
Keep Calm and Carry on with Extra Centrosomes

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Control of Centrosome Biology
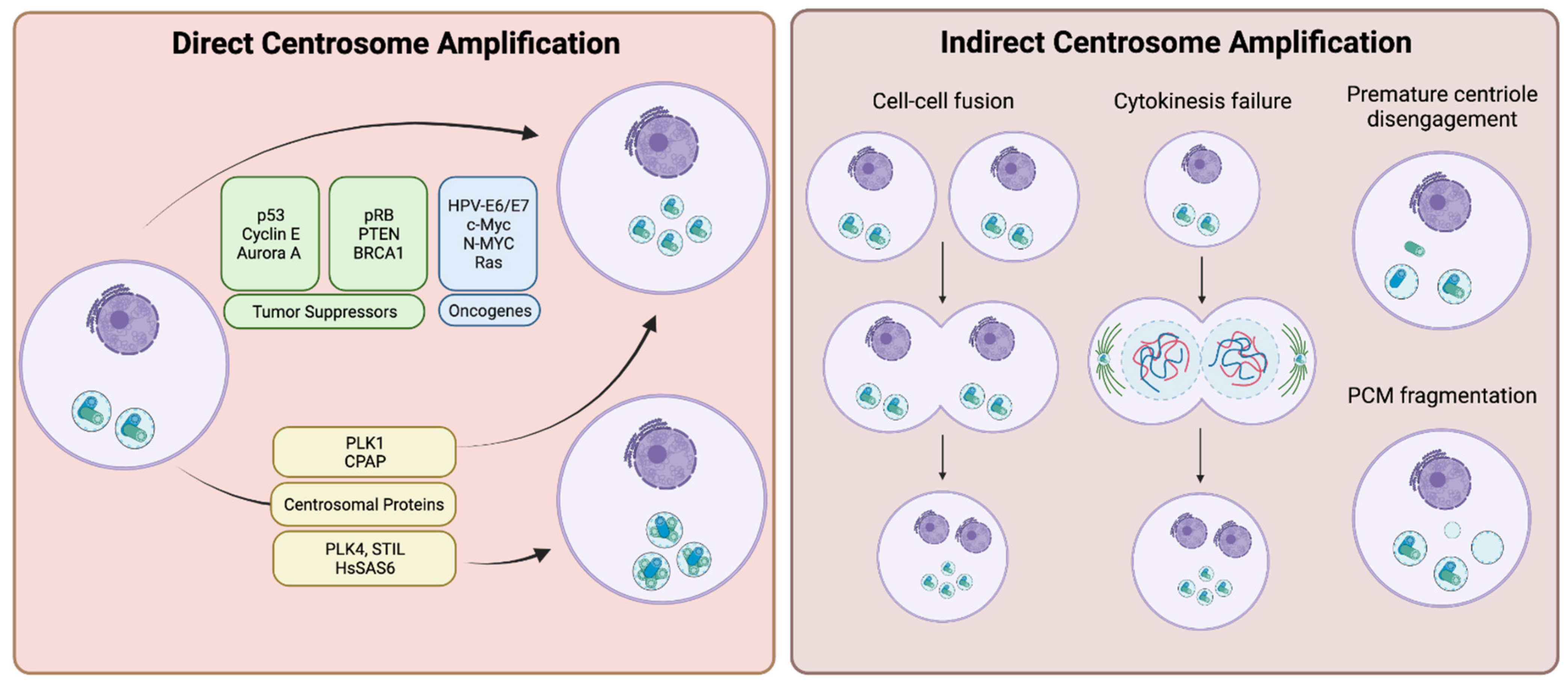
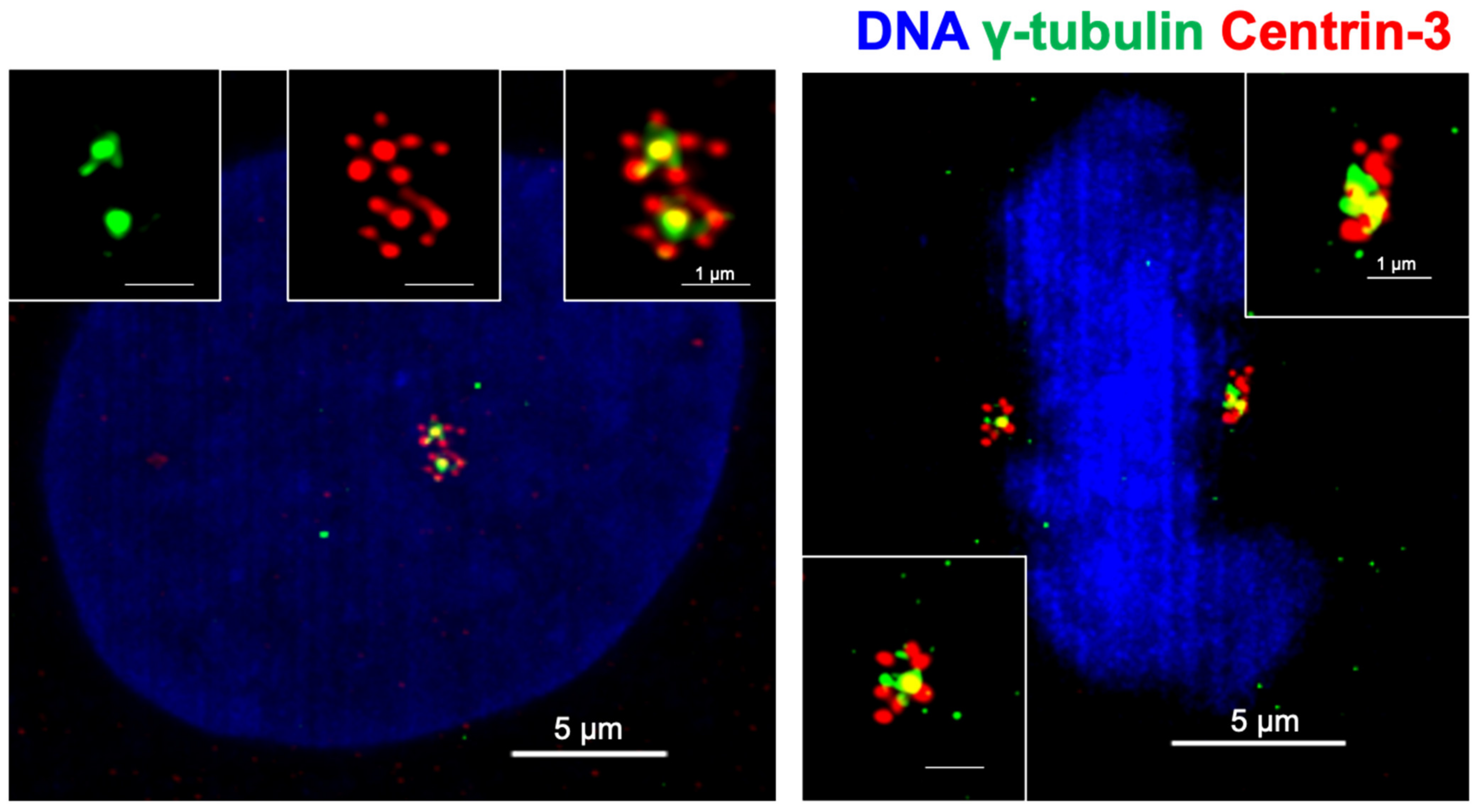
3. Centrosome Amplification in Tumors and the Causes
3.1. Direct Centrosome Amplification
3.1.1. Tumor Suppressors and Proto-Oncogenes
3.1.2. Centrosomal Proteins
3.2. Indirect Centrosome Amplification
3.2.1. Cell Fusion and Failures of Cytokinesis
3.2.2. Premature Centrosome Disengagement and PCM Fragmentation
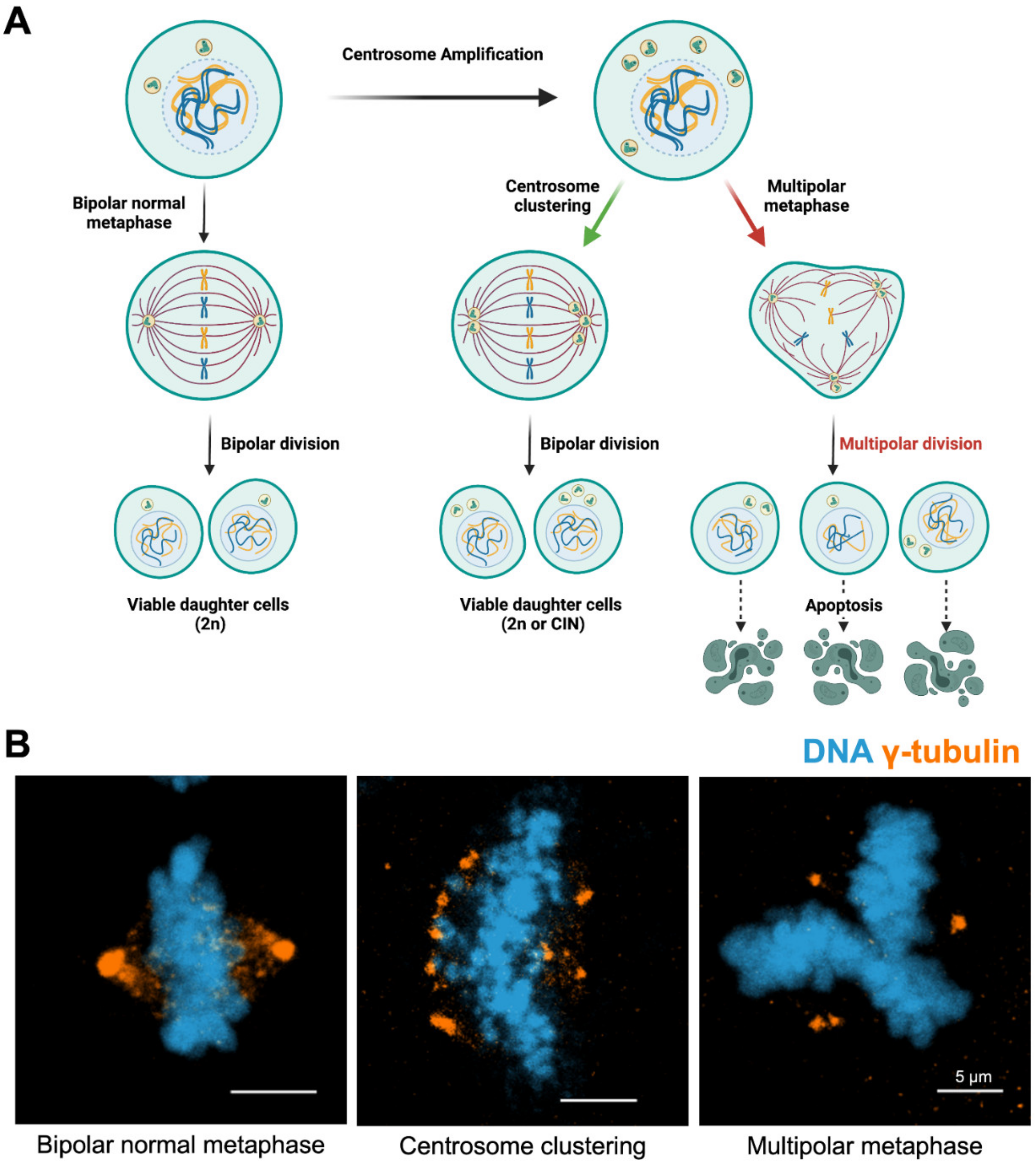
4. Outcomes of Centrosome Amplification
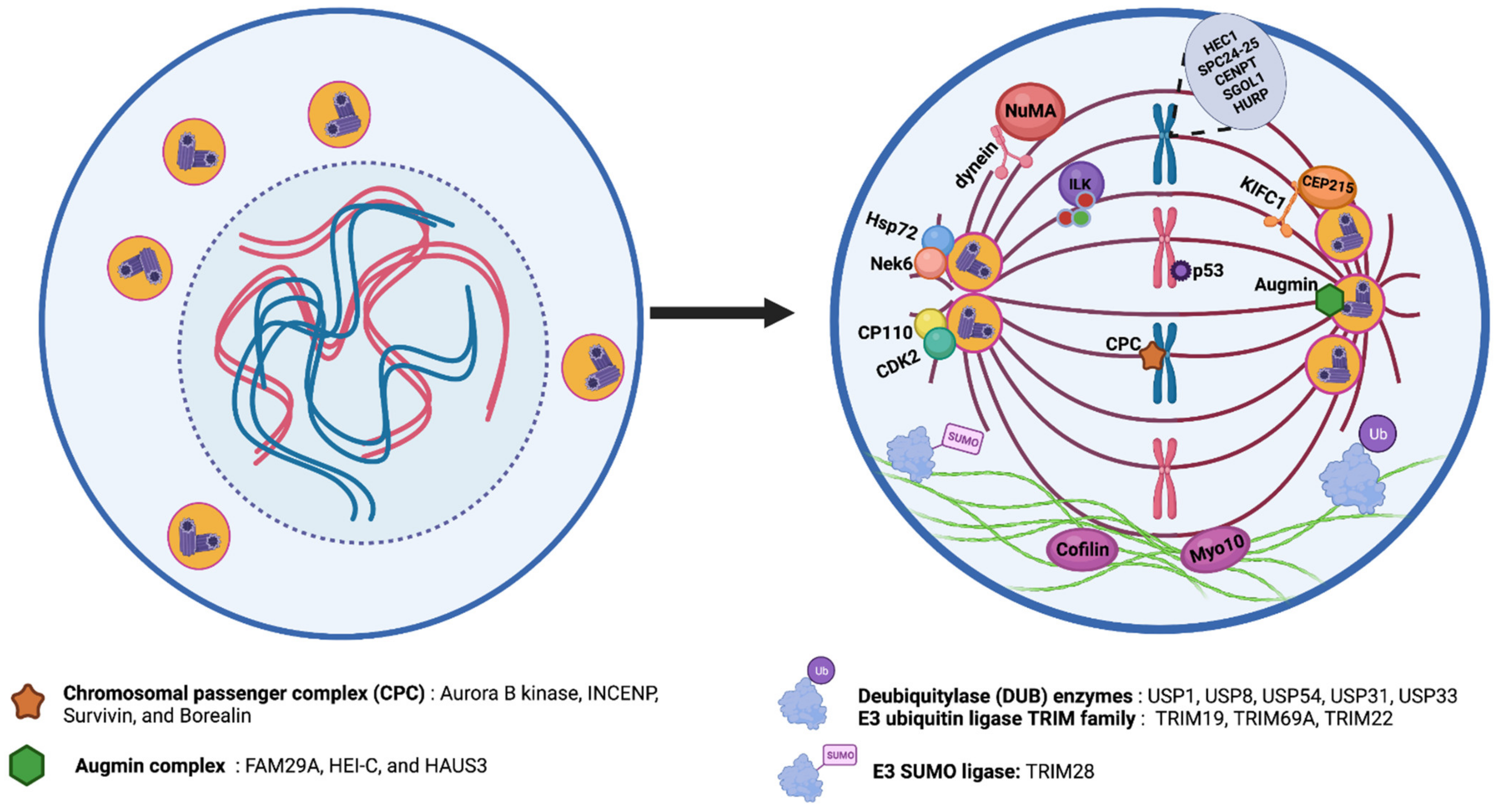
5. Centrosome Clustering and Targeting of Clustering Mechanisms
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bornens, M. The centrosome in cells and organisms. Science 2012, 335, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Godinho, S.A.; Kwon, M.; Pellman, D. Centrosomes and cancer: How cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009, 28, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Dynlacht, B.D. Cilium assembly and disassembly. Nat. Cell Biol. 2016, 18, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balczon, R.; Varden, C.E.; Schroer, T.A. Role for microtubules in centrosome doubling in Chinese hamster ovary cells. Cell Motil. Cytoskelet. 1999, 42, 60–72. [Google Scholar] [CrossRef]
- Hames, R.S.; Crookes, R.E.; Straatman, K.R.; Merdes, A.; Hayes, M.J.; Faragher, A.J.; Fry, A.M. Dynamic recruitment of Nek2 kinase to the centrosome involves microtubules, PCM-1, and localized proteasomal degradation. Mol. Biol. Cell 2005, 16, 1711–1724. [Google Scholar] [CrossRef]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef]
- Qi, F.; Zhou, J. Multifaceted roles of centrosomes in development, health, and disease. J. Mol. Cell Biol. 2021, 13, 611–621. [Google Scholar] [CrossRef]
- Chan, J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144. [Google Scholar] [CrossRef]
- Fukasawa, K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005, 230, 6–19. [Google Scholar] [CrossRef]
- Mahathre, M.M.; Rida, P.C.; Aneja, R. The more the messier: Centrosome amplification as a novel biomarker for personalized treatment of colorectal cancers. J. Biomed. Res. 2016, 30, 441–451. [Google Scholar] [CrossRef]
- Lacey, K.R.; Jackson, P.K.; Stearns, T. Cyclin-dependent kinase control of centrosome duplication. Proc. Natl. Acad. Sci. USA 1999, 96, 2817–2822. [Google Scholar] [CrossRef] [Green Version]
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An integrated view of cyclin E function and regulation. Cell Cycle 2012, 11, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Yam, C.H.; Fung, T.K.; Poon, R.Y. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Hayashi, K.; Nishida, E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 1999, 9, 429–432. [Google Scholar] [CrossRef] [Green Version]
- Okuda, M.; Horn, H.F.; Tarapore, P.; Tokuyama, Y.; Smulian, A.G.; Chan, P.K.; Knudsen, E.S.; Hofmann, I.A.; Snyder, J.D.; Bove, K.E.; et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 2000, 103, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Meraldi, P.; Lukas, J.; Fry, A.M.; Bartek, J.; Nigg, E.A. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat. Cell Biol. 1999, 1, 88–93. [Google Scholar] [CrossRef]
- Blangy, A.; Lane, H.A.; d’Herin, P.; Harper, M.; Kress, M.; Nigg, E.A. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 1995, 83, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- Mardin, B.R.; Schiebel, E. Breaking the ties that bind: New advances in centrosome biology. J. Cell Biol. 2012, 197, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Fry, A.M.; Meraldi, P.; Nigg, E.A. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998, 17, 470–481. [Google Scholar] [CrossRef] [Green Version]
- Mardin, B.R.; Agircan, F.G.; Lange, C.; Schiebel, E. Plk1 controls the Nek2A-PP1gamma antagonism in centrosome disjunction. Curr. Biol. 2011, 21, 1145–1151. [Google Scholar] [CrossRef] [Green Version]
- Habedanck, R.; Stierhof, Y.D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef]
- Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Habedanck, R.; Stierhof, Y.D.; Nigg, E.A. Plk4-induced centriole biogenesis in human cells. Dev. Cell 2007, 13, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Moyer, T.C.; Holland, A.J. PLK4 promotes centriole duplication by phosphorylating STIL to link the procentriole cartwheel to the microtubule wall. eLife 2019, 8, e46054. [Google Scholar] [CrossRef]
- Moyer, T.C.; Clutario, K.M.; Lambrus, B.G.; Daggubati, V.; Holland, A.J. Binding of STIL to Plk4 activates kinase activity to promote centriole assembly. J. Cell Biol. 2015, 209, 863–878. [Google Scholar] [CrossRef] [Green Version]
- Kratz, A.S.; Barenz, F.; Richter, K.T.; Hoffmann, I. Plk4-dependent phosphorylation of STIL is required for centriole duplication. Biol. Open 2015, 4, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Ohta, M.; Ashikawa, T.; Nozaki, Y.; Kozuka-Hata, H.; Goto, H.; Inagaki, M.; Oyama, M.; Kitagawa, D. Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat. Commun. 2014, 5, 5267. [Google Scholar] [CrossRef] [Green Version]
- Ohta, M.; Watanabe, K.; Ashikawa, T.; Nozaki, Y.; Yoshiba, S.; Kimura, A.; Kitagawa, D. Bimodal Binding of STIL to Plk4 Controls Proper Centriole Copy Number. Cell Rep. 2018, 23, 3160–3169.e4. [Google Scholar] [CrossRef]
- Sillibourne, J.E.; Tack, F.; Vloemans, N.; Boeckx, A.; Thambirajah, S.; Bonnet, P.; Ramaekers, F.C.; Bornens, M.; Grand-Perret, T. Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 2010, 21, 547–561. [Google Scholar] [CrossRef] [Green Version]
- Cunha-Ferreira, I.; Bento, I.; Pimenta-Marques, A.; Jana, S.C.; Lince-Faria, M.; Duarte, P.; Borrego-Pinto, J.; Gilberto, S.; Amado, T.; Brito, D.; et al. Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr. Biol. 2013, 23, 2245–2254. [Google Scholar] [CrossRef] [Green Version]
- Guderian, G.; Westendorf, J.; Uldschmid, A.; Nigg, E.A. Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J. Cell Sci. 2010, 123, 2163–2169. [Google Scholar] [CrossRef] [Green Version]
- Arquint, C.; Nigg, E.A. The PLK4-STIL-SAS-6 module at the core of centriole duplication. Biochem. Soc. Trans. 2016, 44, 1253–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vulprecht, J.; David, A.; Tibelius, A.; Castiel, A.; Konotop, G.; Liu, F.; Bestvater, F.; Raab, M.S.; Zentgraf, H.; Izraeli, S.; et al. STIL is required for centriole duplication in human cells. J. Cell Sci. 2012, 125, 1353–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arquint, C.; Sonnen, K.F.; Stierhof, Y.D.; Nigg, E.A. Cell-cycle-regulated expression of STIL controls centriole number in human cells. J. Cell Sci. 2012, 125, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.J.; Marjanovic, M.; Luders, J.; Stracker, T.H.; Costanzo, V. Cep63 and cep152 cooperate to ensure centriole duplication. PLoS ONE 2013, 8, e69986. [Google Scholar] [CrossRef] [Green Version]
- Hatch, E.M.; Kulukian, A.; Holland, A.J.; Cleveland, D.W.; Stearns, T. Cep152 interacts with Plk4 and is required for centriole duplication. J. Cell Biol. 2010, 191, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Park, J.E.; Kim, T.S.; Kim, J.H.; Kwak, M.J.; Ku, B.; Tian, L.; Murugan, R.N.; Ahn, M.; Komiya, S.; et al. Molecular basis for unidirectional scaffold switching of human Plk4 in centriole biogenesis. Nat. Struct. Mol. Biol. 2014, 21, 696–703. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, L.; O’Toole, E.; Schwager, A.; Hyman, A.A.; Muller-Reichert, T. Centriole assembly in Caenorhabditis elegans. Nature 2006, 444, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.I.; Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Lavoie, S.B.; Stierhof, Y.D.; Nigg, E.A. Control of centriole length by CPAP and CP110. Curr. Biol. 2009, 19, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.P.; Sharma, N.K.; Liu, C.; Dong, L.; Li, T.; Swaroop, A. Centrosomal protein CP110 controls maturation of the mother centriole during cilia biogenesis. Development 2016, 143, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.N.; Wu, C.T.; Lin, Y.C.; Hsu, W.B.; Tang, C.J.; Chang, C.W.; Tang, T.K. CEP120 interacts with CPAP and positively regulates centriole elongation. J. Cell Biol. 2013, 202, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Korzeniewski, N.; Cuevas, R.; Duensing, A.; Duensing, S. Daughter centriole elongation is controlled by proteolysis. Mol. Biol. Cell 2010, 21, 3942–3951. [Google Scholar] [CrossRef] [Green Version]
- Boveri, T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. 1), 1–84. [Google Scholar] [CrossRef]
- Fukasawa, K.; Choi, T.; Kuriyama, R.; Rulong, S.; Vande Woude, G.F. Abnormal centrosome amplification in the absence of p53. Science 1996, 271, 1744–1747. [Google Scholar] [CrossRef] [Green Version]
- Tarapore, P.; Fukasawa, K. Loss of p53 and centrosome hyperamplification. Oncogene 2002, 21, 6234–6240. [Google Scholar] [CrossRef] [Green Version]
- Pihan, G.A.; Wallace, J.; Zhou, Y.; Doxsey, S.J. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003, 63, 1398–1404. [Google Scholar]
- Marteil, G.; Guerrero, A.; Vieira, A.F.; de Almeida, B.P.; Machado, P.; Mendonca, S.; Mesquita, M.; Villarreal, B.; Fonseca, I.; Francia, M.E.; et al. Over-elongation of centrioles in cancer promotes centriole amplification and chromosome missegregation. Nat. Commun. 2018, 9, 1258. [Google Scholar] [CrossRef] [Green Version]
- Sabat-Pospiech, D.; Fabian-Kolpanowicz, K.; Prior, I.A.; Coulson, J.M.; Fielding, A.B. Targeting centrosome amplification, an Achilles’ heel of cancer. Biochem. Soc. Trans. 2019, 47, 1209–1222. [Google Scholar] [CrossRef]
- Basto, R.; Brunk, K.; Vinadogrova, T.; Peel, N.; Franz, A.; Khodjakov, A.; Raff, J.W. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008, 133, 1032–1042. [Google Scholar] [CrossRef] [Green Version]
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev. Cell 2017, 40, 313–322.e5. [Google Scholar] [CrossRef] [Green Version]
- Arnandis, T.; Monteiro, P.; Adams, S.D.; Bridgeman, V.L.; Rajeeve, V.; Gadaleta, E.; Marzec, J.; Chelala, C.; Malanchi, I.; Cutillas, P.R.; et al. Oxidative Stress in Cells with Extra Centrosomes Drives Non-Cell-Autonomous Invasion. Dev. Cell 2018, 47, 409–424.e9. [Google Scholar] [CrossRef] [Green Version]
- Blair Zajdel, M.E.; Blair, G.E. The intracellular distribution of the transformation-associated protein p53 in adenovirus-transformed rodent cells. Oncogene 1988, 2, 579–584. [Google Scholar] [PubMed]
- Ciciarello, M.; Mangiacasale, R.; Casenghi, M.; Zaira Limongi, M.; D’Angelo, M.; Soddu, S.; Lavia, P.; Cundari, E. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem. 2001, 276, 19205–19213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinmura, K.; Bennett, R.A.; Tarapore, P.; Fukasawa, K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene 2007, 26, 2939–2944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarapore, P.; Horn, H.F.; Tokuyama, Y.; Fukasawa, K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene 2001, 20, 3173–3184. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Mussman, J.G.; Horn, H.F.; Carroll, P.E.; Okuda, M.; Tarapore, P.; Donehower, L.A.; Fukasawa, K. Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression. Oncogene 2000, 19, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Adon, A.M.; Zeng, X.; Harrison, M.K.; Sannem, S.; Kiyokawa, H.; Kaldis, P.; Saavedra, H.I. Cdk2 and Cdk4 regulate the centrosome cycle and are critical mediators of centrosome amplification in p53-null cells. Mol. Cell. Biol. 2010, 30, 694–710. [Google Scholar] [CrossRef] [Green Version]
- Hollander, M.C.; Sheikh, M.S.; Bulavin, D.V.; Lundgren, K.; Augeri-Henmueller, L.; Shehee, R.; Molinaro, T.A.; Kim, K.E.; Tolosa, E.; Ashwell, J.D.; et al. Genomic instability in Gadd45a-deficient mice. Nat. Genet. 1999, 23, 176–184. [Google Scholar] [CrossRef]
- Kawamura, K.; Izumi, H.; Ma, Z.; Ikeda, R.; Moriyama, M.; Tanaka, T.; Nojima, T.; Levin, L.S.; Fujikawa-Yamamoto, K.; Suzuki, K.; et al. Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res. 2004, 64, 4800–4809. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.L.; Gray, J.W.; Sahin, A.; Brinkley, B.R.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193. [Google Scholar] [CrossRef]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2004, 36, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Fukasawa, K. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer 2007, 7, 911–924. [Google Scholar] [CrossRef]
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J. 2002, 21, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Borel, F.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9819–9824. [Google Scholar] [CrossRef] [Green Version]
- Nigg, E.A. Centrosome aberrations: Cause or consequence of cancer progression? Nat. Rev. Cancer 2002, 2, 815–825. [Google Scholar] [CrossRef]
- Croessmann, S.; Wong, H.Y.; Zabransky, D.J.; Chu, D.; Mendonca, J.; Sharma, A.; Mohseni, M.; Rosen, D.M.; Scharpf, R.B.; Cidado, J.; et al. NDRG1 links p53 with proliferation-mediated centrosome homeostasis and genome stability. Proc. Natl. Acad. Sci. USA 2015, 112, 11583–11588. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.T.; Ongusaha, P.P.; Hong, Y.K.; Kurdistani, S.K.; Nakamura, M.; Lu, K.P.; Lee, S.W. Function of Drg1/Rit42 in p53-dependent mitotic spindle checkpoint. J. Biol. Chem. 2004, 279, 38597–38602. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Duensing, A.; Crum, C.P.; Munger, K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001, 61, 2356–2360. [Google Scholar]
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef] [Green Version]
- Iovino, F.; Lentini, L.; Amato, A.; Di Leonardo, A. RB acute loss induces centrosome amplification and aneuploidy in murine primary fibroblasts. Mol. Cancer 2006, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Weaver, Z.; Linke, S.P.; Li, C.; Gotay, J.; Wang, X.W.; Harris, C.C.; Ried, T.; Deng, C.X. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 1999, 3, 389–395. [Google Scholar] [CrossRef]
- Somasundaram, K.; Zhang, H.; Zeng, Y.X.; Houvras, Y.; Peng, Y.; Zhang, H.; Wu, G.S.; Licht, J.D.; Weber, B.L.; El-Deiry, W.S. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature 1997, 389, 187–190. [Google Scholar] [CrossRef]
- Leonard, M.K.; Hill, N.T.; Bubulya, P.A.; Kadakia, M.P. The PTEN-Akt pathway impacts the integrity and composition of mitotic centrosomes. Cell Cycle 2013, 12, 1406–1415. [Google Scholar] [CrossRef]
- Korzeniewski, N.; Zheng, L.; Cuevas, R.; Parry, J.; Chatterjee, P.; Anderton, B.; Duensing, A.; Munger, K.; Duensing, S. Cullin 1 functions as a centrosomal suppressor of centriole multiplication by regulating polo-like kinase 4 protein levels. Cancer Res. 2009, 69, 6668–6675. [Google Scholar] [CrossRef] [Green Version]
- Preta, G.; de Klark, R.; Gavioli, R.; Glas, R. The Enigma of Tripeptidyl-Peptidase II: Dual Roles in Housekeeping and Stress. J. Oncol. 2010, 2010, 128478. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulou, V.; Xie, J.; Henriksson, M.; Tomkinson, B.; Imreh, S.; Masucci, M.G. Mitotic infidelity and centrosome duplication errors in cells overexpressing tripeptidyl-peptidase II. Cancer Res. 2005, 65, 1361–1368. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Darr, S.; Cuevas, R.; Melquiot, N.; Brickner, A.G.; Duensing, A.; Munger, K. Tripeptidyl Peptidase II Is Required for c-MYC-Induced Centriole Overduplication and a Novel Therapeutic Target in c-MYC-Associated Neoplasms. Genes Cancer 2010, 1, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Slack, A.D.; Chen, Z.; Ludwig, A.D.; Hicks, J.; Shohet, J.M. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res. 2007, 67, 2448–2455. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Shaikh, F.Y.; Harrison, M.K.; Adon, A.M.; Trimboli, A.J.; Carroll, K.A.; Sharma, N.; Timmers, C.; Chodosh, L.A.; Leone, G.; et al. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene 2010, 29, 5103–5112. [Google Scholar] [CrossRef] [Green Version]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef]
- Liao, Z.; Zhang, H.; Fan, P.; Huang, Q.; Dong, K.; Qi, Y.; Song, J.; Chen, L.; Liang, H.; Chen, X.; et al. High PLK4 expression promotes tumor progression and induces epithelialmesenchymal transition by regulating the Wnt/betacatenin signaling pathway in colorectal cancer. Int. J. Oncol. 2019, 54, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhou, D.; Chen, L.; Tian, Y.; Zhong, B.; Cao, Y.; Dong, Q.; Zhou, M.; Yan, J.; Wang, Y.; et al. Polo-like kinase 4 mediates epithelial-mesenchymal transition in neuroblastoma via PI3K/Akt signaling pathway. Cell Death Dis. 2018, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dai, K.; Wang, C.; Song, Y.; Gu, F.; Liu, F.; Fu, L. Expression of Polo-Like Kinase 4(PLK4) in Breast Cancer and Its Response to Taxane-Based Neoadjuvant Chemotherapy. J. Cancer 2016, 7, 1125–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korzeniewski, N.; Hohenfellner, M.; Duensing, S. CAND1 promotes PLK4-mediated centriole overduplication and is frequently disrupted in prostate cancer. Neoplasia 2012, 14, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Shinmura, K.; Kurabe, N.; Goto, M.; Yamada, H.; Natsume, H.; Konno, H.; Sugimura, H. PLK4 overexpression and its effect on centrosome regulation and chromosome stability in human gastric cancer. Mol. Biol. Rep. 2014, 41, 6635–6644. [Google Scholar] [CrossRef]
- Pellegrino, R.; Calvisi, D.F.; Ladu, S.; Ehemann, V.; Staniscia, T.; Evert, M.; Dombrowski, F.; Schirmacher, P.; Longerich, T. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology 2010, 51, 857–868. [Google Scholar] [CrossRef]
- Ward, A.; Sivakumar, G.; Kanjeekal, S.; Hamm, C.; Labute, B.C.; Shum, D.; Hudson, J.W. The deregulated promoter methylation of the Polo-like kinases as a potential biomarker in hematological malignancies. Leuk. Lymphoma 2015, 56, 2123–2133. [Google Scholar] [CrossRef]
- Li, J.; Tan, M.; Li, L.; Pamarthy, D.; Lawrence, T.S.; Sun, Y. SAK, a new polo-like kinase, is transcriptionally repressed by p53 and induces apoptosis upon RNAi silencing. Neoplasia 2005, 7, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130467. [Google Scholar] [CrossRef] [Green Version]
- Korzeniewski, N.; Treat, B.; Duensing, S. The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol. Cancer 2011, 10, 61. [Google Scholar] [CrossRef] [Green Version]
- Sercin, O.; Larsimont, J.C.; Karambelas, A.E.; Marthiens, V.; Moers, V.; Boeckx, B.; Le Mercier, M.; Lambrechts, D.; Basto, R.; Blanpain, C. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat. Cell Biol. 2016, 18, 100–110. [Google Scholar] [CrossRef]
- Coelho, P.A.; Bury, L.; Shahbazi, M.N.; Liakath-Ali, K.; Tate, P.H.; Wormald, S.; Hindley, C.J.; Huch, M.; Archer, J.; Skarnes, W.C.; et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015, 5, 150209. [Google Scholar] [CrossRef] [Green Version]
- Cosenza, M.R.; Cazzola, A.; Rossberg, A.; Schieber, N.L.; Konotop, G.; Bausch, E.; Slynko, A.; Holland-Letz, T.; Raab, M.S.; Dubash, T.; et al. Asymmetric Centriole Numbers at Spindle Poles Cause Chromosome Missegregation in Cancer. Cell Rep. 2017, 20, 1906–1920. [Google Scholar] [CrossRef] [Green Version]
- Dirksen, E.R. Centriole morphogenesis in developing ciliated epithelium of the mouse oviduct. J. Cell Biol. 1971, 51, 286–302. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, R. Centriole assembly in CHO cells expressing Plk4/SAS6/SAS4 is similar to centriogenesis in ciliated epithelial cells. Cell Motil. Cytoskelet. 2009, 66, 588–596. [Google Scholar] [CrossRef]
- Ching, K.; Stearns, T. Centrioles are amplified in cycling progenitors of olfactory sensory neurons. PLoS Biol. 2020, 18, e3000852. [Google Scholar] [CrossRef]
- Holland, A.J.; Cleveland, D.W. Polo-like kinase 4 inhibition: A strategy for cancer therapy? Cancer Cell 2014, 26, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, X. PLK4: A promising target for cancer therapy. J. Cancer Res. Clin. Oncol. 2019, 145, 2413–2422. [Google Scholar] [CrossRef]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef] [Green Version]
- Mason, J.M.; Lin, D.C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176. [Google Scholar] [CrossRef] [Green Version]
- Lohse, I.; Mason, J.; Cao, P.M.; Pintilie, M.; Bray, M.; Hedley, D.W. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget 2017, 8, 3064–3071. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, M.; Mustachio, L.M.; Zheng, L.; Chen, Y.; Rodriguez-Canales, J.; Mino, B.; Kurie, J.M.; Roszik, J.; Villalobos, P.A.; Thu, K.L.; et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 1913–1918. [Google Scholar] [CrossRef] [Green Version]
- Oegema, K.; Davis, R.L.; Lara-Gonzalez, P.; Desai, A.; Shiau, A.K. CFI-400945 is not a selective cellular PLK4 inhibitor. Proc. Natl. Acad. Sci. USA 2018, 115, E10808–E10809. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, M.; Mustachio, L.M.; Zheng, L.; Chen, Y.; Rodriguez-Canales, J.; Mino, B.; Kurie, J.M.; Roszik, J.; Villalobos, P.A.; Thu, K.L.; et al. Reply to Oegema et al.: CFI-400945 and Polo-like kinase 4 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E10810–E10811. [Google Scholar] [CrossRef] [Green Version]
- Meitinger, F.; Ohta, M.; Lee, K.Y.; Watanabe, S.; Davis, R.L.; Anzola, J.V.; Kabeche, R.; Jenkins, D.A.; Shiau, A.K.; Desai, A.; et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 2020, 585, 440–446. [Google Scholar] [CrossRef]
- Macmillan, J.C.; Hudson, J.W.; Bull, S.; Dennis, J.W.; Swallow, C.J. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann. Surg. Oncol. 2001, 8, 729–740. [Google Scholar] [CrossRef]
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.M.; Rubsamen-Waigmann, H.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997, 14, 543–549. [Google Scholar] [CrossRef]
- Tokumitsu, Y.; Mori, M.; Tanaka, S.; Akazawa, K.; Nakano, S.; Niho, Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int. J. Oncol. 1999, 15, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jang, C.; Lee, K.A. Polo-like kinases (plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev. Reprod. 2014, 18, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agircan, F.G.; Schiebel, E. Sensors at centrosomes reveal determinants of local separase activity. PLoS Genet. 2014, 10, e1004672. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Erikson, R.L. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc. Natl. Acad. Sci. USA 2002, 99, 8672–8676. [Google Scholar] [CrossRef] [Green Version]
- de Carcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montanes, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef] [Green Version]
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Matthew, E.M.; Yang, Z.; Peri, S.; Andrake, M.; Dunbrack, R.; Ross, E.; El-Deiry, W.S. Plk2 Loss Commonly Occurs in Colorectal Carcinomas but not Adenomas: Relationship to mTOR Signaling. Neoplasia 2018, 20, 244–255. [Google Scholar] [CrossRef]
- Liu, L.Y.; Wang, W.; Zhao, L.Y.; Guo, B.; Yang, J.; Zhao, X.G.; Song, T.S.; Huang, C.; Xu, J.R. Silencing of polo-like kinase 2 increases cell proliferation and decreases apoptosis in SGC-7901 gastric cancer cells. Mol. Med. Rep. 2015, 11, 3033–3038. [Google Scholar] [CrossRef] [Green Version]
- Ou, B.; Zhao, J.; Guan, S.; Wangpu, X.; Zhu, C.; Zong, Y.; Ma, J.; Sun, J.; Zheng, M.; Feng, H.; et al. Plk2 promotes tumor growth and inhibits apoptosis by targeting Fbxw7/Cyclin E in colorectal cancer. Cancer Lett. 2016, 380, 457–466. [Google Scholar] [CrossRef]
- Yang, Y.; Bai, J.; Shen, R.; Brown, S.A.; Komissarova, E.; Huang, Y.; Jiang, N.; Alberts, G.F.; Costa, M.; Lu, L.; et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res. 2008, 68, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Denkert, C.; Schmidt, M.; Gekeler, V.; Wolf, G.; Kobel, M.; Dietel, M.; Hauptmann, S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br. J. Cancer 2004, 90, 815–821. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561. [Google Scholar] [CrossRef] [Green Version]
- Weichert, W.; Kristiansen, G.; Winzer, K.J.; Schmidt, M.; Gekeler, V.; Noske, A.; Muller, B.M.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase isoforms in breast cancer: Expression patterns and prognostic implications. Virchows Arch. 2005, 446, 442–450. [Google Scholar] [CrossRef]
- Dai, W.; Liu, T.; Wang, Q.; Rao, C.V.; Reddy, B.S. Down-regulation of PLK3 gene expression by types and amount of dietary fat in rat colon tumors. Int. J. Oncol. 2002, 20, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Li, Y.; Ouyang, B.; Pan, H.; Reissmann, P.; Li, J.; Wiest, J.; Stambrook, P.; Gluckman, J.L.; Noffsinger, A.; et al. PRK, a cell cycle gene localized to 8p21, is downregulated in head and neck cancer. Genes Chromosomes Cancer 2000, 27, 332–336. [Google Scholar] [CrossRef]
- Helmke, C.; Becker, S.; Strebhardt, K. The role of Plk3 in oncogenesis. Oncogene 2016, 35, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Strnad, P.; Leidel, S.; Vinogradova, T.; Euteneuer, U.; Khodjakov, A.; Gonczy, P. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev. Cell 2007, 13, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Shinmura, K.; Kato, H.; Kawanishi, Y.; Nagura, K.; Kamo, T.; Okubo, Y.; Inoue, Y.; Kurabe, N.; Du, C.; Iwaizumi, M.; et al. SASS6 overexpression is associated with mitotic chromosomal abnormalities and a poor prognosis in patients with colorectal cancer. Oncol. Rep. 2015, 34, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Kohlmaier, G.; Loncarek, J.; Meng, X.; McEwen, B.F.; Mogensen, M.M.; Spektor, A.; Dynlacht, B.D.; Khodjakov, A.; Gonczy, P. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr. Biol. 2009, 19, 1012–1018. [Google Scholar] [CrossRef] [Green Version]
- Maiato, H.; Logarinho, E. Mitotic spindle multipolarity without centrosome amplification. Nat. Cell Biol. 2014, 16, 386–394. [Google Scholar] [CrossRef]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [Green Version]
- Shekhar, M.P.; Lyakhovich, A.; Visscher, D.W.; Heng, H.; Kondrat, N. Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Res. 2002, 62, 2115–2124. [Google Scholar]
- Lu, X.; Kang, Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc. Natl. Acad. Sci. USA 2009, 106, 9385–9390. [Google Scholar] [CrossRef] [Green Version]
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Prabhu, P.; Juvale, K.; Suares, D.; Yc, M. Cancer cell fusion: A potential target to tackle drug-resistant and metastatic cancer cells. Drug Discov. Today 2019, 24, 1836–1844. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef]
- Millband, D.N.; Campbell, L.; Hardwick, K.G. The awesome power of multiple model systems: Interpreting the complex nature of spindle checkpoint signaling. Trends Cell Biol. 2002, 12, 205–209. [Google Scholar] [CrossRef]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef]
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef]
- Telentschak, S.; Soliwoda, M.; Nohroudi, K.; Addicks, K.; Klinz, F.J. Cytokinesis failure and successful multipolar mitoses drive aneuploidy in glioblastoma cells. Oncol. Rep. 2015, 33, 2001–2008. [Google Scholar] [CrossRef] [Green Version]
- Krzywicka-Racka, A.; Sluder, G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J. Cell Biol. 2011, 194, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z. Genomic instability and cancer: An introduction. J. Mol. Cell Biol. 2011, 3, 1–3. [Google Scholar] [CrossRef]
- McClintock, B. The behavior in successive nuclear divisions of a chromosome broken at meiosis. Proc. Natl. Acad. Sci. USA 1939, 25, 405–416. [Google Scholar] [CrossRef] [Green Version]
- MacKinnon, R.N.; Campbell, L.J. The role of dicentric chromosome formation and secondary centromere deletion in the evolution of myeloid malignancy. Genet. Res. Int. 2011, 2011, 643628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capper, R.; Britt-Compton, B.; Tankimanova, M.; Rowson, J.; Letsolo, B.; Man, S.; Haughton, M.; Baird, D.M. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007, 21, 2495–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acilan, C.; Potter, D.M.; Saunders, W.S. DNA repair pathways involved in anaphase bridge formation. Genes Chromosomes Cancer 2007, 46, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Lv, M.; Tsao, S.W.; Man, C.; Jin, C.; Höglund, M.; Kwong, Y.L.; Jin, Y. Telomere-mediated mitotic disturbances in immortalized ovarian epithelial cells reproduce chromosomal losses and breakpoints from ovarian carcinoma. Genes Chromosomes Cancer 2005, 42, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Jonson, T.; Yu, C.; Martins, C.; Mandahl, N.; Wiegant, J.; Jin, Y.; Mertens, F.; Jin, C. Centrosomal abnormalities, multipolar mitoses, and chromosomal instability in head and neck tumours with dysfunctional telomeres. Br. J. Cancer 2002, 87, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gisselsson, D.; Gorunova, L.; Höglund, M.; Mandahl, N.; Elfving, P. Telomere shortening and mitotic dysfunction generate cytogenetic heterogeneity in a subgroup of renal cell carcinomas. Br. J. Cancer 2004, 91, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Chng, W.J.; Ahmann, G.J.; Henderson, K.; Santana-Davila, R.; Greipp, P.R.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Kumar, S.; Rajkumar, S.V.; et al. Clinical implication of centrosome amplification in plasma cell neoplasm. Blood 2006, 107, 3669–3675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, D.; Feijoo, P.; Bernal, A.; Ercilla, A.; Agell, N.; Genescà, A.; Tusell, L. Centrosome aberrations in human mammary epithelial cells driven by cooperative interactions between p16INK4a deficiency and telomere-dependent genotoxic stress. Oncotarget 2015, 6, 28238–28256. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Sun, Y.; McNutt, M.A.; Sun, Q.; Hou, L.; Liu, H.; Shen, Q.; Ling, Y.; Chi, Y.; Zhang, B. Localization of TEIF in the centrosome and its functional association with centrosome amplification in DNA damage, telomere dysfunction and human cancers. Oncogene 2009, 28, 1549–1560. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zou, Y.; Liu, H.; Wang, H.; Zhang, H.; Hou, W.; Li, X.; Jia, X.; Zhang, J.; Hou, L.; et al. TEIF associated centrosome activity is regulated by EGF/PI3K/Akt signaling. Biochim. Biophys. Acta 2014, 1843, 1851–1864. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhang, B. Expression of TEIF protein in colorectal tumors and its correlation with centrosome abnormality. Chin. J. Cancer 2009, 28, 1277–1282. [Google Scholar] [CrossRef] [Green Version]
- Vlijm, R.; Li, X.; Panic, M.; Ruthnick, D.; Hata, S.; Herrmannsdorfer, F.; Kuner, T.; Heilemann, M.; Engelhardt, J.; Hell, S.W.; et al. STED nanoscopy of the centrosome linker reveals a CEP68-organized, periodic rootletin network anchored to a C-Nap1 ring at centrioles. Proc. Natl. Acad. Sci. USA 2018, 115, E2246–E2253. [Google Scholar] [CrossRef] [Green Version]
- Bahe, S.; Stierhof, Y.D.; Wilkinson, C.J.; Leiss, F.; Nigg, E.A. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 2005, 171, 27–33. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Huang, N.; Bao, Y.; Zhou, H.; Teng, J.; Chen, J. LRRC45 is a centrosome linker component required for centrosome cohesion. Cell Rep. 2013, 4, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- Fang, G.; Zhang, D.; Yin, H.; Zheng, L.; Bi, X.; Yuan, L. Centlein mediates an interaction between C-Nap1 and Cep68 to maintain centrosome cohesion. J. Cell Sci. 2014, 127, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Adamian, M.; Li, T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol. Biol. Cell 2006, 17, 1033–1040. [Google Scholar] [CrossRef]
- Fry, A.M.; Mayor, T.; Meraldi, P.; Stierhof, Y.D.; Tanaka, K.; Nigg, E.A. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J. Cell Biol. 1998, 141, 1563–1574. [Google Scholar] [CrossRef] [Green Version]
- Mayor, T.; Hacker, U.; Stierhof, Y.D.; Nigg, E.A. The mechanism regulating the dissociation of the centrosomal protein C-Nap1 from mitotic spindle poles. J. Cell Sci. 2002, 115, 3275–3284. [Google Scholar] [CrossRef]
- Mayor, T.; Stierhof, Y.D.; Tanaka, K.; Fry, A.M.; Nigg, E.A. The centrosomal protein C-Nap1 is required for cell cycle-regulated centrosome cohesion. J. Cell Biol. 2000, 151, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Hayward, D.G.; Fry, A.M. Nek2 kinase in chromosome instability and cancer. Cancer Lett. 2006, 237, 155–166. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Kaplan, D.D.; Deluca, J.G.; Giddings, T.H., Jr.; O’Toole, E.T.; Winey, M.; Salmon, E.D.; Casey, P.J.; Nelson, W.J.; Barth, A.I. beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008, 22, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Tsou, M.F.; Stearns, T. Controlling centrosome number: Licenses and blocks. Curr. Opin. Cell Biol. 2006, 18, 74–78. [Google Scholar] [CrossRef]
- Karki, M.; Keyhaninejad, N.; Shuster, C.B. Precocious centriole disengagement and centrosome fragmentation induced by mitotic delay. Nat. Commun. 2017, 8, 15803. [Google Scholar] [CrossRef] [Green Version]
- Wirth, K.G.; Wutz, G.; Kudo, N.R.; Desdouets, C.; Zetterberg, A.; Taghybeeglu, S.; Seznec, J.; Ducos, G.M.; Ricci, R.; Firnberg, N.; et al. Separase: A universal trigger for sister chromatid disjunction but not chromosome cycle progression. J. Cell Biol. 2006, 172, 847–860. [Google Scholar] [CrossRef]
- Tsou, M.F.; Stearns, T. Mechanism limiting centrosome duplication to once per cell cycle. Nature 2006, 442, 947–951. [Google Scholar] [CrossRef]
- Thein, K.H.; Kleylein-Sohn, J.; Nigg, E.A.; Gruneberg, U. Astrin is required for the maintenance of sister chromatid cohesion and centrosome integrity. J. Cell Biol. 2007, 178, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Arai, H.; Fujita, N. Centrosomal Aki1 and cohesin function in separase-regulated centriole disengagement. J. Cell Biol. 2009, 187, 607–614. [Google Scholar] [CrossRef]
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393. [Google Scholar] [CrossRef]
- Wilhelm, T.; Olziersky, A.M.; Harry, D.; De Sousa, F.; Vassal, H.; Eskat, A.; Meraldi, P. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 2019, 10, 3585. [Google Scholar] [CrossRef] [Green Version]
- Daum, J.R.; Potapova, T.A.; Sivakumar, S.; Daniel, J.J.; Flynn, J.N.; Rankin, S.; Gorbsky, G.J. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr. Biol. 2011, 21, 1018–1024. [Google Scholar] [CrossRef] [Green Version]
- Brinkley, B.R.; Rao, P.N. Nitrous oxide: Effects on the mitotic apparatus and chromosome movement in HeLa cells. J. Cell Biol. 1973, 58, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Vidair, C.A.; Doxsey, S.J.; Dewey, W.C. Heat shock alters centrosome organization leading to mitotic dysfunction and cell death. J. Cell Physiol. 1993, 154, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Denu, R.A.; Zasadil, L.M.; Kanugh, C.; Laffin, J.; Weaver, B.A.; Burkard, M.E. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016, 16, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asteriti, I.A.; Giubettini, M.; Lavia, P.; Guarguaglini, G. Aurora-A inactivation causes mitotic spindle pole fragmentation by unbalancing microtubule-generated forces. Mol. Cancer 2011, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cizmecioglu, O.; Hoffmann, I. Kizuna takes pole position. Nat. Cell Biol. 2006, 8, 1050–1051. [Google Scholar] [CrossRef]
- Fry, A.M.; Baxter, J.E. Sealed with a Kiz: How Plk1 ensures spindle pole integrity. Dev. Cell 2006, 11, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Oshimori, N.; Ohsugi, M.; Yamamoto, T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat. Cell Biol. 2006, 8, 1095–1101. [Google Scholar] [CrossRef]
- Sapkota, H.; Wren, J.D.; Gorbsky, G.J. CSAG1 maintains the integrity of the mitotic centrosome in cells with defective p53. J. Cell Sci. 2020, 133, jcs239723. [Google Scholar] [CrossRef]
- Leber, B.; Maier, B.; Fuchs, F.; Chi, J.; Riffel, P.; Anderhub, S.; Wagner, L.; Ho, A.D.; Salisbury, J.L.; Boutros, M.; et al. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2010, 2, 33ra38. [Google Scholar] [CrossRef]
- Kubo, A.; Sasaki, H.; Yuba-Kubo, A.; Tsukita, S.; Shiina, N. Centriolar satellites: Molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J. Cell Biol. 1999, 147, 969–980. [Google Scholar] [CrossRef]
- Kubo, A.; Tsukita, S. Non-membranous granular organelle consisting of PCM-1: Subcellular distribution and cell-cycle-dependent assembly/disassembly. J. Cell Sci. 2003, 116, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 2002, 159, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Mimori-Kiyosue, Y.; Grigoriev, I.; Sasaki, H.; Matsui, C.; Akhmanova, A.; Tsukita, S.; Vorobjev, I. Mammalian CLASPs are required for mitotic spindle organization and kinetochore alignment. Genes Cells 2006, 11, 845–857. [Google Scholar] [CrossRef]
- Manning, A.L.; Bakhoum, S.F.; Maffini, S.; Correia-Melo, C.; Maiato, H.; Compton, D.A. CLASP1, astrin and Kif2b form a molecular switch that regulates kinetochore-microtubule dynamics to promote mitotic progression and fidelity. EMBO J. 2010, 29, 3531–3543. [Google Scholar] [CrossRef] [Green Version]
- Logarinho, E.; Maffini, S.; Barisic, M.; Marques, A.; Toso, A.; Meraldi, P.; Maiato, H. CLASPs prevent irreversible multipolarity by ensuring spindle-pole resistance to traction forces during chromosome alignment. Nat. Cell Biol. 2012, 14, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Rhee, K. The pericentriolar satellite protein CEP90 is crucial for integrity of the mitotic spindle pole. J. Cell Sci. 2011, 124, 338–347. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.L.; Compton, D.A. Chromosomes and cancer cells. Chromosome Res. 2011, 19, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of chromosomal instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [Green Version]
- Zyss, D.; Gergely, F. Centrosome function in cancer: Guilty or innocent? Trends Cell Biol. 2009, 19, 334–346. [Google Scholar] [CrossRef]
- Ganem, N.J.; Godinho, S.A.; Pellman, D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009, 460, 278–282. [Google Scholar] [CrossRef] [Green Version]
- D’Assoro, A.B.; Barrett, S.L.; Folk, C.; Negron, V.C.; Boeneman, K.; Busby, R.; Whitehead, C.; Stivala, F.; Lingle, W.L.; Salisbury, J.L. Amplified centrosomes in breast cancer: A potential indicator of tumor aggressiveness. Breast Cancer Res. Treat. 2002, 75, 25–34. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsuyama, H.; Furuya, T.; Oga, A.; Yoshihiro, S.; Okuda, M.; Kawauchi, S.; Sasaki, K.; Naito, K. Centrosome hyperamplification predicts progression and tumor recurrence in bladder cancer. Clin. Cancer Res. 2004, 10, 6449–6455. [Google Scholar] [CrossRef] [Green Version]
- Giehl, M.; Fabarius, A.; Frank, O.; Hochhaus, A.; Hafner, M.; Hehlmann, R.; Seifarth, W. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia 2005, 19, 1192–1197. [Google Scholar] [CrossRef] [Green Version]
- Silkworth, W.T.; Nardi, I.K.; Scholl, L.M.; Cimini, D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS ONE 2009, 4, e6564. [Google Scholar] [CrossRef] [Green Version]
- Cimini, D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim. Biophys. Acta 2008, 1786, 32–40. [Google Scholar] [CrossRef]
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Schvartzman, J.M.; Sotillo, R.; Benezra, R. Mitotic chromosomal instability and cancer: Mouse modelling of the human disease. Nat. Rev. Cancer 2010, 10, 102–115. [Google Scholar] [CrossRef] [Green Version]
- Weaver, B.A.; Silk, A.D.; Montagna, C.; Verdier-Pinard, P.; Cleveland, D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007, 11, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Marthiens, V.; Rujano, M.A.; Pennetier, C.; Tessier, S.; Paul-Gilloteaux, P.; Basto, R. Centrosome amplification causes microcephaly. Nat. Cell Biol. 2013, 15, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Bettencourt-Dias, M.; Glover, D.M. Centrosome biogenesis and function: Centrosomics brings new understanding. Nat. Rev. Mol. Cell Biol. 2007, 8, 451–463. [Google Scholar] [CrossRef]
- Salisbury, J.L.; D’Assoro, A.B.; Lingle, W.L. Centrosome amplification and the origin of chromosomal instability in breast cancer. J. Mammary Gland Biol. Neoplasia 2004, 9, 275–283. [Google Scholar] [CrossRef]
- Tang, N.; Marshall, W.F. Centrosome positioning in vertebrate development. J. Cell Sci. 2012, 125, 4951–4961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Nalbant, P.; Birkenfeld, J.; Chang, Z.F.; Bokoch, G.M. GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA. Mol. Biol. Cell 2008, 19, 2147–2153. [Google Scholar] [CrossRef] [Green Version]
- van Horck, F.P.; Ahmadian, M.R.; Haeusler, L.C.; Moolenaar, W.H.; Kranenburg, O. Characterization of p190RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J. Biol. Chem. 2001, 276, 4948–4956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, E.; Betson, M.; Braga, V.M. Tumor progression: Small GTPases and loss of cell-cell adhesion. BioEssays 2003, 25, 452–463. [Google Scholar] [CrossRef]
- Godinho, S.A.; Picone, R.; Burute, M.; Dagher, R.; Su, Y.; Leung, C.T.; Polyak, K.; Brugge, J.S.; Thery, M.; Pellman, D. Oncogene-like induction of cellular invasion from centrosome amplification. Nature 2014, 510, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Hachet, V.; Canard, C.; Gonczy, P. Centrosomes promote timely mitotic entry in C. elegans embryos. Dev. Cell 2007, 12, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.S.; Wilkinson, C.J.; Mayor, T.; Mortensen, P.; Nigg, E.A.; Mann, M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003, 426, 570–574. [Google Scholar] [CrossRef]
- Mack, G.J.; Ou, Y.; Rattner, J.B. Integrating centrosome structure with protein composition and function in animal cells. Microsc. Res. Tech. 2000, 49, 409–419. [Google Scholar] [CrossRef]
- Jakobsen, L.; Vanselow, K.; Skogs, M.; Toyoda, Y.; Lundberg, E.; Poser, I.; Falkenby, L.G.; Bennetzen, M.; Westendorf, J.; Nigg, E.A.; et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 2011, 30, 1520–1535. [Google Scholar] [CrossRef]
- Fielding, A.B.; Dobreva, I.; McDonald, P.C.; Foster, L.J.; Dedhar, S. Integrin-linked kinase localizes to the centrosome and regulates mitotic spindle organization. J. Cell Biol. 2008, 180, 681–689. [Google Scholar] [CrossRef]
- De Luca, M.; Brunetto, L.; Asteriti, I.A.; Giubettini, M.; Lavia, P.; Guarguaglini, G. Aurora-A and ch-TOG act in a common pathway in control of spindle pole integrity. Oncogene 2008, 27, 6539–6549. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Jenny, A.; Mlodzik, M.; Sokol, S.Y. Centrosomal localization of Diversin and its relevance to Wnt signaling. J. Cell Sci. 2009, 122, 3791–3798. [Google Scholar] [CrossRef] [Green Version]
- Kfoury, Y.; Nasr, R.; Favre-Bonvin, A.; El-Sabban, M.; Renault, N.; Giron, M.L.; Setterblad, N.; Hajj, H.E.; Chiari, E.; Mikati, A.G.; et al. Ubiquitylated Tax targets and binds the IKK signalosome at the centrosome. Oncogene 2008, 27, 1665–1676. [Google Scholar] [CrossRef] [Green Version]
- Holland, A.J.; Fachinetti, D.; Zhu, Q.; Bauer, M.; Verma, I.M.; Nigg, E.A.; Cleveland, D.W. The autoregulated instability of Polo-like kinase 4 limits centrosome duplication to once per cell cycle. Genes Dev. 2012, 26, 2684–2689. [Google Scholar] [CrossRef] [Green Version]
- Brinkley, B.R.; Cox, S.M.; Pepper, D.A.; Wible, L.; Brenner, S.L.; Pardue, R.L. Tubulin assembly sites and the organization of cytoplasmic microtubules in cultured mammalian cells. J. Cell Biol. 1981, 90, 554–562. [Google Scholar] [CrossRef]
- Ring, D.; Hubble, R.; Kirschner, M. Mitosis in a cell with multiple centrioles. J. Cell Biol. 1982, 94, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Marthiens, V.; Piel, M.; Basto, R. Never tear us apart—The importance of centrosome clustering. J. Cell Sci. 2012, 125, 3281–3292. [Google Scholar] [CrossRef] [Green Version]
- Choe, M.H.; Kim, J.; Ahn, J.; Hwang, S.G.; Oh, J.S.; Kim, J.S. Centrosome Clustering Is a Tumor-selective Target for the Improvement of Radiotherapy in Breast Cancer Cells. Anticancer Res. 2018, 38, 3393–3400. [Google Scholar] [CrossRef]
- Milunovic-Jevtic, A.; Mooney, P.; Sulerud, T.; Bisht, J.; Gatlin, J.C. Centrosomal clustering contributes to chromosomal instability and cancer. Curr. Opin. Biotechnol. 2016, 40, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Kramer, A.; Maier, B.; Bartek, J. Centrosome clustering and chromosomal (in)stability: A matter of life and death. Mol. Oncol. 2011, 5, 324–335. [Google Scholar] [CrossRef]
- Quintyne, N.J.; Reing, J.E.; Hoffelder, D.R.; Gollin, S.M.; Saunders, W.S. Spindle multipolarity is prevented by centrosomal clustering. Science 2005, 307, 127–129. [Google Scholar] [CrossRef]
- Kwon, M.; Godinho, S.A.; Chandhok, N.S.; Ganem, N.J.; Azioune, A.; Thery, M.; Pellman, D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008, 22, 2189–2203. [Google Scholar] [CrossRef] [Green Version]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803. [Google Scholar] [CrossRef] [Green Version]
- Uehara, R.; Nozawa, R.S.; Tomioka, A.; Petry, S.; Vale, R.D.; Obuse, C.; Goshima, G. The augmin complex plays a critical role in spindle microtubule generation for mitotic progression and cytokinesis in human cells. Proc. Natl. Acad. Sci. USA 2009, 106, 6998–7003. [Google Scholar] [CrossRef] [Green Version]
- Koffa, M.D.; Casanova, C.M.; Santarella, R.; Kocher, T.; Wilm, M.; Mattaj, I.W. HURP is part of a Ran-dependent complex involved in spindle formation. Curr. Biol. 2006, 16, 743–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breuer, M.; Kolano, A.; Kwon, M.; Li, C.C.; Tsai, T.F.; Pellman, D.; Brunet, S.; Verlhac, M.H. HURP permits MTOC sorting for robust meiotic spindle bipolarity, similar to extra centrosome clustering in cancer cells. J. Cell Biol. 2010, 191, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, Z.Y.; Yang, W.X. Molecular mechanisms of kinesin-14 motors in spindle assembly and chromosome segregation. J. Cell Sci. 2017, 130, 2097–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavali, P.L.; Chandrasekaran, G.; Barr, A.R.; Tatrai, P.; Taylor, C.; Papachristou, E.K.; Woods, C.G.; Chavali, S.; Gergely, F. A CEP215-HSET complex links centrosomes with spindle poles and drives centrosome clustering in cancer. Nat. Commun. 2016, 7, 11005. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.; Weekes, D.; Drosopoulos, K.; Gazinska, P.; Noel, E.; Rashid, M.; Mirza, H.; Quist, J.; Braso-Maristany, F.; Mathew, S.; et al. Integrated genomics and functional validation identifies malignant cell specific dependencies in triple negative breast cancer. Nat. Commun. 2018, 9, 1044. [Google Scholar] [CrossRef]
- Fan, G.; Sun, L.; Meng, L.; Hu, C.; Wang, X.; Shi, Z.; Hu, C.; Han, Y.; Yang, Q.; Cao, L.; et al. The ATM and ATR kinases regulate centrosome clustering and tumor recurrence by targeting KIFC1 phosphorylation. Nat. Commun. 2021, 12, 20. [Google Scholar] [CrossRef]
- Wu, J.; Mikule, K.; Wang, W.; Su, N.; Petteruti, P.; Gharahdaghi, F.; Code, E.; Zhu, X.; Jacques, K.; Lai, Z.; et al. Discovery and mechanistic study of a small molecule inhibitor for motor protein KIFC1. ACS Chem. Biol. 2013, 8, 2201–2208. [Google Scholar] [CrossRef]
- Watts, C.A.; Richards, F.M.; Bender, A.; Bond, P.J.; Korb, O.; Kern, O.; Riddick, M.; Owen, P.; Myers, R.M.; Raff, J.; et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem. Biol. 2013, 20, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhai, L.; Wang, Y.; Boohaker, R.J.; Lu, W.; Gupta, V.V.; Padmalayam, I.; Bostwick, R.J.; White, E.L.; Ross, L.J.; et al. Discovery of a novel inhibitor of kinesin-like protein KIFC1. Biochem. J. 2016, 473, 1027–1035. [Google Scholar] [CrossRef]
- Pannu, V.; Rida, P.C.; Ogden, A.; Turaga, R.C.; Donthamsetty, S.; Bowen, N.J.; Rudd, K.; Gupta, M.V.; Reid, M.D.; Cantuaria, G.; et al. HSET overexpression fuels tumor progression via centrosome clustering-independent mechanisms in breast cancer patients. Oncotarget 2015, 6, 6076–6091. [Google Scholar] [CrossRef] [Green Version]
- Fielding, A.B.; Lim, S.; Montgomery, K.; Dobreva, I.; Dedhar, S. A critical role of integrin-linked kinase, ch-TOG and TACC3 in centrosome clustering in cancer cells. Oncogene 2011, 30, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Fielding, A.B.; Dobreva, I.; Dedhar, S. Beyond focal adhesions: Integrin-linked kinase associates with tubulin and regulates mitotic spindle organization. Cell Cycle 2008, 7, 1899–1906. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Danilov, A.V.; Godek, K.; Orr, B.; Tafe, L.J.; Rodriguez-Canales, J.; Behrens, C.; Mino, B.; Moran, C.A.; Memoli, V.A.; et al. CDK2 Inhibition Causes Anaphase Catastrophe in Lung Cancer through the Centrosomal Protein CP110. Cancer Res. 2015, 75, 2029–2038. [Google Scholar] [CrossRef] [Green Version]
- Sampson, J.; O’Regan, L.; Dyer, M.J.S.; Bayliss, R.; Fry, A.M. Hsp72 and Nek6 Cooperate to Cluster Amplified Centrosomes in Cancer Cells. Cancer Res. 2017, 77, 4785–4796. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, L.; Sampson, J.; Richards, M.W.; Knebel, A.; Roth, D.; Hood, F.E.; Straube, A.; Royle, S.J.; Bayliss, R.; Fry, A.M. Hsp72 is targeted to the mitotic spindle by Nek6 to promote K-fiber assembly and mitotic progression. J. Cell Biol. 2015, 209, 349–358. [Google Scholar] [CrossRef] [Green Version]
- Kwon, M.; Bagonis, M.; Danuser, G.; Pellman, D. Direct Microtubule-Binding by Myosin-10 Orients Centrosomes toward Retraction Fibers and Subcortical Actin Clouds. Dev. Cell 2015, 34, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Kanellos, G.; Frame, M.C. Cellular functions of the ADF/cofilin family at a glance. J. Cell Sci. 2016, 129, 3211–3218. [Google Scholar] [CrossRef] [Green Version]
- Konotop, G.; Bausch, E.; Nagai, T.; Turchinovich, A.; Becker, N.; Benner, A.; Boutros, M.; Mizuno, K.; Kramer, A.; Raab, M.S. Pharmacological Inhibition of Centrosome Clustering by Slingshot-Mediated Cofilin Activation and Actin Cortex Destabilization. Cancer Res. 2016, 76, 6690–6700. [Google Scholar] [CrossRef] [Green Version]
- Rhys, A.D.; Monteiro, P.; Smith, C.; Vaghela, M.; Arnandis, T.; Kato, T.; Leitinger, B.; Sahai, E.; McAinsh, A.; Charras, G.; et al. Loss of E-cadherin provides tolerance to centrosome amplification in epithelial cancer cells. J. Cell Biol. 2018, 217, 195–209. [Google Scholar] [CrossRef]
- Quesada, V.; Diaz-Perales, A.; Gutierrez-Fernandez, A.; Garabaya, C.; Cal, S.; Lopez-Otin, C. Cloning and enzymatic analysis of 22 novel human ubiquitin-specific proteases. Biochem. Biophys. Res. Commun. 2004, 314, 54–62. [Google Scholar] [CrossRef]
- Darling, S.; Fielding, A.B.; Sabat-Pospiech, D.; Prior, I.A.; Coulson, J.M. Regulation of the cell cycle and centrosome biology by deubiquitylases. Biochem. Soc. Trans. 2017, 45, 1125–1136. [Google Scholar] [CrossRef] [Green Version]
- Venuto, S.; Merla, G. E3 Ubiquitin Ligase TRIM Proteins, Cell Cycle and Mitosis. Cells 2019, 8, 510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neo, S.H.; Itahana, Y.; Alagu, J.; Kitagawa, M.; Guo, A.K.; Lee, S.H.; Tang, K.; Itahana, K. TRIM28 Is an E3 Ligase for ARF-Mediated NPM1/B23 SUMOylation That Represses Centrosome Amplification. Mol. Cell Biol. 2015, 35, 2851–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.X.; Zou, W.X.; Lin, P.; Chang, K.S. A role for PML3 in centrosome duplication and genome stability. Mol. Cell 2005, 17, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Sinnott, R.; Winters, L.; Larson, B.; Mytsa, D.; Taus, P.; Cappell, K.M.; Whitehurst, A.W. Mechanisms promoting escape from mitotic stress-induced tumor cell death. Cancer Res. 2014, 74, 3857–3869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersson, J.; Lonnbro, P.; Herr, A.M.; Morgelin, M.; Gullberg, U.; Drott, K. The human IFN-inducible p53 target gene TRIM22 colocalizes with the centrosome independently of cell cycle phase. Exp. Cell Res. 2010, 316, 568–579. [Google Scholar] [CrossRef]
- Rebacz, B.; Larsen, T.O.; Clausen, M.H.; Ronnest, M.H.; Loffler, H.; Ho, A.D.; Kramer, A. Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype-based screen. Cancer Res. 2007, 67, 6342–6350. [Google Scholar] [CrossRef] [Green Version]
- Rathinasamy, K.; Jindal, B.; Asthana, J.; Singh, P.; Balaji, P.V.; Panda, D. Griseofulvin stabilizes microtubule dynamics, activates p53 and inhibits the proliferation of MCF-7 cells synergistically with vinblastine. BMC Cancer 2010, 10, 213. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.S.; Breitkreutz, I.; Anderhub, S.; Ronnest, M.H.; Leber, B.; Larsen, T.O.; Weiz, L.; Konotop, G.; Hayden, P.J.; Podar, K.; et al. GF-15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo. Cancer Res. 2012, 72, 5374–5385. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, E.; Fielding, A.B.; Kannan, N.; Balgi, A.; Eaves, C.J.; Roberge, M.; Dedhar, S. Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget 2013, 4, 1763–1776. [Google Scholar] [CrossRef] [Green Version]
- Yi, Q.; Zhao, X.; Huang, Y.; Ma, T.; Zhang, Y.; Hou, H.; Cooke, H.J.; Yang, D.Q.; Wu, M.; Shi, Q. p53 dependent centrosome clustering prevents multipolar mitosis in tetraploid cells. PLoS ONE 2011, 6, e27304. [Google Scholar] [CrossRef]
- Wang, M.J.; Chen, F.; Lau, J.T.Y.; Hu, Y.P. Hepatocyte polyploidization and its association with pathophysiological processes. Cell Death Dis. 2017, 8, e2805. [Google Scholar] [CrossRef]
- Gentric, G.; Desdouets, C.; Celton-Morizur, S. Hepatocytes polyploidization and cell cycle control in liver physiopathology. Int. J. Hepatol. 2012, 2012, 282430. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalkan, B.M.; Ozcan, S.C.; Quintyne, N.J.; Reed, S.L.; Acilan, C. Keep Calm and Carry on with Extra Centrosomes. Cancers 2022, 14, 442. https://doi.org/10.3390/cancers14020442
Kalkan BM, Ozcan SC, Quintyne NJ, Reed SL, Acilan C. Keep Calm and Carry on with Extra Centrosomes. Cancers. 2022; 14(2):442. https://doi.org/10.3390/cancers14020442
Chicago/Turabian StyleKalkan, Batuhan Mert, Selahattin Can Ozcan, Nicholas J. Quintyne, Samantha L. Reed, and Ceyda Acilan. 2022. "Keep Calm and Carry on with Extra Centrosomes" Cancers 14, no. 2: 442. https://doi.org/10.3390/cancers14020442
APA StyleKalkan, B. M., Ozcan, S. C., Quintyne, N. J., Reed, S. L., & Acilan, C. (2022). Keep Calm and Carry on with Extra Centrosomes. Cancers, 14(2), 442. https://doi.org/10.3390/cancers14020442