.; Montgomery, K.; Zhu, S.; Marinelli, R.J.; De Luca, A.; Downs-Kelly, E.; et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc. Natl. Acad. Sci. USA 2006, 103, 690–695. [Google Scholar] [CrossRef] [Green Version]
- Tap, W.D.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.Y.; Alcindor, T.; Ganjoo, K.; Martin-Broto, J.; Ryan, C.W.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Escamilla, J.; Schokrpur, S.; Liu, C.; Priceman, S.J.; Moughon, D.; Jiang, Z.; Pouliot, F.; Magyar, C.; Sung, J.L.; Xu, J.; et al. CSF1 receptor targeting in prostate cancer reverses macrophage-mediated resistance to androgen blockade therapy. Cancer Res. 2015, 75, 950–962. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autio, K.A.; Klebanoff, C.A.; Schaer, D.; Kauh, J.S.W.; Slovin, S.F.; Adamow, M.; Blinder, V.S.; Brahmachary, M.; Carlsen, M.; Comen, E.; et al. Immunomodulatory Activity of a Colony-stimulating Factor-1 Receptor Inhibitor in Patients with Advanced Refractory Breast or Prostate Cancer: A Phase I Study. Clin. Cancer Res. 2020, 26, 5609–5620. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Meeth, K.M.; Stannard, K.; Barkauskas, D.S.; Bollag, G.; Bosenberg, M.; Smyth, M.J. Co-inhibition of colony stimulating factor-1 receptor and BRAF oncogene in mouse models of BRAF(V600E) melanoma. Oncoimmunology 2016, 5, e1089381. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Falchook, G.S.; Peeters, M.; Rottey, S.; Dirix, L.Y.; Obermannova, R.; Cohen, J.E.; Perets, R.; Frommer, R.S.; Bauer, T.M.; Wang, J.S.; et al. A phase 1a/1b trial of CSF-1R inhibitor LY3022855 in combination with durvalumab or tremelimumab in patients with advanced solid tumors. Investig. New Drugs 2021, 39, 1284–1297. [Google Scholar] [CrossRef]
- Han, X.; Shi, H.; Sun, Y.; Shang, C.; Luan, T.; Wang, D.; Ba, X.; Zeng, X. CXCR2 expression on granulocyte and macrophage progenitors under tumor conditions contributes to mo-MDSC generation via SAP18/ERK/STAT3. Cell Death Dis. 2019, 10, 598. [Google Scholar] [CrossRef] [Green Version]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Rotondi, M.; Coperchini, F.; Latrofa, F.; Chiovato, L. Role of Chemokines in Thyroid Cancer Microenvironment: Is CXCL8 the Main Player? Front. Endocrinol. 2018, 9, 314. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Mo, F.; Li, Q.; Han, X.; Shi, H.; Chen, S.; Wei, Y.; Wei, X. Targeting CXCR2 inhibits the progression of lung cancer and promotes therapeutic effect of cisplatin. Mol. Cancer 2021, 20, 62. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, E.; Ko, E.; Ham, M.; Lee, H.M.; Kim, E.S.; Koh, M.; Lim, H.K.; Jung, J.; Park, S.Y.; et al. Tumor-associated macrophages secrete CCL2 and induce the invasive phenotype of human breast epithelial cells through upregulation of ERO1-alpha and MMP-9. Cancer Lett. 2018, 437, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Lubowicka, E.; Przylipiak, A.; Zajkowska, M.; Piskor, B.M.; Malinowski, P.; Fiedorowicz, W.; Lawicki, S. Plasma Chemokine CCL2 and Its Receptor CCR2 Concentrations as Diagnostic Biomarkers for Breast Cancer Patients. Biomed. Res. Int. 2018, 2018, 2124390. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Noack, A.; Makarevic, J.; Oppermann, E.; Waaga-Gasser, A.M.; Gasser, M.; Borgmann, H.; Huesch, T.; Gust, K.M.; Reiter, M.; et al. CCL2 Chemokine as a Potential Biomarker for Prostate Cancer: A Pilot Study. Cancer Res. Treat. 2015, 47, 306–312. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.; Xia, R.; Wei, Y.; Wei, X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. 2021, 54, e13115. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, H.; Liu, H.; Lin, C.; Cao, Y.; Zhang, W.; Shen, Z.; Xu, J. High expression of C-C chemokine receptor 2 associates with poor overall survival in gastric cancer patients after surgical resection. Oncotarget 2016, 7, 23909–23918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, P.; Thiruppathi, M.; Elshabrawy, H.A.; Alharshawi, K.; Kumar, P.; Prabhakar, B.S. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine 2015, 75, 261–271. [Google Scholar] [CrossRef] [Green Version]
- Small, E.J.; Reese, D.M.; Um, B.; Whisenant, S.; Dixon, S.C.; Figg, W.D. Therapy of advanced prostate cancer with granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 1999, 5, 1738–1744. [Google Scholar]
- Hamilton, J.A. GM-CSF in inflammation. J. Exp. Med. 2020, 217, e20190945. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.L.; Wu, C.C.; Shen, K.Y.; Liu, S.J. Activation of GM-CSF and TLR2 signaling synergistically enhances antigen-specific antitumor immunity and modulates the tumor microenvironment. J. Immunother. Cancer 2021, 9, e002758. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Halstead, E.S.; Umstead, T.M.; Davies, M.L.; Kawasawa, Y.I.; Silveyra, P.; Howyrlak, J.; Yang, L.; Guo, W.; Hu, S.; Hewage, E.K.; et al. GM-CSF overexpression after influenza a virus infection prevents mortality and moderates M1-like airway monocyte/macrophage polarization. Respir Res. 2018, 19, 3. [Google Scholar] [CrossRef]
- Yan, W.L.; Shen, K.Y.; Tien, C.Y.; Chen, Y.A.; Liu, S.J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.L.; Hua, Z.C. Evaluation of effectiveness of granulocyte-macrophage colony-stimulating factor therapy to cancer patients after chemotherapy: A meta-analysis. Oncotarget 2018, 9, 28226–28239. [Google Scholar] [CrossRef] [PubMed]
- Quintero, I.B.; Araujo, C.L.; Pulkka, A.E.; Wirkkala, R.S.; Herrala, A.M.; Eskelinen, E.L.; Jokitalo, E.; Hellstrom, P.A.; Tuominen, H.J.; Hirvikoski, P.P.; et al. Prostatic acid phosphatase is not a prostate specific target. Cancer Res. 2007, 67, 6549–6554. [Google Scholar] [CrossRef] [Green Version]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [Green Version]
- Kyriakopoulos, C.E.; Eickhoff, J.C.; Ferrari, A.C.; Schweizer, M.T.; Wargowski, E.; Olson, B.M.; McNeel, D.G. Multicenter Phase I Trial of a DNA Vaccine Encoding the Androgen Receptor Ligand-binding Domain (pTVG-AR, MVI-118) in Patients with Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 5162–5171. [Google Scholar] [CrossRef] [PubMed]
- McNeel, D.G.; Eickhoff, J.C.; Johnson, L.E.; Roth, A.R.; Perk, T.G.; Fong, L.; Antonarakis, E.S.; Wargowski, E.; Jeraj, R.; Liu, G. Phase II Trial of a DNA Vaccine Encoding Prostatic Acid Phosphatase (pTVG-HP [MVI-816]) in Patients With Progressive, Nonmetastatic, Castration-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 3507–3517. [Google Scholar] [CrossRef]
- Olsson, A.; Nakhle, J.; Sundstedt, A.; Plas, P.; Bauchet, A.L.; Pierron, V.; Bruetschy, L.; Deronic, A.; Torngren, M.; Liberg, D.; et al. Tasquinimod triggers an early change in the polarization of tumor associated macrophages in the tumor microenvironment. J. Immunother. Cancer 2015, 3, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, E.; Dalgleish, A.; Damber, J.E.; Smith, M.; Pili, R. Mechanisms of action of tasquinimod on the tumour microenvironment. Cancer Chemother. Pharmacol. 2014, 73, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.R.; Armstrong, A.J. Tasquinimod in the treatment of castrate-resistant prostate cancer—Current status and future prospects. Ther. Adv. Urol. 2016, 8, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Sundstedt, A.; Ciesielski, M.; Miles, K.M.; Celander, M.; Adelaiye, R.; Orillion, A.; Ciamporcero, E.; Ramakrishnan, S.; Ellis, L.; et al. Tasquinimod modulates suppressive myeloid cells and enhances cancer immunotherapies in murine models. Cancer Immunol. Res. 2015, 3, 136–148. [Google Scholar] [CrossRef] [Green Version]
- Biesenbach, G.; Grafinger, P.; Kaiser, W.; Stuby, U.; Zazgornik, J. Metabolic control in labile type I diabetes with conventional insulin therapy, basal bolus insulin therapy using pens and continuous subcutaneous insulin infusion with the pump. Med. Klin. 1988, 83, 398–401. [Google Scholar]
- Armstrong, A.J.; Humeniuk, M.S.; Healy, P.; Szmulewitz, R.; Winters, C.; Kephart, J.; Harrison, M.R.; Martinez, E.; Mundy, K.; Halabi, S.; et al. Phase Ib Trial of Cabazitaxel and Tasquinimod in Men With Heavily Pretreated Metastatic Castration Resistant Prostate Cancer (mCRPC): The CATCH Trial. Prostate 2017, 77, 385–395. [Google Scholar] [CrossRef]
- Fizazi, K.; Ulys, A.; Sengelov, L.; Moe, M.; Ladoire, S.; Thiery-Vuillemin, A.; Flechon, A.; Guida, A.; Bellmunt, J.; Climent, M.A.; et al. A randomized, double-blind, placebo-controlled phase II study of maintenance therapy with tasquinimod in patients with metastatic castration-resistant prostate cancer responsive to or stabilized during first-line docetaxel chemotherapy. Ann. Oncol. 2017, 28, 2741–2746. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Shen, W.; He, P.; Zhou, Z. Effectiveness and tolerability of targeted drugs for the treatment of metastatic castration-resistant prostate cancer: A network meta-analysis of randomized controlled trials. J. Cancer Res. Clin. Oncol. 2018, 144, 1751–1768. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Haggman, M.; Stadler, W.M.; Gingrich, J.R.; Assikis, V.; Polikoff, J.; Damber, J.E.; Belkoff, L.; Nordle, O.; Forsberg, G.; et al. Long-term survival and biomarker correlates of tasquinimod efficacy in a multicenter randomized study of men with minimally symptomatic metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 6891–6901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pili, R.; Haggman, M.; Stadler, W.M.; Gingrich, J.R.; Assikis, V.J.; Bjork, A.; Nordle, O.; Forsberg, G.; Carducci, M.A.; Armstrong, A.J. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J. Clin. Oncol. 2011, 29, 4022–4028. [Google Scholar] [CrossRef]
- Sternberg, C.; Armstrong, A.; Pili, R.; Ng, S.; Huddart, R.; Agarwal, N.; Khvorostenko, D.; Lyulko, O.; Brize, A.; Vogelzang, N.; et al. Randomized, Double-Blind, Placebo-Controlled Phase III Study of Tasquinimod in Men With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2016, 34, 2636–2643. [Google Scholar] [CrossRef] [PubMed]
- Owen, K.A.; Pixley, F.J.; Thomas, K.S.; Vicente-Manzanares, M.; Ray, B.J.; Horwitz, A.F.; Parsons, J.T.; Beggs, H.E.; Stanley, E.R.; Bouton, A.H. Regulation of lamellipodial persistence, adhesion turnover, and motility in macrophages by focal adhesion kinase. J. Cell Biol. 2007, 179, 1275–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Brooks, M.W.; Inan, M.F.; Reinhardt, F.; Weinberg, R.A. The outgrowth of micrometastases is enabled by the formation of filopodium-like protrusions. Cancer Discov. 2012, 2, 706–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gomez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Bueno, R.; Gill, R.R.; Lizotte, P.H.; Sprott, K.; Jackman, D.M.; Barlow, J.; Sharma, S.; Yeap, B.Y.; Chirieac, L.R.; Lebenthal, A.; et al. Effect of FAK inhibitor defactinib on tumor immune changes and tumor reductions in a phase II window of opportunity study in malignant pleural mesothelioma (MPM). J. Clin. Oncol. 2017, 35 (Suppl. S15), 8555. [Google Scholar] [CrossRef]
- Stresing, V.; Daubine, F.; Benzaid, I.; Monkkonen, H.; Clezardin, P. Bisphosphonates in cancer therapy. Cancer Lett. 2007, 257, 16–35. [Google Scholar] [CrossRef]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Van Acker, H.H.; Anguille, S.; Willemen, Y.; Smits, E.L.; Van Tendeloo, V.F. Bisphosphonates for cancer treatment: Mechanisms of action and lessons from clinical trials. Pharmacol. Ther. 2016, 158, 24–40. [Google Scholar] [CrossRef]
- D’Oronzo, S.; Wood, S.; Brown, J.E. The use of bisphosphonates to treat skeletal complications in solid tumours. Bone 2021, 147, 115907. [Google Scholar] [CrossRef]
- Seider, M.J.; Pugh, S.L.; Langer, C.; Wyatt, G.; Demas, W.; Rashtian, A.; Clausen, C.L.; Derdel, J.D.; Cleary, S.F.; Peters, C.A.; et al. Randomized phase III trial to evaluate radiopharmaceuticals and zoledronic acid in the palliation of osteoblastic metastases from lung, breast, and prostate cancer: Report of the NRG Oncology RTOG 0517 trial. Ann. Nucl. Med. 2018, 32, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Halabi, S.; Ryan, C.J.; Hussain, A.; Vogelzang, N.; Stadler, W.; Hauke, R.J.; Monk, J.P.; Saylor, P.; Bhoopalam, N.; et al. Randomized controlled trial of early zoledronic acid in men with castration-sensitive prostate cancer and bone metastases: Results of CALGB 90202 (alliance). J. Clin. Oncol. 2014, 32, 1143–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himelstein, A.L.; Foster, J.C.; Khatcheressian, J.L.; Roberts, J.D.; Seisler, D.K.; Novotny, P.J.; Qin, R.; Go, R.S.; Grubbs, S.S.; O’Connor, T.; et al. Effect of Longer-Interval vs Standard Dosing of Zoledronic Acid on Skeletal Events in Patients With Bone Metastases: A Randomized Clinical Trial. JAMA 2017, 317, 48–58. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Pirrie, S.J.; Pope, A.M.; Barton, D.; Andronis, L.; Goranitis, I.; Collins, S.; Daunton, A.; McLaren, D.; O’Sullivan, J.; et al. Clinical Outcomes and Survival Following Treatment of Metastatic Castrate-Refractory Prostate Cancer With Docetaxel Alone or With Strontium-89, Zoledronic Acid, or Both: The TRAPEZE Randomized Clinical Trial. JAMA Oncol. 2016, 2, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saad, F.; Gleason, D.M.; Murray, R.; Tchekmedyian, S.; Venner, P.; Lacombe, L.; Chin, J.L.; Vinholes, J.J.; Goas, J.A.; Chen, B.; et al. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J. Natl. Cancer Inst. 2002, 94, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Carducci, M.; Smith, M.; Damiao, R.; Brown, J.; Karsh, L.; Milecki, P.; Shore, N.; Rader, M.; Wang, H.; et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: A randomised, double-blind study. Lancet 2011, 377, 813–822. [Google Scholar] [CrossRef] [Green Version]
- Jakob, T.; Tesfamariam, Y.M.; Macherey, S.; Kuhr, K.; Adams, A.; Monsef, I.; Heidenreich, A.; Skoetz, N. Bisphosphonates or RANK-ligand-inhibitors for men with prostate cancer and bone metastases: A network meta-analysis. Cochrane Database Syst. Rev. 2020, 12, CD013020. [Google Scholar]
- Macherey, S.; Monsef, I.; Jahn, F.; Jordan, K.; Yuen, K.K.; Heidenreich, A.; Skoetz, N. Bisphosphonates for advanced prostate cancer. Cochrane Database Syst. Rev. 2017, 12, CD006250. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, W.; Wu, J.; Zhang, X.; Huang, X.; Lin, R.; Zhu, B.; Jiang, H. Effect of bisphosphonates on overall survival in subgroups of patients with prostate cancer. Clin. Exp. Metastasis 2019, 36, 199–209. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [Green Version]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, M.; Buracchi, C.; Pasqualini, F.; Bergomas, F.; Pesce, S.; Sironi, M.; Grizzi, F.; Mantovani, A.; Belgiovine, C.; Allavena, P. Functional TRAIL receptors in monocytes and tumor-associated macrophages: A possible targeting pathway in the tumor microenvironment. Oncotarget 2016, 7, 41662–41676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, B.J.; Herzog, T.J.; Kaye, S.B.; Krasner, C.N.; Vermorken, J.B.; Muggia, F.M.; Pujade-Lauraine, E.; Park, Y.C.; Parekh, T.V.; Poveda, A.M. Trabectedin plus pegylated liposomal doxorubicin (PLD) versus PLD in recurrent ovarian cancer: Overall survival analysis. Eur. J. Cancer 2012, 48, 2361–2368. [Google Scholar] [CrossRef]
- Barone, A.; Chi, D.C.; Theoret, M.R.; Chen, H.; He, K.; Kufrin, D.; Helms, W.S.; Subramaniam, S.; Zhao, H.; Patel, A.; et al. FDA Approval Summary: Trabectedin for Unresectable or Metastatic Liposarcoma or Leiomyosarcoma Following an Anthracycline-Containing Regimen. Clin. Cancer Res. 2017, 23, 7448–7453. [Google Scholar] [CrossRef] [Green Version]
- Michaelson, M.D.; Bellmunt, J.; Hudes, G.R.; Goel, S.; Lee, R.J.; Kantoff, P.W.; Stein, C.A.; Lardelli, P.; Pardos, I.; Kahatt, C.; et al. Multicenter phase II study of trabectedin in patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2012, 23, 1234–1240. [Google Scholar] [CrossRef]
- Brown, E.J.; Frazier, W.A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar] [CrossRef]
- Okazawa, H.; Motegi, S.; Ohyama, N.; Ohnishi, H.; Tomizawa, T.; Kaneko, Y.; Oldenborg, P.A.; Ishikawa, O.; Matozaki, T. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J. Immunol. 2005, 174, 2004–2011. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Shi, L.; Guo, Y.L.; Lv, Z.; Tang, C.; Niu, S.; Tremblay, A.; Venkataramani, M.; Culpepper, C.; Li, L.; et al. Cd47-Sirpalpha interaction and IL-10 constrain inflammation-induced macrophage phagocytosis of healthy self-cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5434–E5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Wang, J.; Kong, X.; Li, E.; Liu, Y.; Du, X.; Kang, Z.; Tang, Y.; Kuang, Y.; Yang, Z.; et al. CD47 Promotes Tumor Invasion and Metastasis in Non-small Cell Lung Cancer. Sci. Rep. 2016, 6, 29719. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Xiong, M.; Hao, W.; Song, Y.; Liu, R.; Yang, Y.; Yuan, X.; Fan, D.; Zhang, Y.; Hao, M.; et al. A Novel Blockade CD47 Antibody With Therapeutic Potential for Cancer. Front. Oncol. 2020, 10, 615534. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wei, H.; Gao, P.; Yu, H.; Wang, K.; Fu, Z.; Ju, B.; Zhao, M.; Dong, S.; Li, Z.; et al. CD47 promotes ovarian cancer progression by inhibiting macrophage phagocytosis. Oncotarget 2017, 8, 39021–39032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Ni, T.; Wang, J.; Liu, Y.; Fan, Q.; Wang, Y.; Huang, T.; Chu, Y.; Sun, X.; Wang, Y. CD47 Blockade Inhibits Tumor Progression through Promoting Phagocytosis of Tumor Cells by M2 Polarized Macrophages in Endometrial Cancer. J. Immunol. Res. 2018, 2018, 6156757. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chung, H.; Banan, B.; Manning, P.T.; Ott, K.C.; Lin, S.; Capoccia, B.J.; Subramanian, V.; Hiebsch, R.R.; Upadhya, G.A.; et al. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015, 360, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Investig. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Tao, H.; Qian, P.; Wang, F.; Yu, H.; Guo, Y. Targeting CD47 Enhances the Efficacy of Anti-PD-1 and CTLA-4 in an Esophageal Squamous Cell Cancer Preclinical Model. Oncol. Res. 2017, 25, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. eLife 2018, 7. [Google Scholar] [CrossRef]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Cheng, H.C.; Wang, J.; Wang, S.W.; Tai, H.C.; Lin, C.W.; Tang, C.H. Prostate cancer-derived CCN3 induces M2 macrophage infiltration and contributes to angiogenesis in prostate cancer microenvironment. Oncotarget 2014, 5, 1595–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senapati, S.; Rachagani, S.; Chaudhary, K.; Johansson, S.L.; Singh, R.K.; Batra, S.K. Overexpression of macrophage inhibitory cytokine-1 induces metastasis of human prostate cancer cells through the FAK-RhoA signaling pathway. Oncogene 2010, 29, 1293–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Nebozhyn, M.; Cristescu, R.; McClanahan, T.K.; Perini, R.; Rubin, E.; Cheng, J.D.; Kaufman, D.R.; Loboda, A. Molecular Profiling of Cohorts of Tumor Samples to Guide Clinical Development of Pembrolizumab as Monotherapy. Clin. Cancer Res. 2019, 25, 1564–1573. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Han, C.; Xie, B.; Hu, X.; Yu, Q.; Shi, L.; Wang, Q.; Li, D.; Wang, J.; Zheng, P.; et al. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 2013, 152, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abram, C.L.; Lowell, C.A. Shp1 function in myeloid cells. J. Leukoc. Biol. 2017, 102, 657–675. [Google Scholar] [CrossRef] [Green Version]
- Barkal, A.A.; Brewer, R.E.; Markovic, M.; Kowarsky, M.; Barkal, S.A.; Zaro, B.W.; Krishnan, V.; Hatakeyama, J.; Dorigo, O.; Barkal, L.J.; et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019, 572, 392–396. [Google Scholar] [CrossRef]


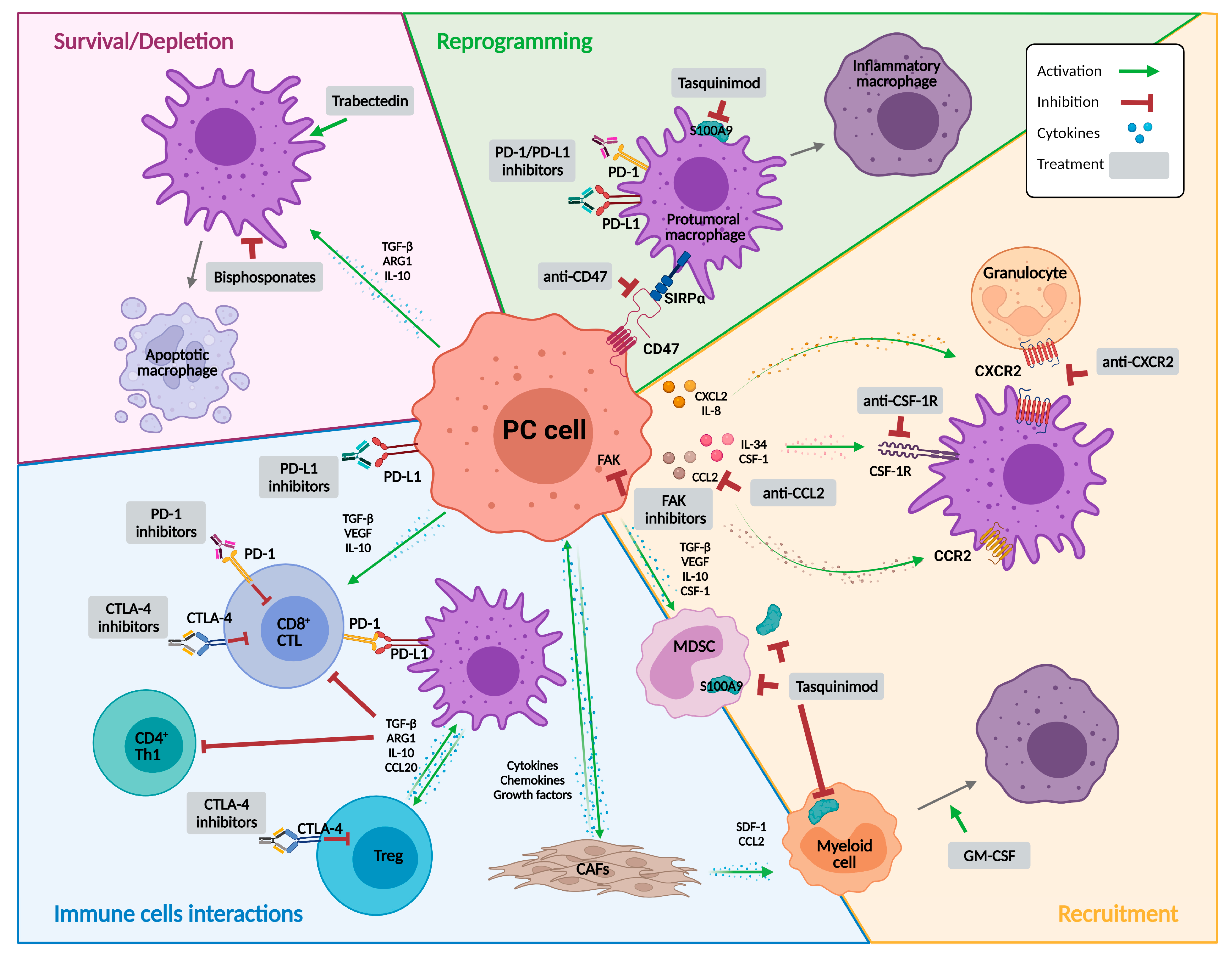{kind=link}
| Intervention 1 | Phase | NCT 2 Number | Indication |
|---|---|---|---|
| CSF-1R inhibitor (JNJ-40346527) | I | NCT03177460 | High-risk localized prostate cancer |
| Enzalutamide + CXCR2 inhibitor (AZD5069) | I/II | NCT03177187 | mCRPC |
| UV1 synthetic peptide vaccine + GM-CSF | I/IIa | NCT01784913 | mPC |
| Carboplatin + GM-CSF Cabazitaxel + GM-CSF | II | NCT04709276 | Metastatic prostate neuroendocrine carcinoma and mPC |
| DNA vaccine pTVG-HP + nivolumab + GM-CSF | II | NCT03600350 | Non-metastatic, non-castrate prostate cancer |
| ProscaVax (PSA/IL-2/GM-CSF) | II | NCT03579654 | Localized prostate cancer |
| Cabazitaxel + prednisone + GM-CSF | III | NCT02961257 | mCRPC previously treated with a docetaxel-containing regimen |
| Sipuleucel-T (APCs loaded with the fusion protein PAP linked to GM-CSF) | III | NCT03686683 | Non-metastatic prostate cancer |
| Enzalutamide and luteinizing hormone-releasing hormone analogue (LHRH-A) + zoledronic acid | II | NCT03336983 | mPC |
| ADT + zoledronic acid ADT + zoledronic acid + docetaxel + prednisolone ADT + zoledronic acid + celecoxib | II/III | NCT00268476 | Hormone-naïve prostate cancer |
| Zoledronic acid | IV | NCT04549207 | Bone metastases from breast cancer and CRPC |
| Pamidronate | |||
| CAR-M (CT-0508) | I | NCT04660929 | HER2 overexpressing solid tumors, including PC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martori, C.; Sanchez-Moral, L.; Paul, T.; Pardo, J.C.; Font, A.; Ruiz de Porras, V.; Sarrias, M.-R. Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance? Cancers 2022, 14, 440. https://doi.org/10.3390/cancers14020440
Martori C, Sanchez-Moral L, Paul T, Pardo JC, Font A, Ruiz de Porras V, Sarrias M-R. Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance? Cancers. 2022; 14(2):440. https://doi.org/10.3390/cancers14020440
Chicago/Turabian StyleMartori, Clara, Lidia Sanchez-Moral, Tony Paul, Juan Carlos Pardo, Albert Font, Vicenç Ruiz de Porras, and Maria-Rosa Sarrias. 2022. "Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance?" Cancers 14, no. 2: 440. https://doi.org/10.3390/cancers14020440
APA StyleMartori, C., Sanchez-Moral, L., Paul, T., Pardo, J. C., Font, A., Ruiz de Porras, V., & Sarrias, M. -R. (2022). Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance? Cancers, 14(2), 440. https://doi.org/10.3390/cancers14020440