
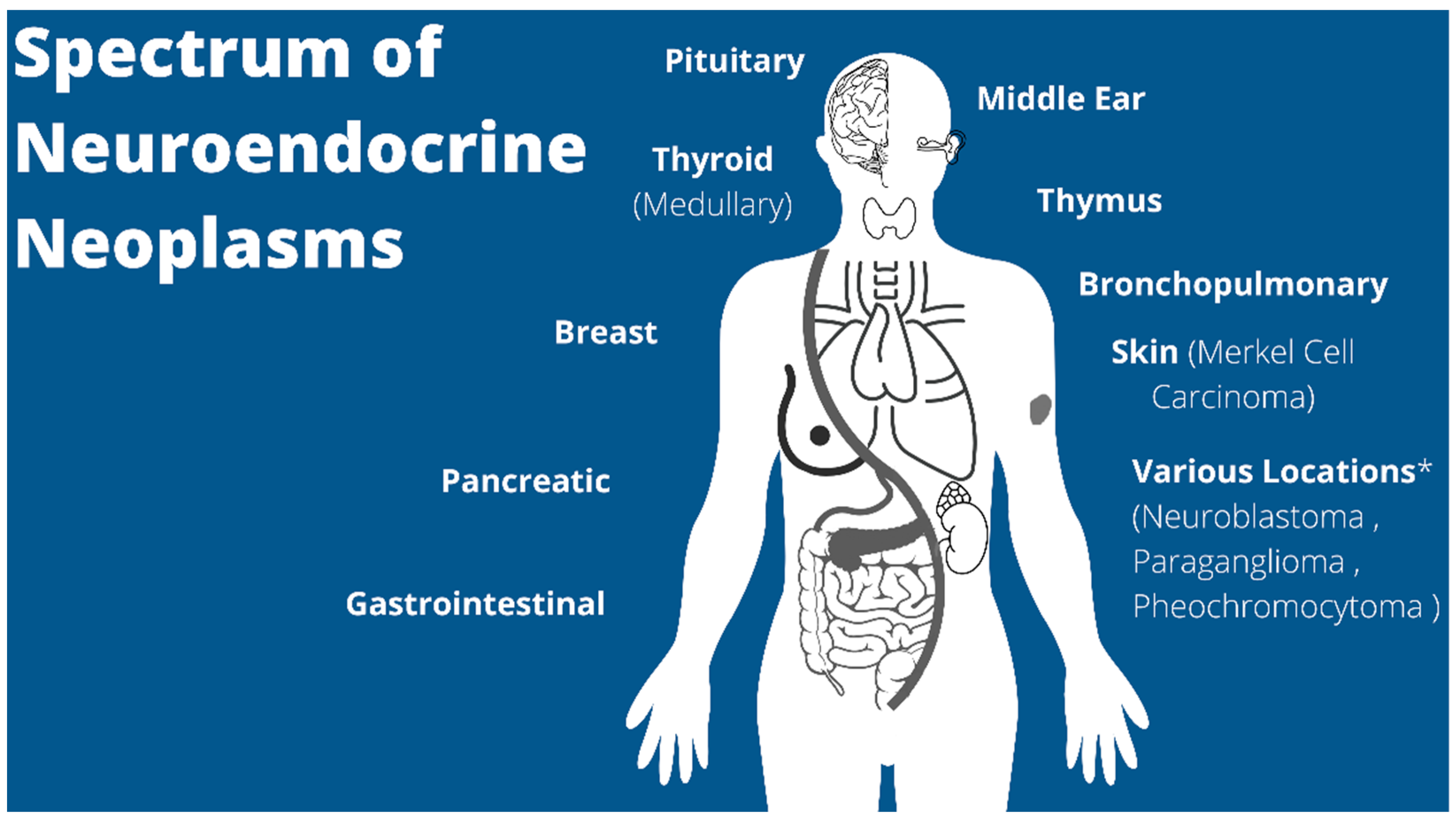
Pediatric Neuroendocrine Neoplasms: Rare Malignancies with Incredible Variability

Abstract
:Simple Summary
Abstract
1. Introduction
2. Epidemiology
3. Diagnosis
3.1. Presentation
3.2. Familial Syndromes
3.3. Biochemical Work Up
3.4. Imaging
3.5. Biopsy
4. How to Treat
4.1. Surgery
4.2. Radiation Therapy
4.3. Medical Management
4.4. Clinical Trials
5. Long-Term Outcomes
6. Challenges and Opportunities
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Man, D.; Wu, J.; Shen, Z.; Zhu, X. Prognosis of patients with neuroendocrine tumor: A SEER database analysis. Cancer Manag. Res. 2018, 10, 5629–5638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.H.; Goldner, W.S.; Benson, A.B.; Bergsland, E.; Blaszkowsky, L.S.; Brock, P.; Chan, J.; Das, S.; Dickson, P.V.; Fanta, P.; et al. Neuroendocrine and Adrenal Tumors, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2021, 19, 839–868. [Google Scholar] [CrossRef]
- Bellizzi, A.M. Immunohistochemistry in the diagnosis and classification of neuroendocrine neoplasms: What can brown do for you? Hum. Pathol. 2020, 96, 8–33. [Google Scholar] [CrossRef]
- Carvão, J.; Dinis-Ribeiro, M.; Pimentel-Nunes, P.; Libânio, D. Neuroendocrine Tumors of the Gastrointestinal Tract: A Focused Review and Practical Approach for Gastroenterologists. GE Port J. Gastroenterol. 2021, 28, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Corbett, V.; Arnold, S.; Anthony, L.; Chauhan, A. Management of Large Cell Neuroendocrine Carcinoma. Front. Oncol. 2021, 11, 653162. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Rindi, G.; Klimistra, D.S.; Abedi-Ardekani, B.; Asa, S.L.; Bosman, F.T.; Brambilla, E.; Busam, K.J.; de Krijger, R.R.; Dietel, M.; El-Naggar, A.K.; et al. A common classification framework for neuroendocrine neoplasms: An International Agency for Research on Cancer (IARC) and World Health Organization (WHO) expert consensus proposal. Mod. Pathol. 2018, 31, 1770–1786. [Google Scholar] [CrossRef] [Green Version]
- Amin, M.B.; Edge, S.B.; Greene, F.L.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Comptom, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual, 8th ed.; Springer: Cham, Switzerland; New York, NY, USA, 2017. [Google Scholar]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Giuseppucci, C.; Reusmann, A.; Giubergia, V.; Barrias, C.; Kruger, A.; Siminovich, M.; Botto, H.; Cadario, M.; Boglione, M.; Strambach, J.; et al. Primary lung tumors in children: 24 years of experience at a referral center. Pediatr. Surg. Int. 2016, 32, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Dishop, M.K.; Kuruvilla, S. Primary and metastatic lung tumors in the pediatric population: A review and 25-year experience at a large children’s hospital. Arch. Pathol. Lab. Med. 2008, 132, 1079–1103. [Google Scholar] [CrossRef]
- Navalkele, P.; O’Dorisio, M.S.; O’Dorisio, T.M.; Zamba, G.K.; Lynch, C.F. Incidence, survival, and prevalence of neuroendocrine tumors versus neuroblastoma in children and young adults: Nine standard SEER registries, 1975–2006. Pediatr. Blood Cancer 2011, 56, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degnan, A.J.; Tocchio, S.; Kurtom, W.; Tadros, S.S. Pediatric neuroendocrine carcinoid tumors: Management, pathology, and imaging findings in a pediatric referral center. Pediatr. Blood Cancer. 2017, 64, e26477. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.I.; Chaya, C. Clinical Presentation and Diagnosis of Neuroendocrine Tumors. Hematol. Oncol. Clin. North Am. 2016, 30, 21–48. [Google Scholar] [CrossRef]
- Khanna, G.; O’Dorisio, S.M.; Menda, Y.; Kirby, P.; Kao, S.; Sato, Y. Gastroenteropancreatic neuroendocrine tumors in children and young adults. Pediatr. Radiol. 2008, 38, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.S.; Kays, D.W.; Larson, S.D.; Islam, S. Appendiceal carcinoids in children--management and outcomes. J. Surg. Res. 2014, 192, 250–253. [Google Scholar] [CrossRef]
- Amr, B.; Froghi, F.; Edmond, M.; Haq, K.; Thengungal-Kochupapy, R. Management and outcomes of appendicular neuroendocrine tumours: Retrospective review with 5-year follow-up. Eur. J. Surg. Oncol. 2015, 41, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Ranaweera, C.; Brar, A.; Somers, G.R.; Sheikh, F.; Pierro, A.; Zani, A. Management of pediatric appendiceal carcinoid: A single institution experience from 5000 appendectomies. Pediatr. Surg. Int. 2019, 35, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Lobeck, I.N.; Jeste, N.; Geller, J.; Pressey, J.; von Allmen, D. Surgical management and surveillance of pediatric appendiceal carcinoid tumor. J. Pediatr. Surg. 2017, 52, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Rojas, Y.; Shi, Y.X.; Zhang, W.; Beierle, E.A.; Doski, J.J.; Goldfarb, M.; Goldin, A.B.; Gow, K.W.; Langer, M.; Vasudevan, S.A.; et al. Primary malignant pulmonary tumors in children: A review of the national cancer data base. J. Pediatr. Surg. 2015, 50, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Madafferi, S.; Catania, V.D.; Accinni, A.; Boldrini, R.; Inserra, A. Endobronchial tumor in children: Unusual finding in recurrent pneumonia, report of three cases. World J. Clin. Pediatr. 2015, 4, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.L.; HaDuong, J.; Okcu, F.; Wu, H.; Chintagumpala, M.; Venkatramani, R. Pediatric Bronchial Carcinoid Tumors: A Case Series and Review of the Literature. J. Pediatr. Hematol. Oncol. 2019, 41, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Lee, L.; Jensen, R.T. Carcinoid-syndrome: Recent advances, current status and controversies. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Ram, P.; Penalver, J.L.; Lo, K.B.U.; Rangaswami, J.; Pressman, G.S. Carcinoid Heart Disease: Review of Current Knowledge. Tex. Heart Inst. J. 2019, 46, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Toumpanakis, C.; Caplin, M.E.; Davar, J. Analysis of 150 patients with carcinoid syndrome seen in a single year at one institution in the first decade of the twenty-first century. Am. J. Cardiol. 2008, 101, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Cogen, J.D.; Swanson, J.; Ong, T. Endobronchial Carcinoid and Concurrent Carcinoid Syndrome in an Adolescent Female. Case Rep. Pediatr. 2016, 2016, 2074970. [Google Scholar] [CrossRef] [Green Version]
- Spunt, S.L.; Pratt, C.B.; Rao, B.N.; Pritchard, M.; Jenkins, J.J.; Hill, D.A.; Cain, A.M.; Pappo, A.S. Childhood carcinoid tumors: The St Jude Children’s Research Hospital experience. J Pediatr. Surg. 2000, 35, 1282–1286. [Google Scholar] [CrossRef]
- Lodish, M.B.; Keil, M.F.; Stratakis, C.A. Cushing’s Syndrome in Pediatrics: An Update. Endocrinol. Metab. Clin. North Am. 2018, 47, 451–462. [Google Scholar] [CrossRef]
- More, J.; Young, J.; Reznik, Y.; Raverot, G.; Borson-Chazot, F.; Rohmer, V.; Baudin, E.; Coutant, R.; Tabarin, A.; Groupe Franҫais des Tumeurs Endocrines (GTE). Ectopic ACTH syndrome in children and adolescents. J. Clin. Endocrinol. Metab. 2011, 96, 1213–1222. [Google Scholar] [CrossRef] [Green Version]
- Saxena, R.; Pathak, M.; Shukla, R.; Sinha, A.; Elhence, P.; Bharti, J.N.; Khera, P. Bronchial Carcinoid Tumour as a Rare Cause of Cushing’s Syndrome in Children: A Case Report and Review of Literature. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 340–346. [Google Scholar] [CrossRef]
- Trouillas, J.; Jaffrain-Rea, M.L.; Vasiljevic, A.; Raverot, G.; Roncaroli, F.; Villa, C. How to Classify the Pituitary Neuroendocrine Tumors (PitNET)s in 2020. Cancers 2020, 12, 514. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2022, 33, 6–26. [Google Scholar] [CrossRef]
- Cholewa, D.; Waldschmidt, J.; Hoffmann, K.; Bäder, M.; Zimmer, T.; Scherübl, H.; Riecken, E.O.; Wiedenmann, B. A 7-year-old child with primary tumour localisation in the distal duodenum--new imaging procedures for an improved diagnosis. Eur. J. Pediatr. 1997, 156, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Jiang, J.; Dai, H.; Hong, X.; Han, X.; Cong, L.; Tong, A.; Li, F.; Luo, Y.; Liu, W.; et al. Robotic enucleation for pediatric insulinoma with MEN1 syndrome: A case report and literature review. BMC Surg. 2018, 18, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padidela, R.; Fiest, M.; Arya, V.; Smith, V.V.; Ashworth, M.; Rampling, D.; Newbould, M.; Batra, G.; James, J.; Wright, N.B.; et al. Insulinoma in childhood: Clinical, radiological, molecular and histological aspects of nine patients. Eur. J. Endocrinol. 2014, 170, 741–747. [Google Scholar] [CrossRef] [Green Version]
- Bonilla Gonzalez, C.; Rusinque, J.; Uribe, C.; Carias, A.; Contreras, M.L. Pancreatic VIPoma as a Differential Diagnosis in Chronic Pediatric Diarrhea: A Case Report and Review of the Literature. J. Med. Cases. 2021, 12, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Garbrecht, N.; Anlauf, M.; Schmitt, A.; Henopp, T.; Sipos, B.; Raffel, A.; Eisenberger, C.F.; Knoefel, W.T.; Pavel, M.; Fottner, C.; et al. Somatostatin-producing neuroendocrine tumors of the duodenum and pancreas: Incidence, types, biological behavior, association with inherited syndromes, and functional activity. Endocr. Relat. Cancer. 2008, 15, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thannberger, P.; Wilhelm, J.M.; Derragui, A.; Saraceni, O.; Kieffer, P. Maladie de Von Recklinghausen associée à un somatostatinome pancréatique [Von Recklinghausen’s disease associated with pancreatic somatostatinoma]. Presse Med. 2001, 30, 1741–1743. [Google Scholar]
- Kindmark, H.; Sundin, A.; Granberg, D.; Dunder, K.; Skogseid, B.; Janson, E.T.; Welin, S.; Oberg, K.; Eriksson, B. Endocrine pancreatic tumors with glucagon hypersecretion: A retrospective study of 23 cases during 20 years. Med. Oncol. 2007, 24, 330–337. [Google Scholar] [CrossRef]
- Luber, A.J.; Ackerman, L.S.; Culpepper, K.S.; Buschmann, C.M.; Koep, L.J. Paediatric necrolytic migratory erythema as a presenting sign of glucagonoma syndrome. Br. J. Dermatol. 2016, 174, 1092–1095. [Google Scholar] [CrossRef]
- Wermers, R.A.; Fatourechi, V.; Wynne, A.G.; Kvols, L.K.; Lloyd, R.V. The glucagonoma syndrome. Clinical and pathologic features in 21 patients. Medicine 1996, 75, 53–63. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic neuroendocrine tumors: Clinical features, diagnosis and medical treatment: Advances. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Bholah, R.; Bunchman, T.E. Review of Pediatric Pheochromocytoma and Paraganglioma. Front Pediatr. 2017, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peard, L.; Cost, N.G.; Saltzman, A.F. Pediatric pheochromocytoma: Current status of diagnostic imaging and treatment procedures. Curr. Opin. Urol. 2019, 29, 493–499. [Google Scholar] [CrossRef]
- Verly, I.R.N.; van Kuilenburg, A.B.P.; Abeling, N.G.G.M.; Goorden, S.M.I.; Fiocco, M.; Vaz, F.M.; van Noesel, M.M.; Zwaan, C.M.; Kaspers, G.L.; Merks, J.H.M.; et al. Catecholamines profiles at diagnosis: Increased diagnostic sensitivity and correlation with biological and clinical features in neuroblastoma patients. Eur. J. Cancer. 2017, 72, 235–243. [Google Scholar] [CrossRef]
- Swift, C.C.; Eklund, M.J.; Kraveka, J.M.; Alazraki, A.L. Updates in Diagnosis, Management, and Treatment of Neuroblastoma. Radiographics 2018, 38, 566–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benya, E. A 13-month-old girl with constipation. Pelvic Neuroblastoma. Pediatr. Ann. 2005, 34, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Marchal, A.L.; Hoeffel, J.C.; Freyd, S.; Schmitt, M.; Olive, D.; Fays, J. Hypertension arterielle par compression extrinseque d’origine tumorale de l’artere renale chez l’enfant [Arterial hypertension caused by extrinsic compression of the renal artery of tumor origin in a child]. Pediatrie 1986, 41, 475–480. [Google Scholar]
- De Martino, L.; Spennato, P.; Vetrella, S.; Capasso, M.; Porfito, C.; Ruotolo, S.; Abate, M.E.; Cinalli, G.; Quaglietta, L. Symptomatic malignant spinal cord compression in children: A single-center experience. Ital. J. Pediatr. 2019, 45, 80. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.; Crook, S.; Horan, J. Suprarenal Neuroblastoma Presenting in a Child With Infantile Scoliosis. Pediatr. Blood Cancer. 2016, 63, 748–749. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, G.J.; Schwab, C.M.; Suarez, E.S.; Carlson, C.L. Neuroblastoma, ganglioneuroblastoma, and ganglioneuroma: Radiologic-pathologic correlation. Radiographics. 2002, 22, 911–934. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers. 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- Köksal, Y.; Toy, H.; Talim, B.; Unal, E.; Akçören, Z.; Cengiz, M. Merkel cell carcinoma in a child. J. Pediatr. Hematol. Oncol. 2009, 31, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Nghiem, P. One in a hundred million: Merkel cell carcinoma in pediatric and young adult patients is rare but more likely to present at advanced stages based on US registry data. J. Am. Acad. Dermatol. 2019, 80, 1758–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandi, M.L.; Agarwal, S.K.; Perrier, N.D.; Lines, K.E.; Valk, G.D.; Thakker, R.V. Multiple Endocrine Neoplasia Type 1: Latest Insights. Endocr. Rev. 2021, 42, 133–170. [Google Scholar] [CrossRef] [PubMed]
- Goudet, P.; Dalac, A.; Le Bras, M.; Cardot-Bauters, C.; Niccoli, P.; Lévy-Bohbot, N.; du Boullay, H.; Bertagna, X.; Ruszniewski, P.; Borson-Chazot, F.; et al. MEN1 disease occurring before 21 years old: A 160-patient cohort study from the Groupe d’etude des Tumeurs Endocrines. J. Clin. Endocrinol. Metab. 2015, 100, 1568–1577. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.M.; Tazelaar, H.D.; Wentzlaff, K.A.; Kosugi, N.S.; Hai, N.; Benson, A.; Miller, D.L.; Yang, P. Familial pulmonary carcinoid tumors. Cancer 2001, 91, 2104–2109. [Google Scholar] [CrossRef]
- Pieterman, C.R.; Valk, G.D. Update on the clinical management of multiple endocrine neoplasia type 1. Clin. Endocrinol. 2022, 97, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Wohllk, N.; Schweizer, H.; Erlic, Z.; Schmid, K.W.; Walz, M.K.; Raue, F.; Neumann, H.P.H. Multiple endocrine neoplasia type 2. Best Pract. Res. Clin. Endocrinol Metab. 2010, 24, 371–387.1. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Launbjerg, K.; Bache, I.; Galanakis, M.; Bisgaard, M.L.; Binderup, M.L.M. von Hippel-Lindau development in children and adolescents. Am. J. Med. Genet. A 2017, 173, 2381–2394. [Google Scholar] [CrossRef]
- Varshney, N.; Kebede, A.A.; Owusu-Dapaah, H.; Lather, J.; Kaushik, M.; Bhullar, J.S. A Review of Von Hippel-Lindau Syndrome. J. Kidney Cancer VHL 2017, 4, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, H.; Sasakura, Y.; Shinozaki, S.; Terauchi, T.; Matsui, J.; Kobayashi, K.; Lefor, A.K.; Ogata, Y. Cystic Pancreatic Neuroendocrine Tumor in a Patient with Neurofibromatosis Type 1. Case Rep. Gastroenterol. 2021, 15, 108–114. [Google Scholar] [CrossRef]
- Gut, P.; Czarnywojtek, A.; Fischbach, J.; Bączyk, M.; Ziemnicka, K.; Wrotkowska, E.; Gryczyńska, M. Ruchala, M. Chromogranin A—Unspecific neuroendocrine marker. Clinical Utility and potential diagnostic pitfalls. Arch. Med. Sci. 2016, 12, 1–9. [Google Scholar] [CrossRef]
- Eriksson, B.; Oberg, K.; Stridsberg, M. Tumor markers in neuroendocrine tumors. Digestion 2000, 62, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Prieur, A.; Kolesar, J.; Arnold, S.; Payen, L.; Mahi, Y.; Vire, B.; Sands, M.; Evers, B.M.; Joubert, D.; et al. hPG80 (Circulating Progastrin), a Novel Blood-Based Biomarker for Detection of Poorly Differentiated Neuroendocrine Carcinoma and Well Differentiated Neuroendocrine Tumors. Cancers 2022, 14, 863. [Google Scholar] [CrossRef] [PubMed]
- Subash, N.; Papali, M.M.; Bahadur, K.P.; Avanthika, C.; Jhaveri, S.; Thannir, S.; Joshi, M.; Valisekka, S.S. Recent Advances in the Diagnosis and Management of Carcinoid Syndrome. Dis. Mon. 2022, 68, 10134. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.E.; Phan, A.T.; Wolin, E.M.; Mirakhur, B.; Liyanage, N.; Lowenthal, S.P.; Fisher, G.A., Jr.; Vinik, A.I.; CLARINET Study Investigators. Effect of Lanreotide Depot/Autogel on Urinary 5-Hydroxyindoleacetic Acid and Plasma Chromogranin A Biomarkers in Nonfunctional Metastatic Enteropancreatic Neuroendocrine Tumors. Oncologist 2019, 24, 463–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berna, M.J.; Hoffmann, K.M.; Serrano, J.; Gibril, F.; Jensen, R.T. Serum gastrin in Zollinger-Ellison syndrome: I. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine 2006, 85, 295–330. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Blau, J.E.; Cochran, C.; Auh, S.; Weinstein, L.S.; Jensen, R.T.; Wank, S. Validity of Secretin Stimulation Testing on Proton Pump Inhibitor Therapy for Diagnosis of Zollinger-Ellison Syndrome. Am. J. Gastroenterol. 2021, 116, 2216–2221. [Google Scholar] [CrossRef] [PubMed]
- Korse, C.M.; Muller, M.; Taal, B.G. Discontinuation of proton pump inhibitors during assessment of chromogranin A levels in patients with neuroendocrine tumours. Br. J. Cancer. 2011, 105, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donegan, D.; Jakubikova, I.; Vella, A. Anthropometric Features Are Not Predictive of 72-Hour Fast Duration in Insulinomas. Endocr. Pract. 2017, 23, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Kanakis, G.; Kaltsas, G. Biochemical markers for gastroenteropancreatic neuroendocrine tumours (GEP-NETs). Best Pract. Res. Clin. Gastroenterol. 2012, 26, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Una Cidon, E. Vasoactive intestinal peptide secreting tumour: An overview. World J. Gastrointest. Oncol. 2022, 14, 808–819. [Google Scholar] [CrossRef]
- Rosenberg, A.M.; Friedmann, P.; Del Rivero, J.; Libutti, S.K.; Laird, A.M. Resection versus expectant management of small incidentally discovered nonfunctional pancreatic neuroendocrine tumors. Surgery 2016, 159, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Bashari, W.A.; Senanayake, R.; Fernández-Pombo, A.; Gillet, D.; Koulouri, O.; Powlson, A.S.; Matys, T.; Scoffings, D.; Cheow, H.; Mendichovszky, I.; et al. Modern imaging of pituitary adenomas. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101278. [Google Scholar] [CrossRef]
- Jacobson, A.F.; Deng, H.; Lombard, J.; Lessig, H.J.; Black, R.R. 123I-meta-iodobenzylguanidine scintigraphy for the detection of neuroblastoma and pheochromocytoma: Results of a meta-analysis. J. Clin. Endocrinol. Metab. 2010, 95, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Haddad, T.; Fard-Esfahani, A.; Vali, R. A review of pediatric neuroendocrine tumors, their detection, and treatment by radioisotopes. Nucl. Med. Commun. 2021, 42, 21–31. [Google Scholar] [CrossRef]
- Shah, S.; Purandare, N.; Agrawal, A.; Rangarajan, V. A pictoral review on somatostatin receptor scintigraphy in neuroendocrine tumors: The role of multimodality imaging with SRS and GLUT receptor imaging with FDG PET-CT. Indian J. Radiol. Imaging 2012, 22, 267–275. [Google Scholar] [CrossRef]
- Hope, T.A.; Bergsland, E.k.; Bozkurt, M.F.; Graham, M.; Heaney, A.P.; Herrman, K.; Howe, J.R.; Kulke, M.H.; Kunz, P.L.; Mailman, J.; et al. Appropriate Use Criteria for Somatostatin Receptor PET Imaging in Neuroendocrine Tumors. J. Nucl. Med. 2018, 59, 66–74. [Google Scholar] [CrossRef]
- Johnbeck, C.B.; Knigge, U.; Loft, A.; Berthelsen, A.K.; Mortensen, J.; Oturai, P.; Langer, S.W.; Elema, D.R.; Kjaer, A. Head-to-Head Comparison of (64)Cu-DOTATATE and (68)Ga-DOTATOC PET/CT: A Prospective Study of 59 Patients with Neuroendocrine Tumors. J. Nucl. Med. 2017, 58, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velikyan, I. Prospective of (68)Ga Radionuclide Contribution to the Development of Imaging Agents for Infection and Inflammation. Contrast Media Mol. Imaging 2018, 9713691. [Google Scholar]
- Kaewput, C.; Vinjamuri, S. Role of Combined 68Ga DOTA-Peptides and 18F FDG PET/CT in the Evaluation of Gastroenteropancreatic Neuroendocrine Neoplasms. Diagnostics 2022, 12, 280. [Google Scholar] [CrossRef]
- Dixon, R.K.; Britt, E.J.; Netzer, G.A.; Afshar, M.; Burke, A.; Liu, S.; Jean, J.; Shah, N.G. Ten-year Single Center Experience of Pulmonary Carcinoid Tumors and Diagnostic Yield of Bronchoscopic Biopsy. Lung 2016, 194, 905–910. [Google Scholar] [CrossRef]
- Yeh, Y.C.; Chou, T.Y. Pulmonary neuroendocrine tumors: Study of 90 cases focusing on clinicopathological characteristics, immunophenotype, preoperative biopsy, and frozen section diagnoses. J. Surg. Oncol. 2014, 109, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M. Gastrointestinal neuroendocrine tumors in 2020. World J. Gastrointest. Oncol. 2020, 12, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Heidsma, C.M.; Tsilimigras, D.I.; Rocha, F.; Abbott, D.E.; Fields, R.; Smith, P.M.; Poultsides, G.A.; Cho, C.; van Eijck, C.; van Dijkum, E.N.; et al. Clinical relevance of performing endoscopic ultrasound-guided fine-needle biopsy for pancreatic neuroendocrine tumors less than 2 cm. J. Surg. Oncol. 2020, 122, 1393–1400. [Google Scholar] [CrossRef]
- Tatsumoto, S.; Kodama, Y.; Sakurai, Y.; Shinohara, T.; Katanuma, A.; Maguchi, H. Pancreatic neuroendocrine neoplasm: Correlation between computed tomography enhancement patterns and prognostic factors of surgical and endoscopic ultrasound-guided fine-needle aspiration biopsy specimens. Abdom. Imaging 2013, 38, 358–366. [Google Scholar] [CrossRef]
- Frilling, A.; Modlin, I.M.; Kidd, M.; Russell, C.; Breitenstein, S.; Salem, R.; Kwekkeboom, D.; Lau, W.; Klersy, C.; Vilgrain, V.; et al. Recommendations for management of patients with neuroendocrine liver metastases. Lancet Oncol. 2014, 15, e8–e21. [Google Scholar] [CrossRef]
- Howe, J.R.; Cardona, K.; Fraker, D.L.; Kebebew, E.; Untch, B.R.; Wang, Y.Z.; Law, C.H.; Liu, E.H.; Kim, M.K.; Menda, Y.; et al. The Surgical Management of Small Bowel Neuroendocrine Tumors: Consensus Guidelines of the North American Neuroendocrine Tumor Society. Pancreas 2017, 46, 715–731. [Google Scholar] [CrossRef]
- NANETS Guidelines 2021 Edition. North American Neuroendocrine Tumor Society. Available online: https://nanets.net/images/guidelines/2021_NANETS_Guidelines_Compendium.pdf (accessed on 14 July 2022).
- Bissonnette, R.T.; Gibney, R.G.; Berry, B.R.; Buckley, A.R. Fatal carcinoid crisis after percutaneous fine-needle biopsy of hepatic metastasis: Case report and literature review. Radiology 1990, 174, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Karmy-Jones, R.; Valliéres, E. Carcinoid crisis after biopsy of a bronchial carcinoid. Ann. Thorac Surg. 1993, 56, 1403–1405. [Google Scholar] [CrossRef]
- Jang, S.; Schmitz, J.J.; Atwell, T.D.; Welch, T.L.; Hobday, T.J.; Adamo, D.A.; Moynagh, M.R. Percutaneous Image-Guided Core Needle Biopsy of Neuroendocrine Tumors: How Common Is Intraprocedural Carcinoid Crisis? J. Vasc. Interv. Radiol. 2021, 32, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Poulet, G.; Massias, J.; Taly, V. Liquid Biopsy: General Concepts. Acta Cytol. 2019, 63, 449–455. [Google Scholar] [CrossRef]
- Modlin, I.M.; Kidd, M.; Malczewska, A.; Drozdov, I.; Bodei, L.; Matar, S.; Chung, K.M. The NETest: The Clinical Utility of Multigene Blood Analysis in the Diagnosis and Management of Neuroendocrine Tumors. Endocrinol. Metab. Clin. North Am. 2018, 47, 485–504. [Google Scholar] [CrossRef]
- Pacak, K.; Kidd, M.; Meuter, L.; Modlin, I.M. A novel liquid biopsy (NETest) identifies paragangliomas and pheochromocytomas with high accuracy. Endocr. Relat. Cancer. 2021, 28, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Filosso, P.L.; Oberg, K.; Malczewska, A.; Lewczuk, A.; Roffinella, M.; Aslanian, H.; Bodei, L. Molecular identification of bronchopulmonary neuroendocrine tumours and neuroendocrine genotype in lung neoplasia using the NETest liquid biopsy. Eur. J. Cardiothorac. Surg. 2020, 57, 1195–1202. [Google Scholar] [CrossRef]
- Malczewska, A.; Witkowska, M.; Makulik, K.; Bocian, A.; Walter, A.; Pilch-Kowalczyk, J.; Zajęcki, W.; Bodei, L.; Oberg, K.E.; Kos-Kudla, B. NETest liquid biopsy is diagnostic of small intestine and pancreatic neuroendocrine tumors and correlates with imaging. Endocr. Connect. 2019, 8, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Bodei, L.; Kidd, M.S.; Singh, A.; van der Zwan, W.A.; Severi, S.; Drozdov, I.A.; Malczewska, A.; Baum, R.P.; Kwekkeboom, D.J.; Paganelli, G.; et al. PRRT neuroendocrine tumor response monitored using circulating transcript analysis: The NETest. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 895–906. [Google Scholar] [CrossRef]
- Partelli, S.; Andreasi, V.; Muffatti, F.; Schiavo Lena, M.; Falconi, M. Circulating Neuroendocrine Gene Transcripts (NETest): A Postoperative Strategy for Early Identification of the Efficacy of Radical Surgery for Pancreatic Neuroendocrine Tumors. Ann. Surg. Oncol. 2020, 27, 3928–3936. [Google Scholar] [CrossRef] [PubMed]
- Nuchtern, J.G.; London, W.B.; Barnewolt, C.E.; Naranjo, A.; McGrady, P.W.; Geiger, J.D.; Diller, L.; Schmidt, M.L.; Maris, J.M.; Cohn, S.L.; et al. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: A Children’s Oncology Group study. Ann. Surg. 2012, 256, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef] [Green Version]
- Sadot, E.; Reidy-Lagunes, D.L.; Tang, L.H.; Do, R.K.G.; Gonen, M.; D’Angelica, M.; DeMatteo, R.P.; Kingham, T.P.; Koerkamp, B.G.; Untch, B.R.; et al. Observation versus Resection for Small Asymptomatic Pancreatic Neuroendocrine Tumors: A Matched Case-Control Study. Ann. Surg. Oncol. 2016, 23, 1361–1370. [Google Scholar] [CrossRef]
- Aziz, H.; Howe, J.R.; Pawlik, T.M. Surgery vs Observation for Patients with Small Pancreatic Neuroendocrine Tumors. JAMA Surg. 2021, 156, 412–413. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J. Management of Appendix Cancer. Clin. Colon. Rectal. Surg. 2015, 28, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Daskalakis, K.; Alexandraki, K.; Kassi, E.; Tsoli, M.; Angelousi, A.; Ragkousi, A.; Kaltsas, G. The risk of lymph node metastases and their impact on survival in patients with appendiceal neuroendocrine neoplasms: A systematic review and meta-analysis of adult and paediatric patients. Endocrine 2020, 67, 20–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, L.; Fehily, C.; Folaranmi, S.; Kelsey, A.; McPartland, J.; Jawaid, W.B.; Craigie, R.; Losty, P.D. Management and outcome of neuroendocrine tumours of the appendix-a two centre UK experience. J. Pediatr. Surg. 2014, 49, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Pawa, N.; Clift, A.K.; Osmani, H.; Drymousis, P.; Cichocki, A.; Flora, R.; Goldin, R.; Patsouras, D.; Baird, A.; Malczewska, A.; et al. Surgical Management of Patients with Neuroendocrine Neoplasms of the Appendix: Appendectomy or More. Neuroendocrinology 2018, 106, 242–251. [Google Scholar] [CrossRef]
- de Lambert, G.; Lardy, H.; Martelli, H.; Orbach, D.; Gauthier, F.; Guérin, F. Surgical Management of Neuroendocrine Tumors of the Appendix in Children and Adolescents: A Retrospective French Multicenter Study of 114 Cases. Pediatr. Blood Cancer 2016, 63, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Fallon, S.C.; Hicks, M.J.; Carpenter, J.L.; Vasudevan, S.A.; Nuchtern, J.G.; Cass, D.L. Management of appendiceal carcinoid tumors in children. J. Surg. Res. 2015, 198, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Malkan, A.D.; Wahid, F.N.; Fernandez-Pineda, I.; Sandoval, J.A. Appendiceal carcinoid tumor in children: Implications for less radical surgery? Clin. Transl. Oncol. 2015, 17, 197–200. [Google Scholar] [CrossRef]
- Zhuge, Y.; Cheung, M.C.; Yang, R.; Eldick, D.; Koniaris, L.G.; Sola, J.E. Pediatric intestinal foregut and small bowel solid tumors: A review of 105 cases. J. Surg. Res. 2009, 156, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Garnier, H.; Loo, C.; Czauderna, P.; Vasudevan, S.A. Pediatric Gastrointestinal Stromal Tumors and Neuroendocrine Tumors: Advances in Surgical Management. Surg. Oncol. Clin. N. Am. 2021, 30, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Suz, P.; Reljic, T.; Are, A.C.; Kumar, A.; Powers, B.; Strosberg, J.; Denbo, J.W.; Fleming, J.B.; Anaya, D.A. Perioperative Carcinoid Crisis: A Systematic Review and Meta-Analysis. Cancers 2022, 14, 2966. [Google Scholar] [CrossRef] [PubMed]
- Seymour, N.; Sawh, S.C. Mega-dose intravenous octreotide for the treatment of carcinoid crisis: A systematic review. Can. J. Anaesth. 2013, 60, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Wonn, S.M.; Ratzlaff, A.N.; Pommier, S.J.; McCully, B.H.; Pommier, R.F. A prospective study of carcinoid crisis with no perioperative octreotide. Surgery 2022, 171, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Mazza, A.; Armigliato, M.; Marzola, M.C.; Schiavon, L.; Montemurro, D.; Vescovo, G.; Zuin, M.; Chondrogiannis, S.; Ravenni, R.; Opocher, G.; et al. Anti-hypertensive treatment in pheochromocytoma and paraganglioma: Current management and therapeutic features. Endocrine 2014, 45, 469–478. [Google Scholar] [CrossRef]
- Reichardt, P.; Apel, T.W.; Domula, M.; Tröbs, R.B.; Krause, I.; Bierbach, U.; Neumann, H.P.H.; Kiess, W. Recurrent polytopic chromaffin paragangliomas in a 9-year-old boy resulting from a novel germline mutation in the von Hippel-Lindau gene. J. Pediatr. Hematol. Oncol. 2002, 24, 145–148. [Google Scholar] [CrossRef]
- Ambarsari, C.G.; Hidayati, E.L.; Tridjaja, B.; Mochtar, C.A.; Wulandari, H.F.; Harahap, A.S.; Grace, A. Silent Hypertensive Crisis in an Adolescent: First Case Report of Pediatric Pheochromocytoma from Indonesia. Glob. Pediatr. Health 2021, 8, 2333794X211015484. [Google Scholar] [CrossRef] [PubMed]
- Raina, R.; Mahajan, Z.; Sharma, A.; Chakraborty, R.; Mahajan, S.; Sethi, S.K.; Kapur, G.; Kaelber, D. Hypertensive Crisis in Pediatric Patients: An Overview. Front. Pediatr. 2020, 8, 588911. [Google Scholar] [CrossRef] [PubMed]
- Havekes, B.; Romijn, J.A.; Eisenhofer, G.; Adams, K.; Pacak, K. Update on pediatric pheochromocytoma. Pediatr. Nephrol. 2009, 24, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Gangi, A.; Howe, J.R. The Landmark Series: Neuroendocrine Tumor Liver Metastases. Ann. Surg. Oncol. 2020, 27, 3270–3280. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, D.I.; Ntanasis-Stathopoulos, I.; Kostakis, I.D.; Moris, D.; Schizas, D.; Cloyd, J.M.; Pawlik, T.M. Is Resection of Primary Midgut Neuroendocrine Tumors in Patients with Unresectable Metastatic Liver Disease Justified? A Systematic Review and Meta-Analysis. J. Gastrointest. Surg. 2019, 23, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Harring, T.R.; Nguyen, N.T.; Goss, J.A.; O’Mahony, C.A. Treatment of liver metastases in patients with neuroendocrine tumors: A comprehensive review. Int. J. Hepatol. 2011, 2011, 154541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kim, J.W.; Han, S.W.; Oh, D.Y.; Lee, S.H.; Kim, D.W.; Im, S.A.; Kim, T.Y.; Seog Heo, D.; Bang, Y.J. Biological characteristics and treatment outcomes of metastatic or recurrent neuroendocrine tumors: Tumor grade and metastatic site are important for treatment strategy. BMC Cancer 2010, 10, 448. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.S.; Lawhn-Heath, C.; Behr, S.; Juarez, R.; Whitman, J.; Paciorek, A.; Nakakura, E.K.; Fidelman, N.; Feng, M.U.S.; Bergsland, E.K.; et al. Outcomes after high-dose radiation in the management of neuroendocrine neoplasms. PLoS ONE 2021, 16, e0252574. [Google Scholar] [CrossRef] [PubMed]
- Fleseriu, M.; Auchus, R.; Bancos, I.; Ben-Shlomo, A.; Bertherat, J.; Biermasz, N.R.; Boguszewski, C.L.; Bronstein, M.D.; Buchfelder, M.; Carmichael, J.D.; et al. Consensus on diagnosis and management of Cushing’s disease: A guideline update. Lancet Diabetes Endocrinol. 2021, 9, 847–875. [Google Scholar] [CrossRef]
- Mehta, G.U.; McCutcheon, I.E. Stereotactic Radiosurgery for Pituitary Carcinoma. Acta Neurochir. Suppl. 2021, 128, 43–49. [Google Scholar]
- van der Lans, R.J.L.; Engel, M.S.D.; Rijken, J.A.; Hensen, E.F.; Bloemena, E.; van der Torn, M.; Leemans, C.R.; Smit, C.F.G.M. Neuroendocrine neoplasms of the middle ear: Unpredictable tumor behavior and tendency for recurrence. Head Neck. 2021, 43, 1848–1853. [Google Scholar]
- Ruffini, E.; Oliaro, A.; Novero, D.; Campisi, P.; Filosso, P.L. Neuroendocrine tumors of the thymus. Thorac. Surg. Clin. 2011, 21, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Thapa, P.; Parghane, R.; Basu, S. (177)Lu-DOTATATE Peptide Receptor Radionuclide Therapy in Metastatic or Advanced and Inoperable Primary Neuroendocrine Tumors of Rare Sites. World J. Nucl. Med. 2017, 16, 223–228. [Google Scholar] [PubMed]
- Chan, D.L.; Thompson, R.; Lam, M.; Pavlakis, N.; Hallet, J.; Law, C.; Singh, S.; Myrehaug, S. External Beam Radiotherapy in the Treatment of Gastroenteropancreatic Neuroendocrine Tumours: A Systematic Review. Clin. Oncol. 2018, 30, 400–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasquillo, J.A.; Chen, C.C.; Jha, A.; Pacak, K.; Pryma, D.A. Lin FI. Systemic Radiopharmaceutical Therapy of Pheochromocytoma and Paraganglioma. J. Nucl. Med. 2021, 62, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Del Rivero, J.; Else, T.; Howe, J.R.; Asa, S.L.; Cohen, D.L.; Dahia, P.L.M.; Fraker, D.L.; Goodman, K.A.; Hope, T.A.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Management of Metastatic and/or Unresectable Pheochromocytoma and Paraganglioma. Pancreas 2021, 50, 469–493. [Google Scholar] [CrossRef]
- Ussowicz, M.; Wieczorek, A.; Dluzniewska, A.; Pieczonka, A.; Dębski, R.; Drabko, K.; Goździk, J.; Balwierz, W.; Handkiewicz-Junak, D.; Wachowiak, J. Factors Modifying Outcome After MIBG Therapy in Children with Neuroblastoma-A National Retrospective Study. Front. Oncol. 2021, 11, 647361. [Google Scholar] [CrossRef]
- Camus, B.; Cottereau, A.S.; Palmieri, L.J.; Dermine, S.; Tenenbaum, F.; Brezault, C.; Coriat, R. Indications of Peptide Receptor Radionuclide Therapy (PRRT) in Gastroenteropancreatic and Pulmonary Neuroendocrine Tumors: An Updated Review. J. Clin. Med. 2021, 10, 1267. [Google Scholar] [CrossRef] [PubMed]
- Nastos, K.; Cheung, V.T.F.; Toumpanakis, C.; Navalkissoor, S.; Quigley, A.M.; Caplin, M.; Khoo, B. Peptide Receptor Radionuclide Treatment and (131)I-MIBG in the management of patients with metastatic/progressive phaeochromocytomas and paragangliomas. J. Surg. Oncol. 2017, 115, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Kong, G.; Hofman, M.S.; Murray, W.K.; Wilson, S.; Wood, P.; Downie, P.; Super, L.; Hogg, A.; Eu, P.; Hicks, R.J. Initial Experience with Gallium-68 DOTA-Octreotate PET/CT and Peptide Receptor Radionuclide Therapy for Pediatric Patients with Refractory Metastatic Neuroblastoma. J. Pediatr. Hematol. Oncol. 2016, 38, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Gains, J.E.; Bomanji, J.B.; Fersht, N.L.; Sullivan, T.; D’Souza, D.; Sullivan, K.P.; Aldridge, M.; Waddington, W.; Gaze, M.N. 177Lu-DOTATATE molecular radiotherapy for childhood neuroblastoma. J. Nucl. Med. 2011, 52, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Fathpour, G.; Jafari, E.; Hashemi, A.; Dadgar, H.; Shahriari, M.; Zareifar, S.; Jenabzade, A.R.; Vali, R.; Ahmadzadehfar, H.; Assadi, M. Feasibility and Therapeutic Potential of Combined Peptide Receptor Radionuclide Therapy with Intensive Chemotherapy for Pediatric Patients With Relapsed or Refractory Metastatic Neuroblastoma. Clin. Nucl. Med. 2021, 46, 540–548. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. (177)Lu-Dotatate plus long-acting octreotide versus highdose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef]
- Bodei, L.; Kidd, M.; Modlin, I.M.; Severi, S.; Drozdov, I.; Nicolini, S.; Kwekkeboom, D.J.; Krenning, E.P.; Baum, R.P.; Paganelli, G. Measurement of circulating transcripts and gene cluster analysis predicts and defines therapeutic efficacy of peptide receptor radionuclide therapy (PRRT) in neuroendocrine tumors. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Dhanani, J.; Pattison, D.A.; Burge, M.; Williams, J.; Riedel, B.; Hicks, R.J.; Reade, M.C. Octreotide for resuscitation of cardiac arrest due to carcinoid crisis precipitated by novel peptide receptor radionuclide therapy (PRRT): A case report. J. Crit. Care. 2020, 60, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Tsushima, Y.; Taketomi-Takahashi, A.; Higuchi, T.; Amanuma, M.; Oriuchi, N.; Endo, K. Hypertensive crisis due to contrast-enhanced computed tomography in a patient with malignant pheochromocytoma. Jpn. J. Radiol. 2011, 29, 449–451. [Google Scholar] [CrossRef]
- Siow Ping, L.; Azraai, B.N.; Subashini, R. Peptide receptor radionuclide therapy induced carcinoid crisis: A case report. Med. J. Malaysia 2022, 77, 128–131. [Google Scholar] [PubMed]
- Tapia Rico, G.; Li, M.; Pavlakis, N.; Cehic, G.; Price, T.J. Prevention and management of carcinoid crises in patients with high-risk neuroendocrine tumours undergoing peptide receptor radionuclide therapy (PRRT): Literature review and case series from two Australian tertiary medical institutions. Cancer Treat. Rev. 2018, 66, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Vainas, I.; Drimonitis, A.; Boudina, M.; Kaprara, A.; Iakovou, I.; Salem, N.; Koussis, C. The therapeutic value of SST-A octreotide alone or with adjuvant treatment in patients with advanced medullary thyroid carcinoma and positive (111)In-octreotide scan. Hell. J. Nucl. Med. 2005, 8, 43–47. [Google Scholar] [PubMed]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Wree, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplin, M.E.; Pavel, M.; Ćwikla, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Müller, H.H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Bläker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef] [PubMed]
- Rinke, A.; Wittenberg, M.; Schade-Brittinger, C.; Aminossadati, B.; Ronicke, E.; Gress, T.M.; Müller, H.H.; Arnold, R.; PROMID Study Group. Placebo-Controlled, Double-Blind, Prospective, Randomized Study on the Effect of Octreotide LAR in the Control of Tumor Growth in Patients with Metastatic Neuroendocrine Midgut Tumors (PROMID): Results of Long-Term Survival. Neuroendocrinology 2017, 104, 26–32. [Google Scholar] [CrossRef]
- Kulke, M.H.; Horsch, D.; Caplin, M.E.; Anthony, L.B.; Bergsland, E.; Öberg, K.; Weliln, S.; Warner, R.R.P.; Lombard-Bohas, C.; Kunz, P.L.; et al. Telotristat Ethyl, a Tryptophan Hydroxylas Inhibitor for the Treatment of Carcinoid Syndrome. J. Clin. Oncol. 2017, 35, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, T.; Heimann, G.; Fink, M. Exposure-response modeling for extrapolation from adult to pediatric patients who differ with respect to prognostic factors: Application to everolimus. CPT Pharmacomet. Syst. Pharmacol. 2021, 10, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Ilanchezhian, M.; Jha, A.; Pacak, K.; Del Rivero, J. Emerging Treatments for Advanced/Metastatic Pheochromocytoma and Paraganglioma. Curr. Treat Options Oncol. 2020, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Koumarianou, A.; Kaltsas, G.; Kulke, M.H.; Oberg, K.; Strosberg, J.R.; Spada, F.; Galdy, S.; Barberis, M.; Fumagalli, C.; Berruti, A.; et al. Temozolomide in Advanced Neuroendocrine Neoplasms: Pharmacological and Clinical Aspects. Neuroendocrinology 2015, 101, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.D.; Noonan, K.L.; Pavlovich, P.; Kennecke, H.F.; Lim, H.J. Outcomes of patients treated with capecitabine and temozolamide for advanced pancreatic neuroendocrine tumors (PNETs) and non-PNETs. J. Gastrointest. Oncol. 2014, 5, 247–252. [Google Scholar] [CrossRef]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Hörsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): A randomised, placebo-controlled, phase 3 study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Lombard-Bohas, C.; Wolin, E.M.; Cutsem, E.V.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.E.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef]
- Lacombe, C.; De Ryke, O.; Couvelard, A.; Turpin, A.; Cazes, A.; Hentic, O.; Gounant, V.; Zalcman, G.; Ruszniewski, P.; Cros, J.; et al. Biomarkers of Response to Etoposide-Platinum Chemotherapy in Patients with Grade 3 Neuroendocrine Neoplasms. Cancers 2021, 13, 643. [Google Scholar] [CrossRef]
- National Cancer Institute-Children’s Oncology Group Pediatric MATCH Trial. Children’s Oncology Group. Available online: https://childrensoncologygroup.org/pediatricmatch (accessed on 14 July 2022).
- Sundquist, F.; Georgantzi, K.; Jarvis, K.B.; Brok, J.; Koskenvuo, M.; Rascon, J.; van Noesel, M.; Grybäck, P.; Nilsson, J.; Braat, A.; et al. A Phase II Trial of a Personalized, Dose-Intense Administration Schedule of (177)Lutetium-DOTATATE in Children With Primary Refractory or Relapsed High-Risk Neuroblastoma-LuDO-N. Front Pediatr. 2022, 10, 836230. [Google Scholar] [CrossRef] [PubMed]
- Advanced Accelerator Applications. A Multicenter Open-Label Study to Evaluate Safety and Dosimetry of Lutathera in Adolescent Patients With Somatostatin Receptor Positive Gastroenteropancreatic Neuroendocrine (GEP-NET) Tumors, Pheochromocytoma and Paragangliomas (PPGL). NCT04711135. Available online: https://clinicaltrials.gov/ct2/show/NCT04711135 (accessed on 20 September 2022).
- Institut Claudius Regaud. PHASE I Clinical Study: Safety Evaluation of Peptide Receptor Radionuclide Therapy (PRRT) With 177Lu-DOTA0-Tyr3-Octreotate for Refractory or Recurrent Metastatic Neuroblastoma Expressing Somatostatin Receptors. NCT03966651. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03966651 (accessed on 20 September 2022).
- Menda, Y.; O’Dorisio, M.S.; Kao, S.; Khanna, G.; Michael, S.; Connolly, M.; Babich, J.; O’Dorisio, T.; Bushnell, D.; Madsen, M. Phase I trial of 90Y-DOTATOC therapy in children and young adults with refractory solid tumors that express somatostatin receptors. J. Nucl. Med. 2010, 51, 1524–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gains, J.E.; Moroz, V.; Aldridge, M.D.; Wan, S.; Wheatley, K.; Laidler, J.; Peet, C.; Bomanji, J.B.; Gaze, M.N. A phase IIa trial of molecular radiotherapy with 177-lutetium DOTATATE in children with primary refractory or relapsed high-risk neuroblastoma. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 2348–2357. [Google Scholar] [CrossRef]
- Erdmann, F.; Frederiksen, L.E.; Bonaventure, A.; Mader, L.; Hasle, H.; Robison, L.L.; Winther, J.F. Childhood cancer: Survival, treatment modalities, late effects and improvements over time. Cancer Epidemiol. 2021, 71, 101733. [Google Scholar] [CrossRef] [PubMed]
- Panek, M.; Szymczak, M.; Stepaniuk, M.; Górecki, W.; Gawłowska-Marciniak, A.; Wolak, P.; Zbyrad, D.; Rybkiewicz, M.; Chrobak, K.; Noparlik, R.; et al. Radical surgical treatment of neuroendocrine tumors of the appendix in children—A Polish multicenter study. Arch. Med. Sci. 2021, 17, 1128–1131. [Google Scholar] [CrossRef]
- Ramsey, M.J.; Nadol, J.B., Jr.; Pilch, B.Z.; McKenna, M.J. Carcinoid tumor of the middle ear: Clinical features, recurrences, and metastases. Laryngoscope 2005, 115, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.K.; Landreneau, R.J.; Hazelrigg, S.R.; Altorki, N.K.; Naunheim, K.S.; Zwischenberger, J.B.; Kent, M.; Yim, A.P. Long-term outcome after resection for bronchial carcinoid tumors. Eur. J. Cardiothorac. Surg. 2000, 18, 156–161. [Google Scholar] [CrossRef] [Green Version]
- American Thyroid Association Guidelines Task Force; Kloos, R.T.; Eng, C.; Evans, D.B.; Francis, G.L.; Gagel, R.F.; Gharib, H.; Moley, J.F.; Pacini, F.; Ringel, M.D.; et al. American Thyroid Association Guidelines—Medullary thyroid cancer: Management guidelines of the American Thyroid Association. Thyroid 2009, 19, 565–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidi, O.; Young, W.F., Jr.; Iniguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Bancos, I. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J. Clin. Endocrinol. Metab. 2017, 102, 3296–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robison, L.L.; Armstrong, G.T.; Boice, J.D.; Chow, E.J.; Davies, S.M.; Donaldson, S.S.; Green, D.M.; Hammond, S.; Meadows, A.T.; Mertens, A.C.; et al. The Childhood Cancer Survivor Study: A National Cancer Institute-supported resource for outcome and intervention research. J. Clin. Oncol. 2009, 27, 2308–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, M.M.; Lancashire, E.R.; Winter, D.L.; Frobisher, C.; Reulen, R.C.; Taylor, A.J.; Stevens, M.C.G.; Jenney, M. The British Childhood Cancer Survivor Study: Objectives, methods, population structure, response rates and initial descriptive information. Pediatr. Blood Cancer 2008, 50, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.L.; Whitton, J.; Leisenring, W.; Mertens, A.C.; Hammond, S.; Stovall, M.; Donaldson, S.S.; Meadows, A.T.; Robison, L.L.; Neglia, J.P. Subsequent neoplasms in 5-year survivors of childhood cancer: The Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2010, 102, 1083–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunnes, M.W.; Lie, R.T.; Bjørge, T.; Syse, A.; Ruud, E.; Wesenberg, F.; Moster, D. Economic independence in survivors of cancer diagnosed at a young age: A Norwegian national cohort study. Cancer 2016, 122, 3873–3882. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, T.M.; Recklitis, C.J.; Michel, G.; Grootenhuis, M.A.; Klosky, J.L. Psychological Symptoms, Social Outcomes, Socioeconomic Attainment, and Health Behaviors Among Survivors of Childhood Cancer: Current State of the Literature. J. Clin. Oncol. 2018, 36, 2190–2197. [Google Scholar] [CrossRef]
- Crochet, E.; Tyc, V.L.; Wang, M.; Srivastava, D.K.; Van Sickle, K.; Nathan, P.C.; Leisenring, W.; Gibson, T.M.; Armstrong, G.T.; Krull, K. Posttraumatic stress as a contributor to behavioral health outcomes and healthcare utilization in adult survivors of childhood cancer: A report from the Childhood Cancer Survivor Study. J. Cancer Surviv. 2019, 13, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Kohn, E.; Del Rivero, J. Neuroendocrine Tumors-Less Well Known, Often Misunderstood, and Rapidly Growing in Incidence. JAMA Oncol. 2020, 6, 21–22. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Neuroendocrine Neoplasms | Grade | Terminology/Differentiation/ Location | Mitotic Rate (per 2 mm2) | Ki-67 (%) |
| Low | Grade 1, well-differentiated NET (extra-thoracic) | <2 | <3 | |
| Typical Carcinoid (Thoracic) | <2 | – | ||
| Intermediate | Grade 2, well-differentiated NET (extra-thoracic) | 2–20 | 3–20 | |
| Atypical Carcinoid (Thoracic) | 2–10 | – | ||
| High | Grade 2, well-differentiated NET | >20 | >20 | |
| Poorly differentiated Carcinoma (small cell and large cell) | >20 (thoracic > 10) | >20 |
| Familial Syndromes | Syndrome | NEN | NEN Percent Occurrence | Screening Recommendations | |
| MEN 1 | Pancreatic (gastrinoma, insulinoma) | 20–80% | – chromogranin-A, pancreatic polypeptide, glucagon, VIP annually starting at 8 years old – fasting gastrin annually starting at 20 years old – consider abdominal CT, MRI, or EUS every 3–5 years starting at 20 years old | ||
| Pituitary Adenoma (prolactinoma) | 30–40% | – serum prolactin, IGF-1, fasting glucose and insulin annually starting at 5 years old – head MRI every 3–5 years starting at 5 years old | |||
| MEN 2A/2B | Medullary Thyroid Cancer | ≥98% | Highest Risk | Prophylactic thyroidectomy at or before 1 year old with physical exam, neck ultrasound, serum calcitonin/CEA every 6 months for 1 year and annually thereafter (*serum calcitonin confounding in infants as normally elevated) | |
| High Risk | Physical exam, neck ultrasound, serum calcitonin annually starting at 3 years old; Prophylactic thyroidectomy at or before 5 years old based on serum calcitonin followed by physical exam, neck ultrasound, serum calcitonin, and CEA every 6 months for 1 year and annually thereafter | ||||
| Moderate Risk | Physical exam, serum calcitonin every 6 months for 1 year and annually thereafter if calcitonin remains normal; prophylactic thyroidectomy when calcitonin levels elevated | ||||
| Pheochromocytoma | ≥50% | Highest/ High Risk | Free plasma metanephrines/normetanephrines or 24 h urine fractionated metanephrines annually starting at 11 years old. Adrenal imaging with CT/MRI if elevated | ||
| Moderate Risk | Free plasma metanephrines/normetanephrines or 24 h urine fractionated metanephrines annually starting at 16 years old. Adrenal imaging with CT/MRI if elevated | ||||
| Von-Hippel Lindau | Pheochromocytoma | 10–20% | – Blood pressure at all medical visits starting at 2 years old – Free plasma metanephrines/normetanephrines or 24 h urine fractionated metanephrines annually starting at 5 years old – abdominal MRI or CT with and without IV contrast every 2 years starting at 15 years old | ||
| Paraganglioma | 10–20% | ||||
| Pancreatic | 5–17% | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castle, J.T.; Levy, B.E.; Chauhan, A. Pediatric Neuroendocrine Neoplasms: Rare Malignancies with Incredible Variability. Cancers 2022, 14, 5049. https://doi.org/10.3390/cancers14205049
Castle JT, Levy BE, Chauhan A. Pediatric Neuroendocrine Neoplasms: Rare Malignancies with Incredible Variability. Cancers. 2022; 14(20):5049. https://doi.org/10.3390/cancers14205049
Chicago/Turabian StyleCastle, Jennifer T., Brittany E. Levy, and Aman Chauhan. 2022. "Pediatric Neuroendocrine Neoplasms: Rare Malignancies with Incredible Variability" Cancers 14, no. 20: 5049. https://doi.org/10.3390/cancers14205049
APA StyleCastle, J. T., Levy, B. E., & Chauhan, A. (2022). Pediatric Neuroendocrine Neoplasms: Rare Malignancies with Incredible Variability. Cancers, 14(20), 5049. https://doi.org/10.3390/cancers14205049