
Functional Implications of Epstein-Barr Virus Lytic Genes in Carcinogenesis



Abstract
:Simple Summary
Abstract
1. Introduction
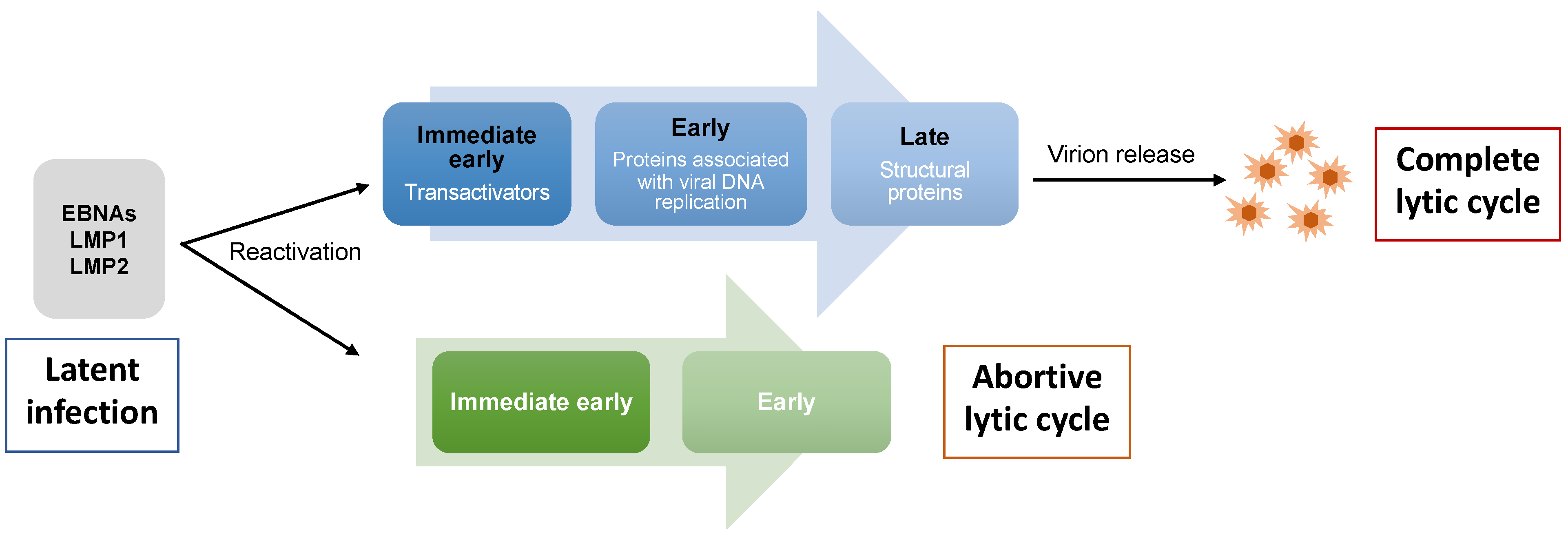
2. EBV Lytic Cycle
3. Expression of EBV Lytic Genes in Tumors
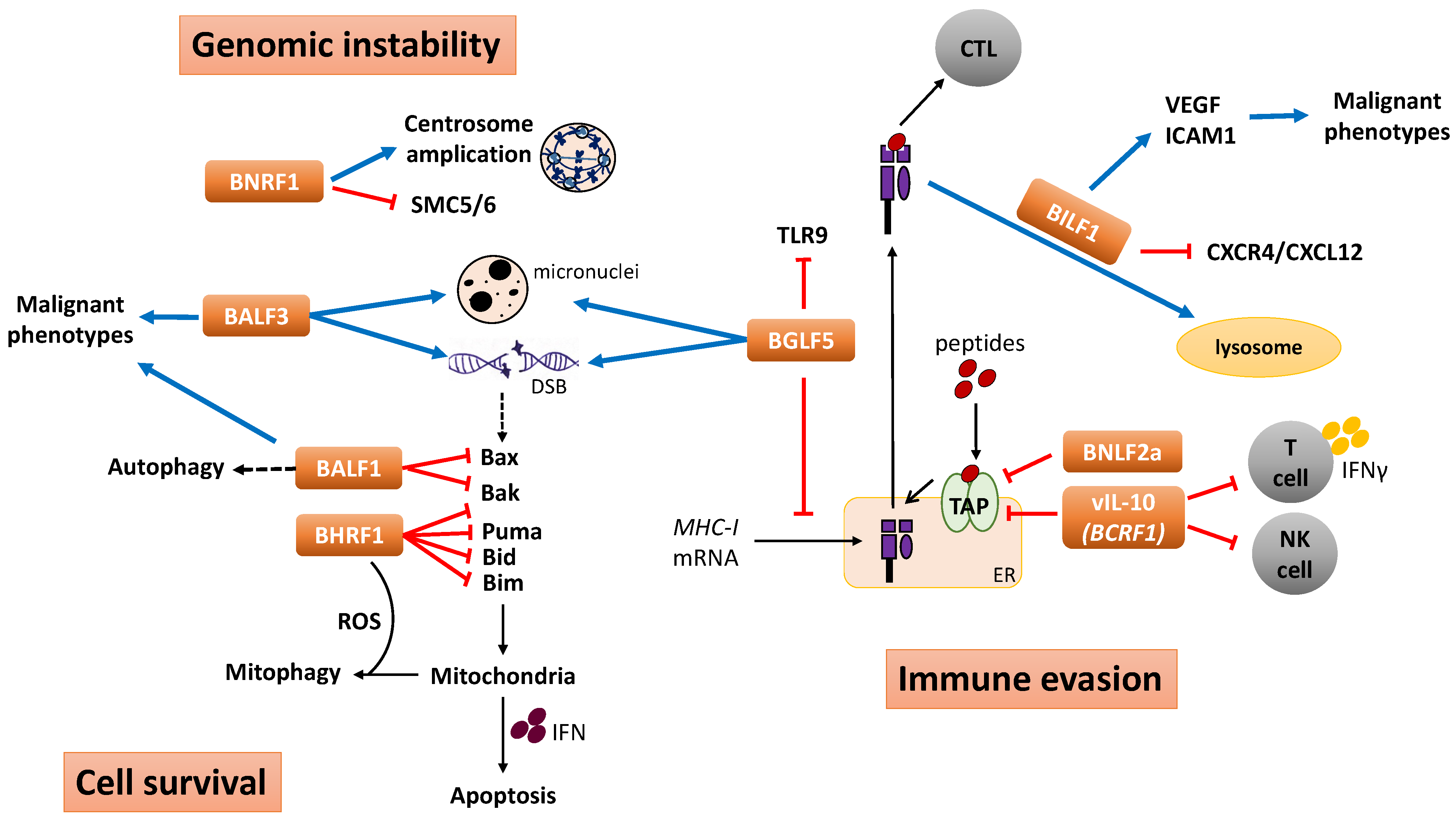
4. Functional Implication of EBV Lytic Genes in Tumorigenesis
4.1. Immunomodulation and Immune Evasion
4.1.1. BNLF2a
4.1.2. BGLF5
4.1.3. BILF1
4.1.4. BCRF1
4.2. Genomic Instability
4.2.1. BGLF5 and BALF3
4.2.2. BNRF1
4.3. Cell Survival
4.3.1. BHRF1
4.3.2. BALF1
5. Conclusions Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Henle, G.; Henle, W.; Diehl, V. Relation of Burkitt’s tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc. Natl. Acad. Sci. USA 1968, 59, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babcock, G.J.; Decker, L.L.; Volk, M.; Thorley-Lawson, D.A. EBV persistence in memory B cells in vivo. Immunity 1998, 9, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein–Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Nakhoul, H.; Lin, Z.; Wang, X.; Roberts, C.; Dong, Y.; Flemington, E. High-throughput sequence analysis of peripheral T-Cell lymphomas indicates subtype-specific viral gene expression patterns and immune cell microenvironments. Msphere 2019, 4, e00248-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Lim, Y.; Im, H.; Bae, J.M.; Kang, G.H.; Ahn, J.; Baek, D.; Kim, T.-Y.; Yoon, S.-S.; Koh, Y. Interpretation of EBV infection in pan-cancer genome considering viral life cycle: LiEB (Life cycle of Epstein-Barr virus). Sci. Rep. 2019, 9, 3465. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Strong, M.J.; Baddoo, M.; Lin, Z.; Wang, Y.-P.; Flemington, E.K.; Liu, Y.-Z. Interaction of Epstein-Barr virus genes with human gastric carcinoma transcriptome. Oncotarget 2017, 8, 38399. [Google Scholar] [CrossRef] [Green Version]
- Abate, F.; Ambrosio, M.R.; Mundo, L.; Laginestra, M.A.; Fuligni, F.; Rossi, M.; Zairis, S.; Gazaneo, S.; De Falco, G.; Lazzi, S. Distinct viral and mutational spectrum of endemic Burkitt lymphoma. PLoS Pathog. 2015, 11, e1005158. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Morgan, D.R.; Meyers, M.O.; Dominguez, R.L.; Martinez, E.; Kakudo, K.; Kuan, P.F.; Banet, N.; Muallem, H.; Woodward, K. Epstein-barr virus infected gastric adenocarcinoma expresses latent and lytic viral transcripts and has a distinct human gene expression profile. Infect. Agent. Cancer 2012, 7, 21. [Google Scholar] [CrossRef]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein-Barr virus genomes and expression profiles in gastric adenocarcinoma. J. Virol. 2018, 92, e01239-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Lin, Z.; Wu, Y.; Dong, J.; Zhao, B.; Cheng, Y.; Huang, P.; Xu, L.; Xia, T.; Xiong, D. Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired-end transcriptome sequencing. Front. Med. 2016, 10, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.-J.; Han, B.-W.; Cai, Q.-Q.; Zuo, X.-Y.; Xia, T.; Chen, J.-R.; Feng, L.-N.; Lim, J.Q.; Chen, S.-W.; Zeng, M.-S. Genomic and transcriptomic landscapes of Epstein-Barr virus in extranodal natural killer T-cell lymphoma. Leukemia 2019, 33, 1451–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaymaz, Y.; Oduor, C.I.; Yu, H.; Otieno, J.A.; Ong’echa, J.M.; Moormann, A.M.; Bailey, J.A. Comprehensive transcriptome and mutational profiling of endemic Burkitt lymphoma reveals EBV type–specific differences. Mol. Cancer Res. 2017, 15, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; O’Grady, T.; Lin, Z.; Xu, G.; Baddoo, M.; Parsons, C.; Zhang, K.; Taylor, C.M.; Flemington, E.K. Epstein-Barr virus and human herpesvirus 6 detection in a non-Hodgkin’s diffuse large B-cell lymphoma cohort by using RNA sequencing. J. Virol. 2013, 87, 13059–13062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayda, N.; Tilloy, V.; Chaunavel, A.; Bahri, R.; Halabi, M.A.; Feuillard, J.; Jaccard, A.; Ranger-Rogez, S. Comprehensive Epstein-Barr virus transcriptome by RNA-sequencing in angioimmunoblastic T cell lymphoma (AITL) and other lymphomas. Cancers 2021, 13, 610. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Li, R.; Chen, M.-Y.; Yu, C.; Tang, L.-Q.; Liu, Y.-M.; Li, J.-P.; Liu, Y.-N.; Luo, Y.-L.; Zhao, Y. Single-cell transcriptomic analysis defines the interplay between tumor cells, viral infection, and the microenvironment in nasopharyngeal carcinoma. Cell Res. 2020, 30, 950–965. [Google Scholar] [CrossRef]
- Xiong, J.; Cui, B.-W.; Wang, N.; Dai, Y.-T.; Zhang, H.; Wang, C.-F.; Zhong, H.-J.; Cheng, S.; Ou-Yang, B.-S.; Hu, Y. Genomic and transcriptomic characterization of natural killer T cell lymphoma. Cancer Cell 2020, 37, 403–419.e406. [Google Scholar] [CrossRef]
- Murata, T. Encyclopedia of EBV-encoded lytic genes: An update. Hum. Herpesviruses 2018, 395–412. [Google Scholar] [CrossRef]
- Hausen, H.Z.; O’NEILL, F.J.; Freese, U.K.; HECKER, E. Persisting oncogenic herpesvirus induced by the tumour promoter TPA. Nature 1978, 272, 373–375. [Google Scholar] [CrossRef]
- Kallin, B.; Luka, J.; Klein, G. Immunochemical characterization of Epstein-Barr virus-associated early and late antigens in n-butyrate-treated P3HR-1 cells. J. Virol. 1979, 32, 710–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fixman, E.D.; Hayward, G.; Hayward, S. trans-acting requirements for replication of Epstein-Barr virus ori-Lyt. J. Virol. 1992, 66, 5030–5039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scepers, A.; Pich, D.; Hammerschmidt, W. Activation oforiLyt, the Lytic Origin of DNA Replication of Epstein–Barr Virus, by BZLF1. Virology 1996, 220, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenspan, J.S.; Greenspan, D.; Lennette, E.T.; Abrams, D.I.; Conant, M.A.; Petersen, V.; Freese, U.K. Replication of Epstein-Barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 1985, 313, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of Epstein–Barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalla, M.; Hammerschmidt, W. Human B cells on their route to latent infection–early but transient expression of lytic genes of Epstein-Barr virus. Eur. J. Cell Biol. 2012, 91, 65–69. [Google Scholar] [CrossRef]
- Dickerson, S.J.; Xing, Y.; Robinson, A.R.; Seaman, W.T.; Gruffat, H.; Kenney, S.C. Methylation-dependent binding of the epstein-barr virus BZLF1 protein to viral promoters. PLoS Pathog. 2009, 5, e1000356. [Google Scholar] [CrossRef] [Green Version]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein–Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J. Gen. Virol. 1995, 76, 1401–1408. [Google Scholar] [CrossRef]
- Ramayanti, O.; Juwana, H.; Verkuijlen, S.A.; Adham, M.; Pegtel, M.D.; Greijer, A.E.; Middeldorp, J.M. Epstein-Barr virus mRNA profiles and viral DNA methylation status in nasopharyngeal brushings from nasopharyngeal carcinoma patients reflect tumor origin. Int. J. Cancer 2017, 140, 149–162. [Google Scholar] [CrossRef]
- Xue, S.a.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of Epstein-Barr virus genes in Burkitt’s lymphoma from the central African country Malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef]
- Djavadian, R.; Hayes, M.; Johannsen, E. CAGE-seq analysis of Epstein-Barr virus lytic gene transcription: 3 kinetic classes from 2 mechanisms. PLoS Pathog. 2018, 14, e1007114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic properties of the EBV ZEBRA protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef] [PubMed]
- Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Brousset, P.; Knecht, H.; Rubin, B.; Drouet, E.; Chittal, S.; Meggetto, F.; Al Saati, T.; Bachmann, E.; Denoyel, G.; Sergeant, A. Demonstration of Epstein-Barr virus replication in Reed-Sternberg cells of Hodgkin’s disease. Blood 1993, 82, 872–876. [Google Scholar] [CrossRef] [Green Version]
- Xue, S.-A.; Lu, Q.-L.; Poulsom, R.; Karran, L.; Jones, M.; Griffin, B.E. Expression of two related viral early genes in Epstein-Barr virus-associated tumors. J. Virol. 2000, 74, 2793–2803. [Google Scholar] [CrossRef] [Green Version]
- Adamson, A.L.; Darr, D.; Holley-Guthrie, E.; Johnson, R.A.; Mauser, A.; Swenson, J.; Kenney, S.C. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 2000, 74, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Song, M.; Camargo, M.C.; Van Duine, J.; Williams, S.; Chung, Y.; Kim, K.-M.; Lissowska, J.; Sivins, A.; Gao, W. Identification of anti-Epstein-Barr virus (EBV) antibody signature in EBV-associated gastric carcinoma. J. Gastric Cancer 2021, 24, 858–867. [Google Scholar] [CrossRef]
- Zhu, Y.-H.; Wei, Y.-S.; Li, H.; Liang, W.-B.; Du, B.; Zhang, G.-Q.; Zhang, L. Construction and characterization of monoclonal antibodies specific for the R transactivator 185 of Epstein-Barr virus. J. Virol. Methods 2007, 144, 12–16. [Google Scholar] [CrossRef]
- Bentz, G.L.; Liu, R.; Hahn, A.M.; Shackelford, J.; Pagano, J.S. Epstein–Barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-β. J. Virol. 2010, 402, 121–128. [Google Scholar] [CrossRef]
- Yokoyama, N.; Fujii, K.; Hirata, M.; Tamai, K.; Kiyono, T.; Kuzushima, K.; Nishiyama, Y.; Fujita, M.; Tsurumi, T. Assembly of the Epstein–Barr virus BBLF4, BSLF1 and BBLF2/3 proteins and their interactive properties. J. Gen. Virol. 1999, 80, 2879–2887. [Google Scholar] [CrossRef] [PubMed]
- Cook, I.; Shanahan, F.; Farrell, P. Epstein-Barr virus SM protein. J. Virol. 1994, 205, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.-M.; Levine, A.J. Characterization of proteins encoded by the Epstein-Barr virus transactivator gene BMLF1. Virol. J. 1989, 168, 101–111. [Google Scholar] [CrossRef]
- Kondo, S.; Okuno, Y.; Murata, T.; Dochi, H.; Wakisaka, N.; Mizokami, H.; Moriyama-Kita, M.; Kobayashi, E.; Kano, M.; Komori, T. EBV genome variations enhance clinicopathological features of nasopharyngeal carcinoma in a non-endemic region. Cancer Sci. 2022, 113, 2446. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.; Hardwick, J.; Hayward, S. Responsiveness of the Epstein-Barr virus NotI repeat promoter to the Z transactivator is mediated in a cell-type-specific manner by two independent signal regions. J. Virol. 1989, 63, 3040–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.Y.; Shih, Y.Y.; Li, L.Y.; Chou, S.P.; Sheen, T.S.; Chen, C.L.; Yang, C.S.; Chen, J.Y. Expression of the Epstein-Barr virus BHRF1 gene, a homologue of Bcl-2, in nasopharyngeal carcinoma tissue. J. Med. Virol. 2000, 61, 241–250. [Google Scholar] [CrossRef]
- Nicholls, J.; Kremmer, E.; Meseda, C.A.; Mackett, M.; Hahn, P.; Gulley, M.L.; Brink, A.; Swinnen, L.J.; Greenspan, J.; De Souza, Y. Comparative analysis of the expression of the epstein-barr virus (EBV) anti-apoptotic gene BHRF1 in nasopharyngeal carcinoma and EBV-related lymphoid diseases. J. Med. Virol. 2001, 65, 105–113. [Google Scholar] [CrossRef]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Epstein-Barr virus lytic cycle involvement in diffuse large B cell lymphoma. Hematol. Oncol. 2018, 36, 98–103. [Google Scholar] [CrossRef]
- Nadala, E.; Tan, T.; Wong, H.; Ting, R. ELISA for the detection of serum and saliva IgA against the BMRFI gene product of Epstein-Barr virus. J. Med. Virol. 1996, 50, 93–96. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, H.; Sun, G.; Xu, X.; Sun, X.; Cao, L. Trichloromethane fraction of Incarvillea compacta induces lytic cytotoxicity and apoptosis in Epstein-Barr virus-positive gastric cancer AGS cells. BMC Complement. Altern. Med. 2016, 16, 344. [Google Scholar] [CrossRef]
- Tsurumi, T.; Kobayashi, A.; Tamai, K.; Daikoku, T.; Kurachi, R.; Nishiyama, Y. Functional expression and characterization of the Epstein-Barr virus DNA polymerase catalytic subunit. J. Virol. 1993, 67, 4651–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, E.; Yang, L.; Middeldorp, J.; Sheen, T.S.; Chen, J.Y.; Fukayama, M.; Eizuru, Y.; Ooka, T.; Takada, K. Epstein–Barr virus (EBV)-encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J. Med. Virol. 2005, 76, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Horst, D.; Van Leeuwen, D.; Croft, N.P.; Garstka, M.A.; Hislop, A.D.; Kremmer, E.; Rickinson, A.B.; Wiertz, E.J.; Ressing, M.E. Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J. Immunol. 2009, 182, 2313–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, M.J.; Laskow, T.; Nakhoul, H.; Blanchard, E.; Liu, Y.; Wang, X.; Baddoo, M.; Lin, Z.; Yin, Q.; Flemington, E.K. Latent expression of the Epstein-Barr virus (EBV)-encoded major histocompatibility complex class I TAP inhibitor, BNLF2a, in EBV-positive gastric carcinomas. J. Virol. 2015, 89, 10110–10114. [Google Scholar] [CrossRef] [Green Version]
- Gore, M.; Hutt-Fletcher, L.M. The BDLF2 protein of Epstein–Barr virus is a type II glycosylated envelope protein whose processing is dependent on coexpression with the BMRF2 protein. J. Virol. 2009, 383, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Itano, H.; Zhang, W.; Ritter, J.H.; McCarthy, T.J.; Yew, N.S.; Mohanakumar, T.; Patterson, G.A. Endobronchial transfection of naked viral interleukin-10 gene in rat lung allotransplantation. Ann. Thorac. Surg. 2001, 71, 1126–1133. [Google Scholar] [CrossRef]
- Tanner, J.E.; Mitoma, F.D.; Rooney, C.M.; Alfieri, C. Anti-interleukin-10 antibodies in patients with chronic active Epstein-Barr virus infection. J. Infect. Dis. 1997, 176, 1454–1461. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Hong, G.; Li, Z.; Xu, J.; Zhu, Z.; Li, L. Expression of VCA (viral capsid antigen) and EBNA1 (Epstein—Barr-virus-encoded nuclear antigen 1) genes of Epstein–Barr virus in Pichia pastoris and application of the products in a screening test for patients with nasopharyngeal carcinoma. Biotechnol. Appl. Biochem. 2007, 47, 59–69. [Google Scholar]
- Yaswen, L.R.; Stephens, E.B.; Davenport, L.C.; Hutt-Fletcher, L.M. Epstein-Barr virus glycoprotein gp85 associates with the BKRF2 gene product and is incompletely processed as a recombinant protein. J. Virol. 1993, 195, 387–396. [Google Scholar] [CrossRef]
- Nuebling, C.M.; Buck, M.; Boos, H.; Von Deimling, A.; Mueller-Lantzsch, N. Expression of Epstein-Barr virus membrane antigen gp350/220 in E. coli and in insect cells. J. Virol. 1992, 191, 443–447. [Google Scholar] [CrossRef]
- Bertoni, G.; Kostyal, D.A.; Reisert, P.S.; Humphreys, R.E.; Sairenji, T. Synthetic peptides to identify antigenic determinants on Epstein-Barr virus gp350/220. Intervirology 1990, 31, 290–294. [Google Scholar] [CrossRef]
- Ge, J.; Huang, Y.; Hu, X.; Zhong, J. A surface-modified baculovirus vector with improved gene delivery to B-lymphocytic cells. J. Biotechnol. 2007, 129, 367–372. [Google Scholar] [CrossRef]
- Tsai, K.; Chan, L.; Gibeault, R.; Conn, K.; Dheekollu, J.; Domsic, J.; Marmorstein, R.; Schang, L.M.; Lieberman, P.M. Viral reprogramming of the Daxx histone H3. 3 chaperone during early Epstein-Barr virus infection. J. Virol. 2014, 88, 14350–14363. [Google Scholar] [CrossRef] [Green Version]
- Ressing, M.E.; Keating, S.E.; van Leeuwen, D.; Koppers-Lalic, D.; Pappworth, I.Y.; Wiertz, E.J.; Rowe, M. Impaired transporter associated with antigen processing-dependent peptide transport during productive EBV infection. J. Immunol. 2005, 174, 6829–6838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, S.; Prince, S.; Jones, M.; Rowe, M. The lytic cycle of Epstein-Barr virus is associated with decreased expression of cell surface major histocompatibility complex class I and class II molecules. J. Virol. 2002, 76, 8179–8188. [Google Scholar] [CrossRef] [Green Version]
- Hislop, A.D.; Ressing, M.E.; Van Leeuwen, D.; Pudney, V.A.; Horst, D.l.; Koppers-Lalic, D.; Croft, N.P.; Neefjes, J.J.; Rickinson, A.B.; Wiertz, E.J. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J. Exp. Med. 2007, 204, 1863–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pudney, V.A.; Leese, A.M.; Rickinson, A.B.; Hislop, A.D. CD8+ immunodominance among Epstein-Barr virus lytic cycle antigens directly reflects the efficiency of antigen presentation in lytically infected cells. J. Exp. Med. 2005, 201, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Griekspoor, A.C.; Neefjes, J. How does TAP pump peptides? Insights from DNA repair and traffic ATPases. Immunol. Today 2000, 21, 598–600. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.J.; Momburg, F.; Hämmerling, G.J. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science 1993, 261, 769–771. [Google Scholar] [CrossRef]
- Horst, D.; Favaloro, V.; Vilardi, F.; Van Leeuwen, H.C.; Garstka, M.A.; Hislop, A.D.; Rabu, C.; Kremmer, E.; Rickinson, A.B.; High, S. EBV protein BNLF2a exploits host tail-anchored protein integration machinery to inhibit TAP. J. Immunol. 2011, 186, 3594–3605. [Google Scholar] [CrossRef] [Green Version]
- Croft, N.P.; Shannon-Lowe, C.; Bell, A.I.; Horst, D.; Kremmer, E.; Ressing, M.E.; Wiertz, E.J.; Middeldorp, J.M.; Rowe, M.; Rickinson, A.B. Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog. 2009, 5, e1000490. [Google Scholar] [CrossRef] [PubMed]
- Almohammed, R.; Osborn, K.; Ramasubramanyan, S.; Perez-Fernandez, I.B.N.; Godfrey, A.; Mancini, E.J.; Sinclair, A.J. Mechanism of activation of the BNLF2a immune evasion gene of Epstein-Barr virus by Zta. J. Gen. Virol. 2018, 99, 805. [Google Scholar] [CrossRef] [PubMed]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef] [Green Version]
- Bruce, J.P.; To, K.-F.; Lui, V.W.; Chung, G.T.; Chan, Y.-Y.; Tsang, C.M.; Yip, K.Y.; Ma, B.B.; Woo, J.K.; Hui, E.P. Whole-genome profiling of nasopharyngeal carcinoma reveals viral-host co-operation in inflammatory NF-κB activation and immune escape. Nat. Commun. 2021, 12, 4193. [Google Scholar] [CrossRef]
- Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr virus infection in non-small cell lung cancer. Cancers 2019, 11, 759. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.X.; Decaussin, G.; Tessier, M.d.T.; Daillie, J.; Ooka, T. Identification of an Epstein-Barr virus-specific desoxyribonuclease gene using complementary DNA. Nucleic Acids Res. 1987, 15, 2707–2717. [Google Scholar] [CrossRef] [Green Version]
- Baylis, S.A.; Purifoy, D.J.; Littler, E. The characterization of the EBV alkaline deoxyribonuclease cloned and expressed in E. coli. Nucleic Acids Res. 1989, 17, 7609–7622. [Google Scholar] [CrossRef]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J. Exp. Med. 2004, 200, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Chavez, L.; Ganem, D. The exonuclease and host shutoff functions of the SOX protein of Kaposi’s sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005, 79, 7396–7401. [Google Scholar] [CrossRef] [Green Version]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive Epstein–Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. Viral modulation of antigen presentation: Manipulation of cellular targets in the ER and beyond. Immunol. Rev. 2005, 207, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.; Ressing, M.E.; Rowe, M. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horst, D.; Burmeister, W.P.; Boer, I.G.; van Leeuwen, D.; Buisson, M.; Gorbalenya, A.E.; Wiertz, E.J.; Ressing, M.E. The “Bridge” in the Epstein-Barr virus alkaline exonuclease protein BGLF5 contributes to shutoff activity during productive infection. J. Virol. 2012, 86, 9175–9187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, R.; Miller, G. Epstein-Barr virus-induced nodules on viral replication compartments contain RNA processing proteins and a viral long noncoding RNA. J. Virol. 2018, 92, e01254-18. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, M.; Griffin, B.D.; Berkhoff, E.G.; van Leeuwen, D.; Boer, I.G.; Buisson, M.; Hartgers, F.C.; Burmeister, W.P.; Wiertz, E.J.; Ressing, M.E. EBV lytic-phase protein BGLF5 contributes to TLR9 downregulation during productive infection. J. Immunol. 2011, 186, 1694–1702. [Google Scholar] [CrossRef] [Green Version]
- Van Gent, M.; Gram, A.M.; Boer, I.G.; Geerdink, R.J.; Lindenbergh, M.F.; Lebbink, R.J.; Wiertz, E.J.; Ressing, M.E. Silencing the shutoff protein of Epstein–Barr virus in productively infected B cells points to (innate) targets for immune evasion. J. Gen. Virol. 2015, 96, 858–865. [Google Scholar] [CrossRef]
- Feederle, R.; Bannert, H.; Lips, H.; Muller-Lantzsch, N.; Delecluse, H.-J. The Epstein-Barr virus alkaline exonuclease BGLF5 serves pleiotropic functions in virus replication. J. Virol. 2009, 83, 4952–4962. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-Y.; Liu, M.-Y.; Hsu, T.-Y.; Cho, S.-M.; Yang, C.-S. Use of bacterially-expressed antigen for detection of antibodies to the EBV-specific deoxyribonuclease in sera from patients with nasopharyngeal carcinoma. J. Virol. Methods 1993, 45, 49–66. [Google Scholar] [CrossRef]
- Sbih-Lammali, F.; Berger, F.; Busson, P.; Ooka, T. Expression of the DNase encoded by the BGLF5 gene of Epstein–Barr virus in nasopharyngeal carcinoma epithelial cells. Virology 1996, 222, 64–74. [Google Scholar] [CrossRef] [PubMed]
- DAVIS-POYNTER, N.J.; Farrell, H.E. Masters of deception: A review of herpesvirus immune evasion strategies. Immunol. Cell Biol. 1996, 74, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Beisser, P.S.; Verzijl, D.; Gruijthuijsen, Y.K.; Beuken, E.; Smit, M.J.; Leurs, R.; Bruggeman, C.A.; Vink, C. The Epstein-Barr virus BILF1 gene encodes a G protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J. Virol. 2005, 79, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, S.J.; Rosenkilde, M.M.; Eugen-Olsen, J.; Kledal, T.N. Epstein-Barr virus-encoded BILF1 is a constitutively active G protein-coupled receptor. J. Virol. 2005, 79, 536–546. [Google Scholar] [CrossRef] [Green Version]
- Griffin, B.D.; Gram, A.M.; Mulder, A.; Van Leeuwen, D.; Claas, F.H.; Wang, F.; Ressing, M.E.; Wiertz, E. EBV BILF1 evolved to downregulate cell surface display of a wide range of HLA class I molecules through their cytoplasmic tail. J. Immunol. 2013, 190, 1672–1684. [Google Scholar] [CrossRef] [Green Version]
- Chakravorty, S.; Yan, B.; Wang, C.; Wang, L.; Quaid, J.T.; Lin, C.F.; Briggs, S.D.; Majumder, J.; Canaria, D.A.; Chauss, D. Integrated Pan-Cancer Map of EBV-Associated Neoplasms Reveals Functional Host–Virus InteractionsPan-Cancer Analysis of EBV-Associated Neoplasms. Cancer Res. 2019, 79, 6010–6023. [Google Scholar] [CrossRef] [Green Version]
- Tierney, R.J.; Shannon-Lowe, C.D.; Fitzsimmons, L.; Bell, A.I.; Rowe, M. Unexpected patterns of Epstein–Barr virus transcription revealed by a high throughput PCR array for absolute quantification of viral mRNA. Virology 2015, 474, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.; Rowe, M. The Epstein-Barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Quinn, L.L.; Tamblyn, J.; Thomas, W.A.; Feederle, R.; Delecluse, H.-J.; Hislop, A.D.; Rowe, M. The Epstein-Barr virus-encoded BILF1 protein modulates immune recognition of endogenously processed antigen by targeting major histocompatibility complex class I molecules trafficking on both the exocytic and endocytic pathways. J. Virol. 2011, 85, 1604–1614. [Google Scholar] [CrossRef] [Green Version]
- Fares, S.; Spiess, K.; Olesen, E.T.; Zuo, J.; Jackson, S.; Kledal, T.N.; Wills, M.R.; Rosenkilde, M.M. Distinct roles of extracellular domains in the epstein-barr virus-encoded BILF1 receptor for signaling and major histocompatibility complex class I downregulation. MBio 2019, 10, e01707–e01718. [Google Scholar] [CrossRef] [Green Version]
- Milligan, G. G-protein-coupled receptor heterodimers: Pharmacology, function and relevance to drug discovery. Drug Discov. Today 2006, 11, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Vischer, H.F.; Nijmeijer, S.; Smit, M.J.; Leurs, R. Viral hijacking of human receptors through heterodimerization. Biochem. Biophys. Res. Commun. 2008, 377, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Nijmeijer, S.; Leurs, R.; Smit, M.J.; Vischer, H.F. The Epstein-Barr Virus-encoded G Protein-coupled Receptor BILF1 Hetero-oligomerizes with Human CXCR4, Scavenges Gαi Proteins, and Constitutively Impairs CXCR4 Functioning [S]. J. Biol. Chem. 2010, 285, 29632–29641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, L.; Qiao, A.; Watson, K.; Zhang, J.; Fan, G.-H. Activation of CXCR4 triggers ubiquitination and down-regulation of major histocompatibility complex class I (MHC-I) on epithelioid carcinoma HeLa cells. J. Biol. Chem. 2008, 283, 3951–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyngaa, R.; Nørregaard, K.; Kristensen, M.; Kubale, V.; Rosenkilde, M.; Kledal, T.N. Cell transformation mediated by the Epstein–Barr virus G protein-coupled receptor BILF1 is dependent on constitutive signaling. Oncogene 2010, 29, 4388–4398. [Google Scholar] [CrossRef] [Green Version]
- Murono, S.; Inoue, H.; Tanabe, T.; Joab, I.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Induction of cyclooxygenase-2 by Epstein–Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 6905–6910. [Google Scholar] [CrossRef] [Green Version]
- Paydas, S.; Ergin, M.; Erdogan, S.; Seydaoglu, G. Prognostic significance of EBV-LMP1 and VEGF-A expressions in non-Hodgkin’s lymphomas. Leuk. Res. 2008, 32, 1424–1430. [Google Scholar] [CrossRef]
- Guo, Q.; Gao, J.; Cheng, L.; Yang, X.; Li, F.; Jiang, G. The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 upregulates ICAM-1 through a mechanism involving the NF-қB pathway. Biosci. Biotechnol. Biochem. 2020, 84, 1810–1819. [Google Scholar] [CrossRef]
- Hubbard, A.K.; Rothlein, R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic. Biol. Med. 2000, 28, 1379–1386. [Google Scholar] [CrossRef]
- Hayes, S.H.; Seigel, G.M. Immunoreactivity of ICAM-1 in human tumors, metastases and normal tissues. Int. J. Clin. Exp. Pathol. 2009, 2, 553–560. [Google Scholar]
- Quinn, L.L.; Zuo, J.; Abbott, R.J.; Shannon-Lowe, C.; Tierney, R.J.; Hislop, A.D.; Rowe, M. Cooperation between Epstein-Barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog. 2014, 10, e1004322. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; Vieira, P.; Fiorentino, D.F.; Trounstine, M.L.; Khan, T.A.; Mosmann, T.R. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science 1990, 248, 1230–1234. [Google Scholar] [CrossRef] [PubMed]
- De Vieira, P.; de Waal-Malefyt, R.; Dang, M.; Johnson, K.; Kastelein, R.; Fiorentino, D.; DeVries, J.; Roncarolo, M.G.; Mosmann, T.; Moore, K. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: Homology to Epstein-Barr virus open reading frame BCRFI. Proc. Natl. Acad. Sci. USA 1991, 88, 1172–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, I.; Cheung, R.K.; Dosch, H.-M. Viral interleukin 10 is critical for the induction of B cell growth transformation by Epstein-Barr virus. J. Exp. Med. 1993, 178, 439–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubry, V.; Mure, F.; Mariamé, B.; Deschamps, T.; Wyrwicz, L.S.; Manet, E.; Gruffat, H. Epstein-Barr virus late gene transcription depends on the assembly of a virus-specific preinitiation complex. J. Virol. 2014, 88, 12825–12838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, J.; Lopez-Giraldez, F.; Delecluse, H.-J.; Walsh, A.; El-Guindy, A. The Epstein-Barr virus immunoevasins BCRF1 and BPLF1 are expressed by a mechanism independent of the canonical late pre-initiation complex. PLoS Pathog. 2016, 12, e1006008. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S.; Hesselton, R.; Sullivan, J.; Kieff, E. Epstein-Barr virus recombinants with specifically mutated BCRF1 genes. J. Virol. 1993, 67, 7406–7413. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Iwatsuki, K.; Oyama, N.; Ohtsuka, M.; Satoh, M.; Kikuchi, S.; Akiba, H.; Kaneko, F. The latency pattern of Epstein–Barr virus infection and viral IL-10 expression in cutaneous natural killer/T-cell lymphomas. Br. J. Cancer 2001, 84, 920–925. [Google Scholar] [CrossRef] [Green Version]
- Kurilla, M.G.; Swaminathan, S.; Welsh, R.M.; Kieff, E.; Brutkiewicz, R.R. Effects of virally expressed interleukin-10 on vaccinia virus infection in mice. J. Virol. 1993, 67, 7623–7628. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.-H.; Malefyt, R.d.W.; Fiorentino, D.F.; Dang, M.-N.; Vieira, P.; Devries, J.; Spits, H.; Mosmann, T.R.; Moore, K.W. Expression of interleukin-10 activity by Epstein-Barr virus protein BCRF1. Science 1990, 250, 830–832. [Google Scholar] [CrossRef]
- Zeidler, R.; Eissner, G.n.; Meissner, P.; Uebel, S.; Tampé, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus–encoded interleukin-10. Am. J. Hematol. 1997, 90, 2390–2397. [Google Scholar]
- De Waal Malefyt, R.; Haanen, J.; Spits, H.; Roncarolo, M.-G.; te Velde, A.; Figdor, C.; Johnson, K.; Kastelein, R.; Yssel, H.; De Vries, J. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J. Exp. Med. 1991, 174, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Salek-Ardakani, S.; Arrand, J.R.; Mackett, M. Epstein–Barr virus encoded interleukin-10 inhibits HLA-class I, ICAM-1, and B7 expression on human monocytes: Implications for immune evasion by EBV. Virology 2002, 304, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Bejarano, M.; Masucci, M. Interleukin-10 Abrogates the Inhibition of Epstein-Barr Virus–Induced B-Cell Transformation by Memory T-Cell Responses. Am. J. Hematol. 1998, 92, 4256–4262. [Google Scholar]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruhne, B.; Kamranvar, S.A.; Masucci, M.G.; Sompallae, R. EBV and genomic instability—A new look at the role of the virus in the pathogenesis of Burkitt’s lymphoma. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2009; Volume 19, pp. 394–400. [Google Scholar]
- Fang, C.Y.; Lee, C.H.; Wu, C.C.; Chang, Y.T.; Yu, S.L.; Chou, S.P.; Huang, P.T.; Chen, C.L.; Hou, J.W.; Chang, Y. Recurrent chemical reactivations of EBV promotes genome instability and enhances tumor progression of nasopharyngeal carcinoma cells. Int. J. Cancer 2009, 124, 2016–2025. [Google Scholar] [CrossRef]
- Shumilov, A.; Tsai, M.-H.; Schlosser, Y.T.; Kratz, A.-S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J. Epstein–Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-H.; Fang, C.-Y.; Sheu, J.J.-C.; Chang, Y.; Takada, K.; Chen, J.-Y. Amplicons on chromosome 3 contain oncogenes induced by recurrent exposure to 12-O-tetradecanoylphorbol-13-acetate and sodium n-butyrate and Epstein–Barr virus reactivation in a nasopharyngeal carcinoma cell line. Cancer Genet. 2008, 185, 1–10. [Google Scholar] [CrossRef]
- Wu, C.-C.; Liu, M.-T.; Chang, Y.-T.; Fang, C.-Y.; Chou, S.-P.; Liao, H.-W.; Kuo, K.-L.; Hsu, S.-L.; Chen, Y.-R.; Wang, P.-W. Epstein–Barr Virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef]
- Chiu, S.-H.; Wu, M.-C.; Wu, C.-C.; Chen, Y.-C.; Lin, S.-F.; Hsu, J.T.-A.; Yang, C.-S.; Tsai, C.-H.; Takada, K.; Chen, M.-R. Epstein-Barr virus BALF3 has nuclease activity and mediates mature virion production during the lytic cycle. J. Virol. 2014, 88, 4962–4975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, S.-H.; Wu, C.-C.; Fang, C.-Y.; Yu, S.-L.; Hsu, H.-Y.; Chow, Y.-H.; Chen, J.-Y. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlmann, F. SMC complexes: From DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016, 17, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Yiu, S.P.T.; Guo, R.; Zerbe, C.; Weekes, M.P.; Gewurz, B.E. Epstein-Barr virus BNRF1 destabilizes SMC5/6 cohesin complexes to evade its restriction of replication compartments. Cell Rep. 2022, 38, 110411. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.J.; Quinn, L.L.; Leese, A.M.; Scholes, H.M.; Pachnio, A.; Rickinson, A.B. CD8+ T cell responses to lytic EBV infection: Late antigen specificities as subdominant components of the total response. J. Immunol. 2013, 191, 5398–5409. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.-J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Deng, Z.; Vladimirova, O.; Wiedmer, A.; Lu, F.; Lieberman, P.M.; Patel, D.J. Structural basis underlying viral hijacking of a histone chaperone complex. Nat. Commun. 2016, 7, 12707. [Google Scholar] [CrossRef] [Green Version]
- Milosevic, S.; Behrends, U.; Adhikary, D.; Mautner, J. Identification of major histocompatibility complex class II-restricted antigens and epitopes of the Epstein-Barr virus by a novel bacterial expression cloning approach. J. Virol. 2006, 80, 10357–10364. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, D.; Damaschke, J.; Mautner, J.; Behrends, U. The Epstein-Barr virus major tegument protein BNRF1 is a common target of cytotoxic CD4+ T cells. J. Virol. 2020, 94, e00284-20. [Google Scholar] [CrossRef]
- Xue, W.-Q.; Wang, T.-M.; Huang, J.-W.; Zhang, J.-B.; He, Y.-Q.; Wu, Z.-Y.; Liao, Y.; Yuan, L.-L.; Mu, J.; Jia, W.-H. A comprehensive analysis of genetic diversity of EBV reveals potential high-risk subtypes associated with nasopharyngeal carcinoma in China. Virus Evol. 2021, 7, veab010. [Google Scholar] [CrossRef]
- Khanim, F.; Dawson, C.; Meseda, C.; Dawson, J.; Mackett, M.; Young, L.S. BHRF1, a viral homologue of the Bcl-2 oncogene, is conserved at both the sequence and functional level in different Epstein-Barr virus isolates. J. Gen. Virol. 1997, 78, 2987–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, A.K.; Tao, Q.; Srivastava, G.; Ho, F.C. Nasal NK-and T-cell lymphomas share the same type of Epstein-Barr virus latency as nasopharyngeal carcinoma and Hodgkin’s disease. Int. J. Cancer 1996, 68, 285–290. [Google Scholar] [CrossRef]
- Hayes, D.; Brink, A.; Vervoort, M.; Middeldorp, J.; Meijer, C.; van Den Brule, A. Expression of Epstein-Barr virus (EBV) transcripts encoding homologues to important human proteins in diverse EBV associated diseases. Mol. Pathol. 1999, 52, 97. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- Zhu, S.; Sun, P.; Zhang, Y.; Yan, L.; Luo, B. Expression of c-myc and PCNA in Epstein-Barr virus-associated gastric carcinoma. Exp. Ther. Med. 2013, 5, 1030–1034. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Jiang, Z.; Spezia-Lindner, D.E.; Liang, T.; Xu, C.; Wang, H.; Tian, Y.; Bai, Y. BHRF1 Enhances EBV Mediated Nasopharyngeal Carcinoma Tumorigenesis through Modulating Mitophagy Associated with Mitochondrial Membrane Permeabilization Transition. Cells 2020, 9, 1158. [Google Scholar] [CrossRef]
- Henderson, S.; Huen, D.; Rowe, M.; Dawson, C.; Johnson, G.; Rickinson, A. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. USA 1993, 90, 8479–8483. [Google Scholar] [CrossRef] [Green Version]
- Tarodi, B.; Subramanian, T.; Chinnadurai, G. Epstein-Barr virus BHRF1 protein protects against cell death induced by DNA-damaging agents and heterologous viral infection. Virology 1994, 201, 404–407. [Google Scholar] [CrossRef]
- Dawson, C.W.; Eliopoulos, A.G.; Dawson, J.; Young, L.S. BHRF1, a viral homologue of the Bcl-2 oncogene, disturbs epithelial cell differentiation. Oncogene 1995, 10, 69–77. [Google Scholar] [PubMed]
- Foghsgaard, L.; Jäättelä, M. The ability of BHRF1 to inhibit apoptosis is dependent on stimulus and cell type. J. Virol. 1997, 71, 7509–7517. [Google Scholar] [CrossRef] [Green Version]
- Kawanishi, M. Epstein-Barr virus BHRF1 protein protects intestine 407 epithelial cells from apoptosis induced by tumor necrosis factor alpha and anti-Fas antibody. J. Virol. 1997, 71, 3319–3322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbien, A.L.; Kappler, J.W.; Marrack, P. The Epstein–Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc. Natl. Acad. Sci. USA 2009, 106, 5663–5668. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Wei, A.H.; Fletcher, J.I.; Willis, S.N.; Chen, L.; Roberts, A.W.; Huang, D.C.; Colman, P.M. Structural basis for apoptosis inhibition by Epstein-Barr virus BHRF1. PLoS Pathog. 2010, 6, e1001236. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, L.; Cartlidge, R.; Chang, C.; Sejic, N.; Galbraith, L.C.; Suraweera, C.D.; Croom-Carter, D.; Dewson, G.; Tierney, R.J.; Bell, A.I. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020, 27, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Granville, D.J.; Gottlieb, R.A. Mitochondria: Regulators of cell death and survival. Sci. World J. 2002, 2, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Zhou, R.; Ma, Z. Autophagy—Cell survival and death. In Autophagy: Biology and Diseases; Science Press and Springer Nature Singapore Pte Ltd.: Singapore, 2019; pp. 667–696. [Google Scholar]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Milián, E.; Prats, E.; Cairó, J.J.; Gòdia, F.; Vives, J. BHRF1 exerts an antiapoptotic effect and cell cycle arrest via Bcl-2 in murine hybridomas. J. Biotechnol. 2015, 209, 58–67. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilmen, G.; Glon, D.; Siracusano, G.; Lussignol, M.; Shao, Z.; Hernandez, E.; Perdiz, D.; Quignon, F.; Mouna, L.; Poüs, C. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy 2021, 17, 1296–1315. [Google Scholar] [CrossRef] [PubMed]
- Glon, D.; Vilmen, G.; Perdiz, D.; Hernandez, E.; Beauclair, G.; Quignon, F.; Berlioz-Torrent, C.; Maréchal, V.; Poüs, C.; Lussignol, M. Essential role of hyperacetylated microtubules in innate immunity escape orchestrated by the EBV-encoded BHRF1 protein. PLoS Pathog. 2022, 18, e1010371. [Google Scholar] [CrossRef]
- Granato, M.; Santarelli, R.; Farina, A.; Gonnella, R.; Lotti, L.V.; Faggioni, A.; Cirone, M. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 2014, 88, 12715–12726. [Google Scholar] [CrossRef] [PubMed]
- Cirone, M. EBV and KSHV infection dysregulates autophagy to optimize viral replication, prevent immune recognition and promote tumorigenesis. Viruses 2018, 10, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellows, D.S.; Howell, M.; Pearson, C.; Hazlewood, S.A.; Hardwick, J.M. Epstein-Barr virus BALF1 is a BCL-2-like antagonist of the herpesvirus antiapoptotic BCL-2 proteins. J. Virol. 2002, 76, 2469–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabras, G.; Decaussin, G.; Zeng, Y.; Djennaoui, D.; Melouli, H.; Broully, P.; Bouguermouh, A.; Ooka, T. Epstein–Barr virus encoded BALF1 gene is transcribed in Burkitt’s lymphoma cell lines and in nasopharyngeal carcinoma’s biopsies. J. Clin. Virol. 2005, 34, 26–34. [Google Scholar] [CrossRef]
- Hsu, W.-L.; Chung, P.-J.; Tsai, M.-H.; Chang, C.L.-T.; Liang, C.-L. A role for Epstein–Barr viral BALF1 in facilitating tumor formation and metastasis potential. Virus Res. 2012, 163, 617–627. [Google Scholar] [CrossRef]
- Shao, Z.; Borde, C.; Quignon, F.; Escargueil, A.; Maréchal, V. Epstein–Barr Virus BALF0 and BALF1 Modulate Autophagy. Viruses 2019, 11, 1099. [Google Scholar] [CrossRef] [Green Version]
- Münz, C. Autophagy beyond intracellular MHC class II antigen presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef]
- Arias-Calvachi, C.; Blanco, R.; Calaf, G.M.; Aguayo, F. Epstein–Barr Virus Association with Breast Cancer: Evidence and Perspectives. Biology 2022, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yao, Y.; Chen, H.; Zhang, S.; Cao, S.-M.; Zhang, Z.; Luo, B.; Liu, Z.; Li, Z.; Xiang, T. Genome sequencing analysis identifies Epstein–Barr virus subtypes associated with high risk of nasopharyngeal carcinoma. Nat. Genet. 2019, 51, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.H.; Lo, K.W.; Au, F.W.; Huang, D.P.; To, K.-F. Re: Discrete alterations in the BZLF1 promoter in tumor and non-tumor-associated Epstein–Barr virus. J. Natl. Cancer Inst. 2003, 95, 1008–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef] [PubMed]
- Dheekollu, J.; Malecka, K.; Wiedmer, A.; Delecluse, H.-J.; Chiang, A.K.; Altieri, D.C.; Messick, T.E.; Lieberman, P.M. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr virus episomal latency. Oncotarget 2017, 8, 7248. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-H.; Raykova, A.; Klinke, O.; Bernhardt, K.; Gärtner, K.; Leung, C.S.; Geletneky, K.; Sertel, S.; Münz, C.; Feederle, R. Spontaneous lytic replication and epitheliotropism define an Epstein-Barr virus strain found in carcinomas. Cell Rep. 2013, 5, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Y.; Zhang, L.; Wu, Y.; Huang, Y.; Huang, N.; Li, J.; Wang, Y.; Jiang, M.; Fang, Z.; Meng, N. Prospective studies on nasopharyngeal carcinoma in epstein-barr virus IgA/VCA antibody-positne persons in Wuzhou city, china. Int. J. Cancer 1985, 36, 545–547. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene Name | Kinetics | Lytic Function | Cancer Types | References |
|---|---|---|---|---|
| BZLF1 | IE | Transactivator | GC, NPC, COAD, BL, DLBCL, PTCL | [6,7,8,9,10] |
| BRLF1 | IE | Transactivator | GC, NPC, DLBCL | [7,8,10,11,12] |
| BORF2 | E | Ribonucleotide reductase large subunit | BL, ENKTCL | [13,14] |
| BSLF1 | E | Primase | GC, DLBCL | [7,15] |
| BSLF2/BMLF1 | E | mRNA export factor ICP27 homolog | PTCL, DLBCL, AITL | [6,15,16] |
| BALF1 | E | vBcl-2 | GC, NPC | [8,11,15,17] |
| BALF2 | E | Single-stranded DNA-binding protein | GC, NPC, COAD, BL, DLBCL, ENKTCL | [7,8,9,11,14,15,17,18] |
| BALF3 | E | Terminase large subunit | GC, NPC, BL, DLBCL, AITL, ENKTCL | [11,12,14,15,16,17,18] |
| BHLF1 | E | Involved in viral DNA synthesis | AITL, BL, DLBCL, | [14,15,16] |
| BHRF1 | E | vBcl-2 | GC, NPC, BL, DLBCL | [7,9,10,12,14,15] |
| BMRF1 | E | DNA polymerase processivity factor | GC, COAD, BL, DLBCL, ENKTCL | [7,9,13,14,15] |
| BALF5 | E | DNA polymerase catalytic subunit | GC, NPC, BL, DLBCL, ENKTCL | [7,8,11,12,14,15,18] |
| BARF1 | E | Soluble decoy for CSF-1 | GC, ENKTCL | [8,10,11,13] |
| BBLF4 | E | Helicase | NPC, GC | [7,12] |
| BILF1 | E | gp64, vGPCR | GC, NPC, BL, DLBCL, ENKTCL, | [9,11,12,15,17,18] |
| BNLF2a | E | Inhibtor of TAP | GC, NPC, BL, DLBCL, PTCL, AITL, ENKTCL | [6,9,11,13,15,16,17] |
| BNLF2b | E | Not reported | GC, NPC, DLBCL, PTCL, ENKTCL | [6,11,13,17,18] |
| LF3 | E | Involves in viral DNA synthesis | GC, NPC, BL, AITL | [11,14,16,17] |
| BMRF2 | E/L | Membrane proteins | BL, DLBCL, ENKTCL | [13,14,15] |
| BCRF1 | L | vIL-10 | GC, NPC, AITL, BL, | [7,9,10,12,16] |
| BALF4 | L | Envelope glycoprotein B | GC, NPC, BL, DLBCL, ENKTCL | [7,8,9,11,12,14,15,17,18] |
| BKRF2 | L | gL, gp25 | BL, DLBCL | [14,15] |
| BLLF1 | L | gp350/220 | GC | [7,10] |
| BNRF1 | L | Major tegument protein | GC, NPC, BL, AITL, ENKTCL | [8,11,12,13,14,16,17,18] |
| BCLF1 | L | Major Capsid Protein | NPC, GC | [7,12] |
| LF1 | Unknown | Not reported | GC, NPC, DLBCL | [8,11,12,15,17,19] |
| LF2 | Unknown | Protein that binds Rta | GC, NPC, BL, DLBCL | [8,9,11,12,14,15,17] |
| Gene Name | Samples | Assays | References |
|---|---|---|---|
| BZLF1 | Biopsy, FNA, cell line | IHC, ICC, WB | [31,35,36,37] |
| BRLF1 | Plasma, cell line | ELISA, WB, IF | [37,38,39,40] |
| BORF2 | Plasma | ELISA | [38] |
| BSLF1 | Cell line | WB | [41] |
| BSLF2/BMLF1 | Plasma, cell line | Protein array, WB | [38,42,43] |
| BALF2 | Biopsy, plasma | IHC, ELISA | [38,44] |
| BHLF1 | Cell line | IF | [45] |
| BHRF1 | Biopsy, plasma | IHC, WB, protein array | [38,46,47] |
| BMRF1 | Biopsy, serum, saliva, cell line | IHC, ELISA, WB | [37,48,49,50] |
| BALF5 | Plasma, cell line | Protein array, WB | [38,51] |
| BARF1 | Biopsy | WB | [52] |
| BBLF4 | Cell line | WB | [41] |
| BNLF2a | Cell line | WB, IF | [53,54] |
| LF3 | FNA, biopsy | ICC, IHC | [31,36] |
| BMRF2 | Cell line | WB, IF | [55] |
| BCRF1 | Allograft, serum | IHC, ELISA | [56,57] |
| BALF4 | Plasma, serum | Protein array, ELISA | [38,58] |
| BKRF2 | Cell line | IP | [59] |
| BLLF1 | Serum, cell line | IF, WB | [60,61,62] |
| BNRF1 | Cell line | WB | [63] |
| LF2 | Plasma | ELISA | [38] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yap, L.F.; Wong, A.K.C.; Paterson, I.C.; Young, L.S. Functional Implications of Epstein-Barr Virus Lytic Genes in Carcinogenesis. Cancers 2022, 14, 5780. https://doi.org/10.3390/cancers14235780
Yap LF, Wong AKC, Paterson IC, Young LS. Functional Implications of Epstein-Barr Virus Lytic Genes in Carcinogenesis. Cancers. 2022; 14(23):5780. https://doi.org/10.3390/cancers14235780
Chicago/Turabian StyleYap, Lee Fah, Anna Kang Chee Wong, Ian C. Paterson, and Lawrence S. Young. 2022. "Functional Implications of Epstein-Barr Virus Lytic Genes in Carcinogenesis" Cancers 14, no. 23: 5780. https://doi.org/10.3390/cancers14235780
APA StyleYap, L. F., Wong, A. K. C., Paterson, I. C., & Young, L. S. (2022). Functional Implications of Epstein-Barr Virus Lytic Genes in Carcinogenesis. Cancers, 14(23), 5780. https://doi.org/10.3390/cancers14235780