
Contribution of Epstein–Barr Virus Lytic Proteins to Cancer Hallmarks and Implications from Other Oncoviruses


Abstract
:Simple Summary
Abstract
1. Introduction
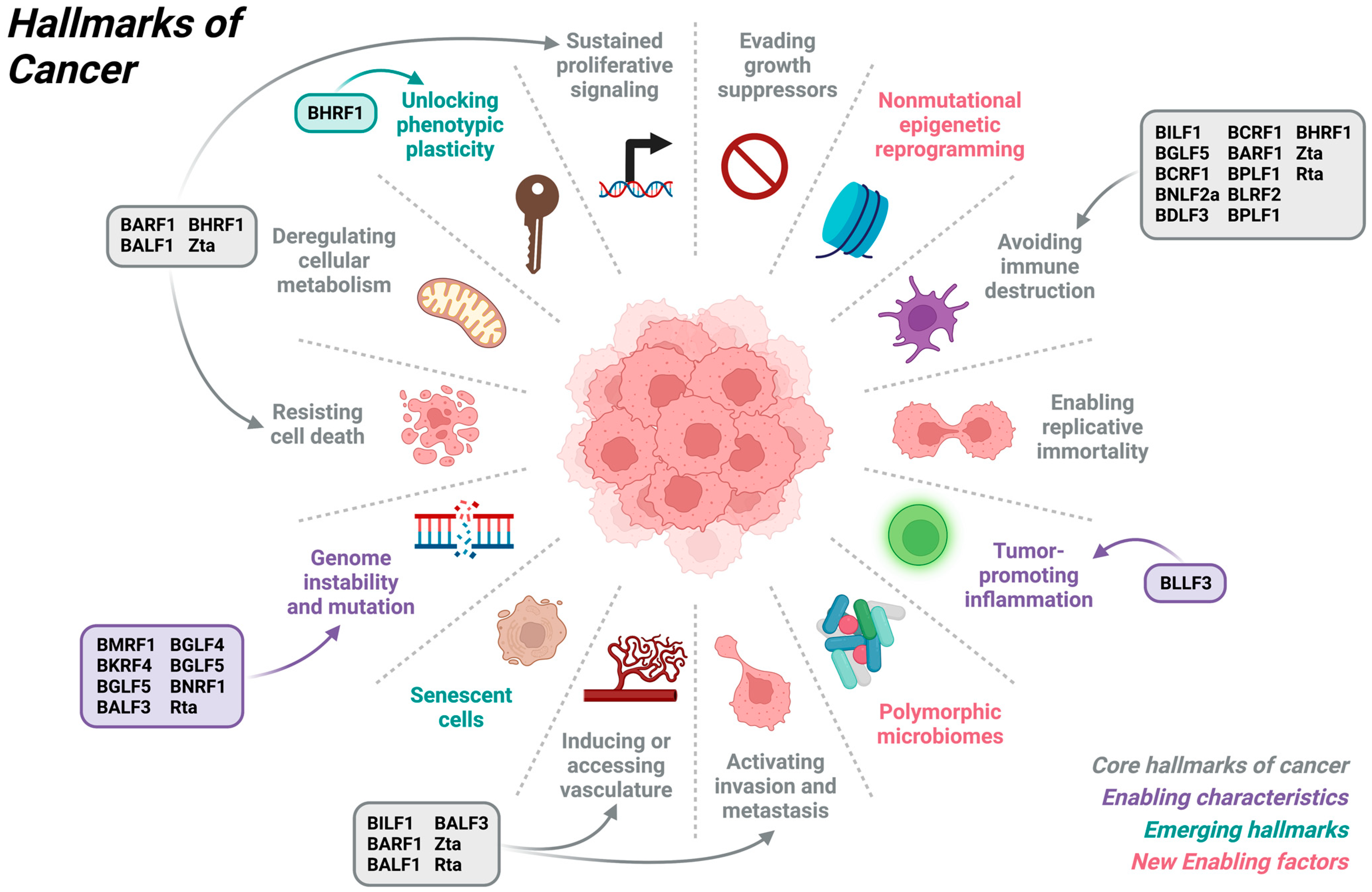
2. Overview of Cancer Hallmarks
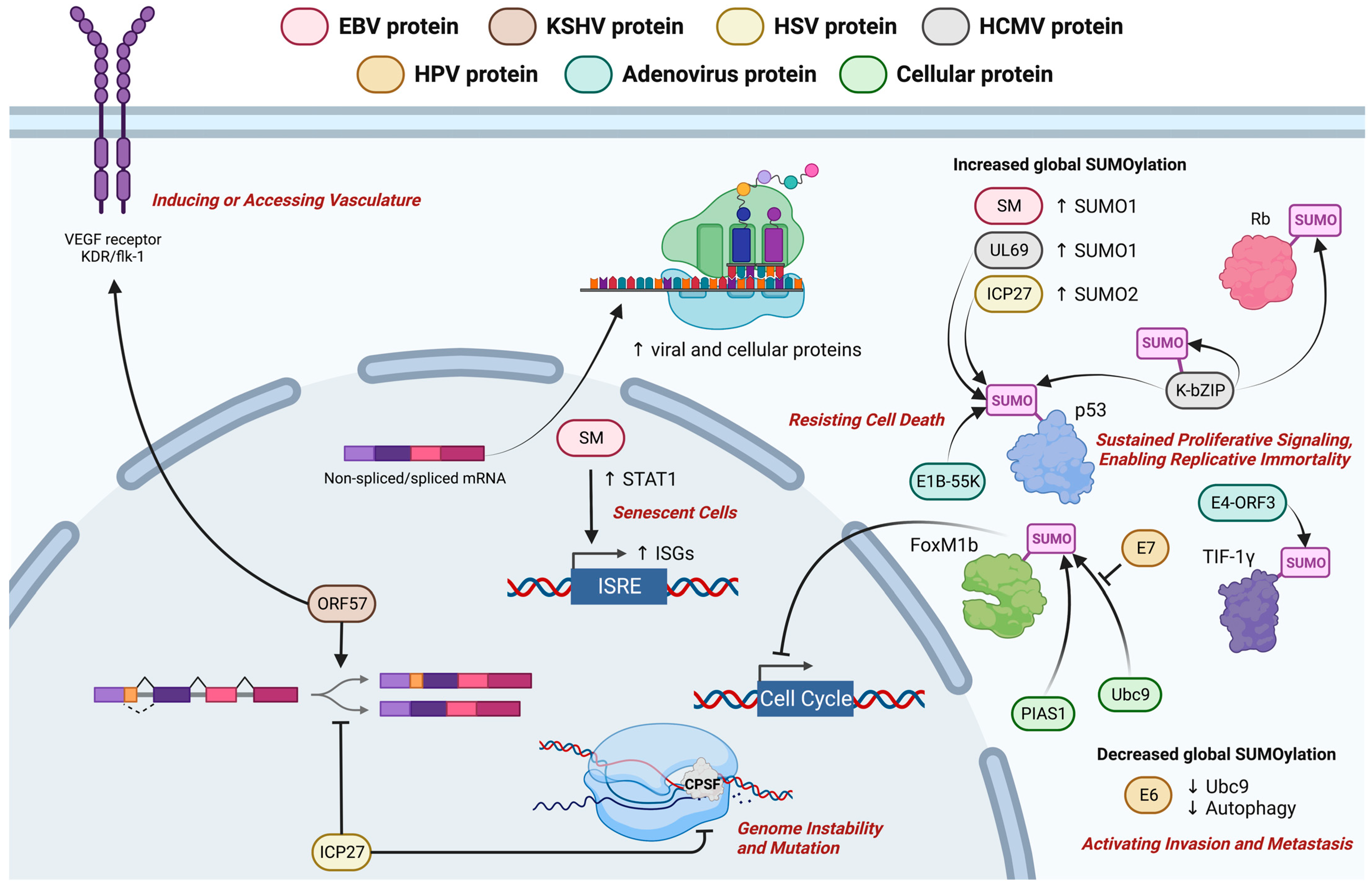
3. Contribution of EBV to Hallmarks of Cancers
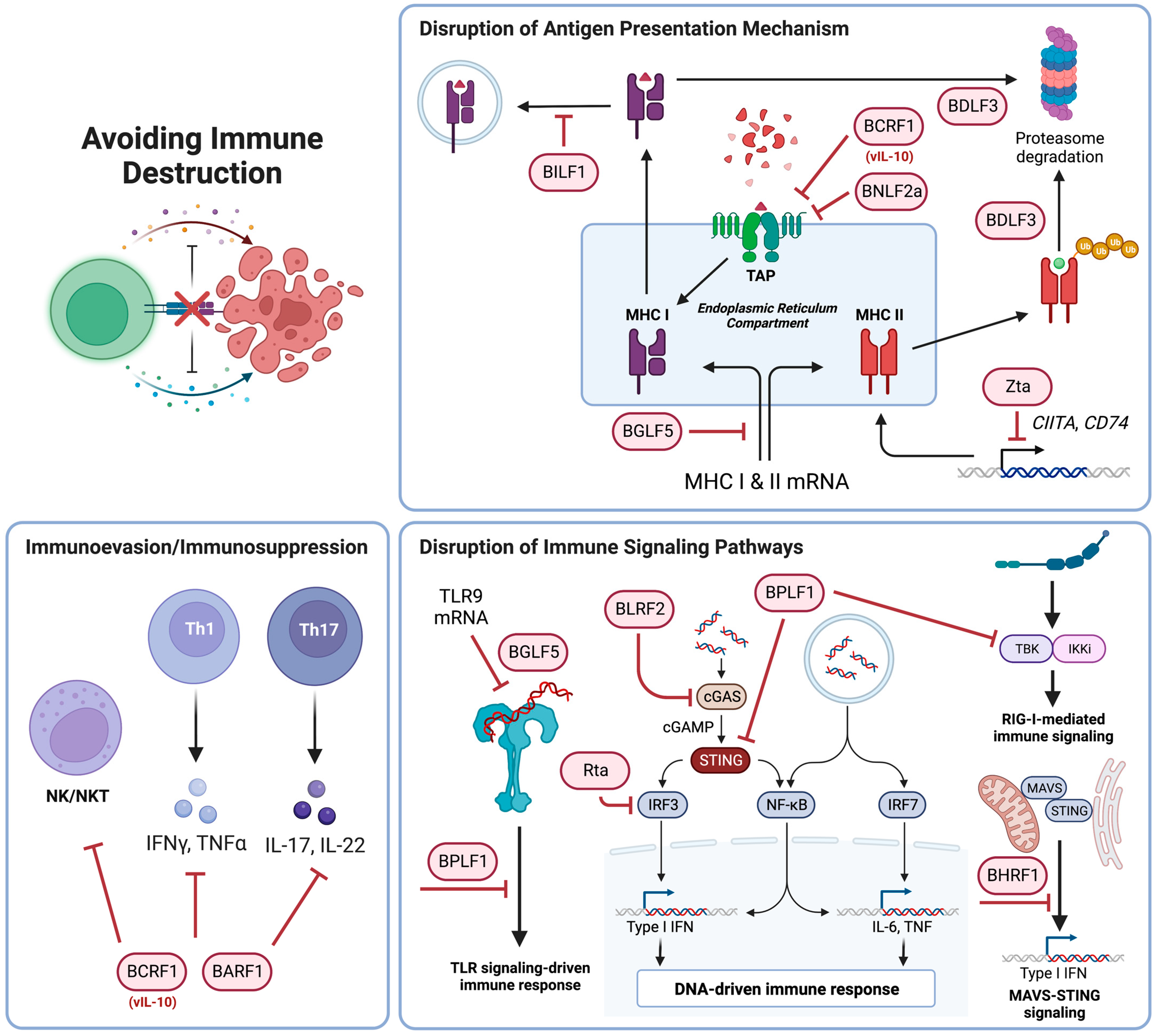
3.1. Avoiding Immune Destruction
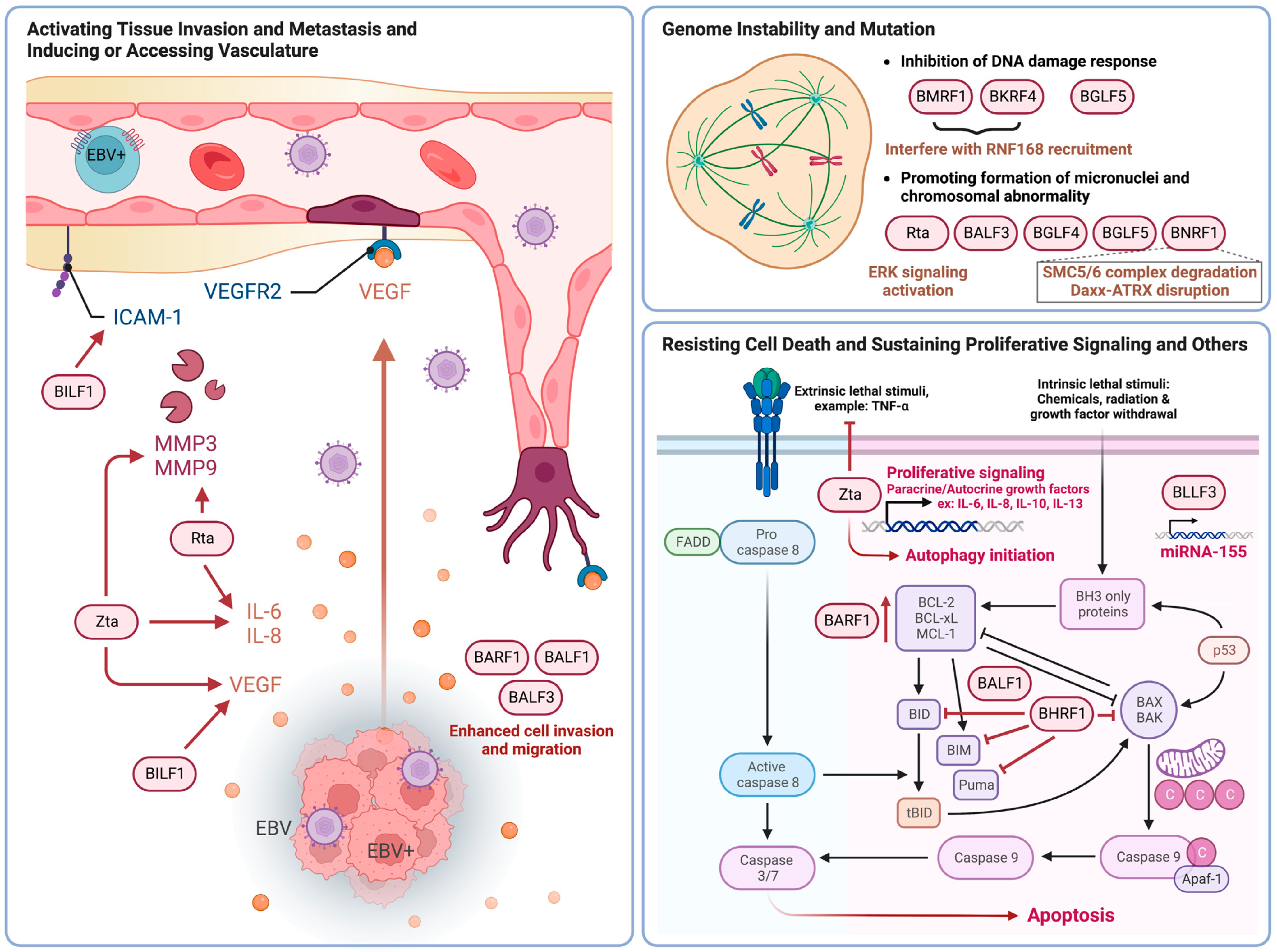
3.2. Activating Tissue Invasion and Metastasis and Inducing or Accessing Vasculature
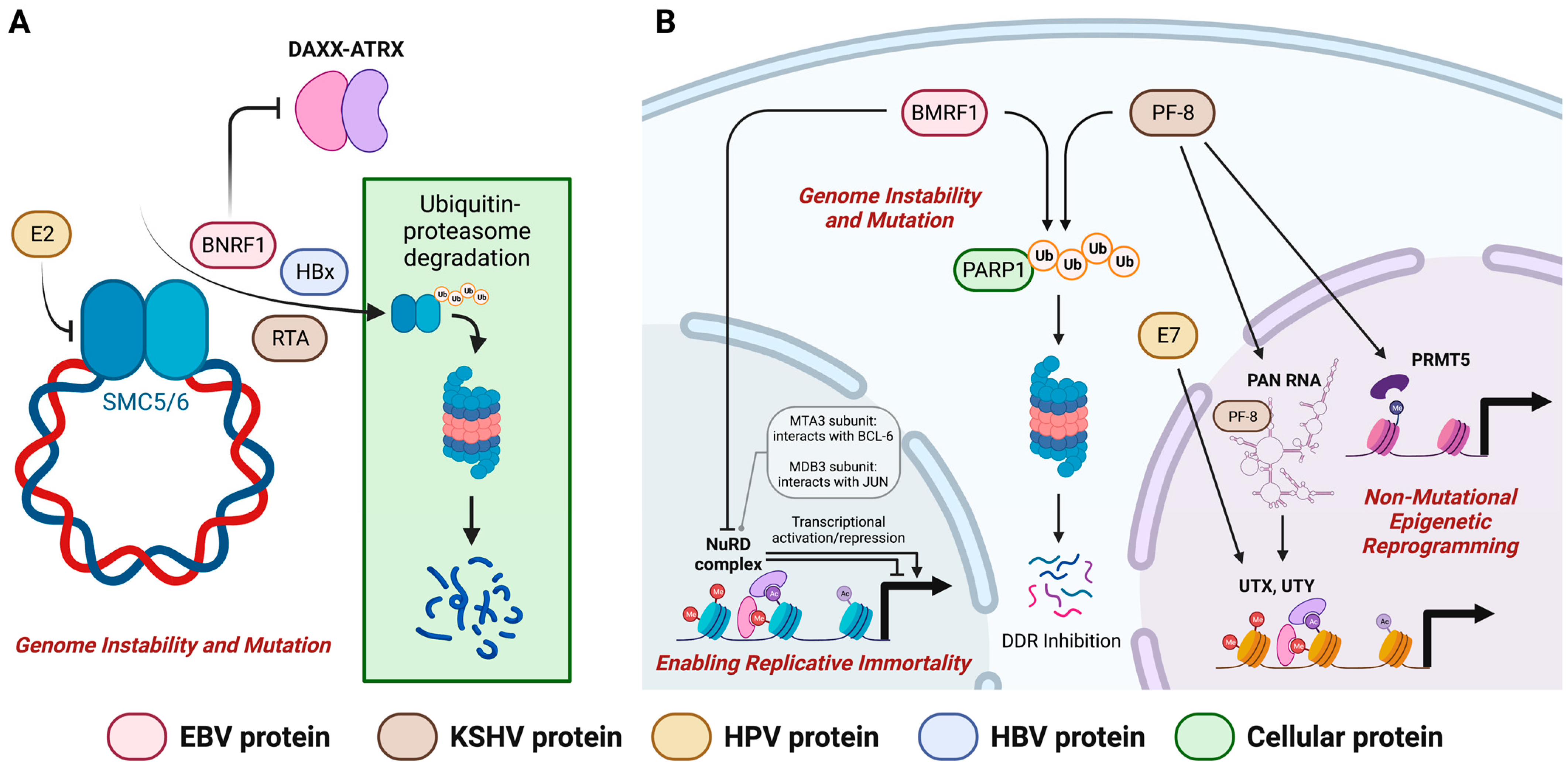
3.3. Genome Instability and Mutation
3.4. Resisting Cell Death, Sustaining Proliferative Signaling and Other Cancer Hallmarks
4. Postulated Oncogenic Roles of EBV Lytic Proteins
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Epstein, M.; Achong, B.; Barr, Y. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Yan, B.; Wang, C.; Wang, L.; Quaid, J.T.; Lin, C.F.; Briggs, S.D.; Majumder, J.; Canaria, D.A.; Chauss, D.; et al. Integrated Pan-Cancer Map of EBV-Associated Neoplasms Reveals Functional Host–Virus Interactions. Cancer Res. 2019, 79, 6010–6023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein–Barr Virus Epidemiology, Serology, and Genetic Variability of LMP-1 Oncogene Among Healthy Population: An Update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 2005, 15, 3–15. [Google Scholar] [CrossRef]
- Hui, K.F.; Yiu, S.P.T.; Tam, K.P.; Chiang, A.K.S. Viral-Targeted Strategies Against EBV-Associated Lymphoproliferative Diseases. Front. Oncol. 2019, 9, 81. [Google Scholar] [CrossRef]
- Young, L.S.; Dawson, C.W.; Eliopoulos, A.G. The expression and function of Epstein-Barr virus encoded latent genes. Mol. Pathol. 2000, 53, 238–247. [Google Scholar] [CrossRef] [Green Version]
- Kempkes, B.; Robertson, E.S. Epstein-Barr virus latency: Current and future perspectives. Curr. Opin. Virol. 2015, 14, 138–144. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.-S.; Kieff, E. Epstein–Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- El-Sharkawy, A.; Al Zaidan, L.; Malki, A. Epstein–Barr Virus-Associated Malignancies: Roles of Viral Oncoproteins in Carcinogenesis. Front. Oncol. 2018, 8, 265. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, W.; Sugden, B. Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 1988, 55, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.; Ho, D.N.; Tsang, C.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [Green Version]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Sato, Y.; Ito, J.; Takaki, M.; Okuno, Y.; Yaguchi, M.; Al Masud, H.M.A.; Watanabe, T.; Sato, K.; Iwami, S.; et al. Direct Evidence of Abortive Lytic Infection-Mediated Establishment of Epstein-Barr Virus Latency During B-Cell Infection. Front. Microbiol. 2020, 11, 575255. [Google Scholar] [CrossRef]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef] [Green Version]
- Traylen, C.; Ramasubramanyan, S.; Zuo, J.; Rowe, M.; Almohammad, R.; Heesom, K.; Sweet, S.M.M.; Matthews, D.A.; Sinclair, A.J. Identification of Epstein-Barr Virus Replication Proteins in Burkitt’s Lymphoma Cells. Pathogens 2015, 4, 739–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ersing, I.; Nobre, L.; Wang, L.W.; Soday, L.; Ma, Y.; Paulo, J.A.; Narita, Y.; Ashbaugh, C.W.; Jiang, C.; Grayson, N.E.; et al. A Temporal Proteomic Map of Epstein-Barr Virus Lytic Replication in B Cells. Cell Rep. 2017, 19, 1479–1493. [Google Scholar] [CrossRef] [Green Version]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of Epstein-Barr Virus Genomes and Expression Profiles in Gastric Adenocarcinoma. J. Virol. 2018, 92, e01239-17. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Sato, Y.; Kimura, H. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev. Med. Virol. 2014, 24, 242–253. [Google Scholar] [CrossRef]
- Ma, S.-D.; Yu, X.; Mertz, J.E.; Gumperz, J.E.; Reinheim, E.; Zhou, Y.; Tang, W.; Burlingham, W.J.; Gulley, M.L.; Kenney, S.C. An Epstein-Barr Virus (EBV) Mutant with Enhanced BZLF1 Expression Causes Lymphomas with Abortive Lytic EBV Infection in a Humanized Mouse Model. J. Virol. 2012, 86, 7976–7987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.; Iwakiri, D.; Yamamoto, K.; Maruo, S.; Kanda, T.; Takada, K. Epstein-Barr Virus BZLF1 Gene, a Switch from Latency to Lytic Infection, Is Expressed as an Immediate-Early Gene after Primary Infection of B Lymphocytes. J. Virol. 2007, 81, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.-J.; Rickinson, A.B.; Rowe, M. Features Distinguishing Epstein-Barr Virus Infections of Epithelial Cells and B Cells: Viral Genome Expression, Genome Maintenance, and Genome Amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.M.; Zhang, G.; Seto, E.; Takada, K.; Deng, W.; Yip, Y.L.; Man, C.; Hau, P.M.; Chen, H.; Cao, Y.; et al. Epstein-Barr virus infection in immortalized nasopharyngeal epithelial cells: Regulation of infection and phenotypic characterization. Int. J. Cancer 2010, 127, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A New Model of Epstein-Barr Virus Infection Reveals an Important Role for Early Lytic Viral Protein Expression in the Development of Lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Katsumura, K.R.; Maruo, S.; Takada, K. EBV lytic infection enhances transformation of B-lymphocytes infected with EBV in the presence of T-lymphocytes. J. Med. Virol. 2012, 84, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Morales-Sánchez, A.; Fuentes-Panana, E.M. The Immunomodulatory Capacity of an Epstein-Barr Virus Abortive Lytic Cycle: Potential Contribution to Viral Tumorigenesis. Cancers 2018, 10, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef]
- Münz, C. Tumor Microenvironment Conditioning by Abortive Lytic Replication of Oncogenic γ-Herpesviruses. Adv. Exp. Med. Biol. 2020, 1225, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, M.; Longnecker, R. Latent Membrane Protein 2A Inhibits Transforming Growth Factor-β1-Induced Apoptosis through the Phosphatidylinositol 3-Kinase/Akt Pathway. J. Virol. 2004, 78, 1697–1705. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.-M.; Kim, Y.S.; Hur, D.Y. LMP1 and 2A Induce the Expression of Nrf2 Through Akt Signaling Pathway in Epstein-Barr Virus–Transformed B Cells. Transl. Oncol. 2019, 12, 775–783. [Google Scholar] [CrossRef]
- Hong, Y.-K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma–associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [Green Version]
- Charette, S.T.; McCance, D.J. The E7 protein from human papillomavirus type 16 enhances keratinocyte migration in an Akt-dependent manner. Oncogene 2007, 26, 7386–7390. [Google Scholar] [CrossRef] [Green Version]
- Olagnier, D.; Sze, A.; Hadj, S.B.; Chiang, C.; Steel, C.; Han, X.; Routy, J.-P.; Lin, R.; Hiscott, J.; Van Grevenynghe, J. HTLV-1 Tax-Mediated Inhibition of FOXO3a Activity Is Critical for the Persistence of Terminally Differentiated CD4+ T Cells. PLoS Pathog. 2014, 10, e1004575. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qian, L.; Chen, C.; Shi, M.; Yu, M.; Hu, M.; Song, L.; Shen, B.; Guo, N. Down-Regulation of MHC Class II Expression through Inhibition of CIITA Transcription by Lytic Transactivator Zta during Epstein-Barr Virus Reactivation. J. Immunol. 2009, 182, 1799–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, J.; Thomas, W.A.; Haigh, T.A.; Fitzsimmons, L.; Long, H.M.; Hislop, A.D.; Taylor, G.S.; Rowe, M. Epstein-Barr Virus Evades CD4+ T Cell Responses in Lytic Cycle through BZLF1-mediated Downregulation of CD74 and the Cooperation of vBcl-2. PLoS Pathog. 2011, 7, e1002455. [Google Scholar] [CrossRef] [Green Version]
- Bentz, G.L.; Liu, R.; Hahn, A.M.; Shackelford, J.; Pagano, J.S. Epstein–Barr virus BRLF1 inhibits transcription of IRF3 and IRF7 and suppresses induction of interferon-β. Virology 2010, 402, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Vilmen, G.; Glon, D.; Siracusano, G.; Lussignol, M.; Shao, Z.; Hernandez, E.; Perdiz, D.; Quignon, F.; Mouna, L.; Poüs, C.; et al. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy 2021, 17, 1296–1315. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.H.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.H.J.; Ressing, M.E.; Rowe, M. The DNase of Gammaherpesviruses Impairs Recognition by Virus-Specific CD8 + T Cells through an Additional Host Shutoff Function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef] [Green Version]
- van Gent, M.; Griffin, B.D.; Berkhoff, E.G.; van Leeuwen, D.; Boer, I.G.J.; Buisson, M.; Hartgers, F.C.; Burmeister, W.P.; Wiertz, E.J.; Ressing, M.E. EBV Lytic-Phase Protein BGLF5 Contributes to TLR9 Downregulation during Productive Infection. J. Immunol. 2011, 186, 1694–1702. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.H.J.; Rowe, M. The Epstein-Barr Virus G-Protein-Coupled Receptor Contributes to Immune Evasion by Targeting MHC Class I Molecules for Degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Quinn, L.L.; Tamblyn, J.; Thomas, W.A.; Feederle, R.; Delecluse, H.-J.; Hislop, A.D.; Rowe, M. The Epstein-Barr Virus-Encoded BILF1 Protein Modulates Immune Recognition of Endogenously Processed Antigen by Targeting Major Histocompatibility Complex Class I Molecules Trafficking on both the Exocytic and Endocytic Pathways. J. Virol. 2011, 85, 1604–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, B.D.; Gram, A.M.; Mulder, A.; Van Leeuwen, D.; Claas, F.H.J.; Wang, F.; Ressing, M.E.; Wiertz, E. EBV BILF1 Evolved To Downregulate Cell Surface Display of a Wide Range of HLA Class I Molecules through Their Cytoplasmic Tail. J. Immunol. 2013, 190, 1672–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hislop, A.D.; Ressing, M.E.; van Leeuwen, D.; Pudney, V.A.; Horst, D.; Koppers-Lalic, D.; Croft, N.P.; Neefjes, J.J.; Rickinson, A.B.; Wiertz, E.J. A CD8+ T cell immune evasion protein specific to Epstein-Barr virus and its close relatives in Old World primates. J. Exp. Med. 2007, 204, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Quinn, L.L.; Williams, L.R.; White, C.; Forrest, C.; Zuo, J.; Rowe, M. The Missing Link in Epstein-Barr Virus Immune Evasion: The BDLF3 Gene Induces Ubiquitination and Downregulation of Major Histocompatibility Complex Class I (MHC-I) and MHC-II. J. Virol. 2016, 90, 356–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.W.; Vieira, P.; Fiorentino, D.F.; Trounstine, M.L.; Khan, T.A.; Mosmann, T.R. Homology of Cytokine Synthesis Inhibitory Factor (IL-10) to the Epstein-Barr Virus Gene BCRFI. Science 1990, 248, 1230–1234. [Google Scholar] [CrossRef]
- Vieira, P.; de Waal-Malefyt, R.; Dang, M.N.; E Johnson, K.; Kastelein, R.; Fiorentino, D.F.; E Devries, J.; Roncarolo, M.G.; Mosmann, T.R.; Moore, K.W. Isolation and expression of human cytokine synthesis inhibitory factor cDNA clones: Homology to Epstein-Barr virus open reading frame BCRFI. Proc. Natl. Acad. Sci. USA 1991, 88, 1172–1176. [Google Scholar] [CrossRef] [Green Version]
- Zeidler, R.; Eissner, G.; Meissner, P.; Uebel, S.; Tampe, R.; Lazis, S.; Hammerschmidt, W. Downregulation of TAP1 in B lymphocytes by cellular and Epstein-Barr virus-encoded interleukin-10. Blood 1997, 90, 2390–2397. [Google Scholar] [CrossRef]
- Slobedman, B.; Barry, P.A.; Spencer, J.V.; Avdic, S.; Abendroth, A. Virus-Encoded Homologs of Cellular Interleukin-10 and Their Control of Host Immune Function. J. Virol. 2009, 83, 9618–9629. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, M.T.; Masucci, M.G. Interleukin-10 abrogates the inhibition of Epstein-Barr virus-induced B-cell transformation by memory T-cell responses. Blood 1998, 92, 4256–4262. [Google Scholar] [CrossRef]
- Lo, A.K.-F.; Dawson, C.W.; Lung, H.L.; Wong, K.L.; Young, L.S. The Therapeutic Potential of Targeting BARF1 in EBV-Associated Malignancies. Cancers 2020, 12, 1940. [Google Scholar] [CrossRef]
- Strockbine, L.D.; Cohen, J.I.; Farrah, T.; Lyman, S.D.; Wagener, F.; DuBose, R.F.; Armitage, R.J.; Spriggs, M.K. The Epstein-Barr Virus BARF1 Gene Encodes a Novel, Soluble Colony-Stimulating Factor-1 Receptor. J. Virol. 1998, 72, 4015–4021. [Google Scholar] [CrossRef] [Green Version]
- van Gent, M.; Braem, S.G.E.; De Jong, A.; Delagic, N.; Peeters, J.; Boer, I.G.J.; Moynagh, P.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-Barr Virus Large Tegument Protein BPLF1 Contributes to Innate Immune Evasion through Interference with Toll-Like Receptor Signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-J.; Li, W.; Shao, Y.; Avey, D.; Fu, B.; Gillen, J.; Hand, T.; Ma, S.; Liu, X.; Miley, W.; et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 2015, 18, 333–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Liu, C.; Zhou, S.; Li, Q.; Feng, Y.; Sun, P.; Feng, H.; Gao, Y.; Zhu, J.; Luo, X.; et al. Viral tegument proteins restrict cGAS-DNA phase separation to mediate immune evasion. Mol. Cell 2021, 81, 2823–2837 e2829. [Google Scholar] [CrossRef]
- Lui, W.-Y.; Bharti, A.; Wong, N.-H.M.; Jangra, S.; Botelho, M.G.; Yuen, K.-S.; Jin, D.-Y. Suppression of cGAS- and RIG-I-mediated innate immune signaling by Epstein-Barr virus deubiquitinase BPLF1. PLoS Pathog. 2023, 19, e1011186. [Google Scholar] [CrossRef]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic Properties of the EBV ZEBRA Protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef] [PubMed]
- Hong, G.K.; Kumar, P.; Wang, L.; Damania, B.; Gulley, M.L.; Delecluse, H.-J.; Polverini, P.J.; Kenney, S.C. Epstein-Barr Virus Lytic Infection Is Required for Efficient Production of the Angiogenesis Factor Vascular Endothelial Growth Factor in Lymphoblastoid Cell Lines. J. Virol. 2005, 79, 13984–13992. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.-Y.; Yeh, T.-H.; Lin, W.-H.; Wu, S.-Y.; Lai, H.-C.; Chang, F.-H.; Takada, K.; Chang, Y. Epstein-Barr Virus Zta Upregulates Matrix Metalloproteinases 3 and 9 That Synergistically Promote Cell Invasion In Vitro. PLoS ONE 2013, 8, e56121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizaki, T.; Sato, H.; Murono, S.; Pagano, J.S.; Furukawa, M. Matrix metalloproteinase 9 is induced by the Epstein–Barr virus BZLF1 transactivator. Clin. Exp. Metastasis 1999, 17, 431–436. [Google Scholar] [CrossRef]
- Hong, G.K.; Gulley, M.L.; Feng, W.-H.; Delecluse, H.-J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr Virus Lytic Infection Contributes to Lymphoproliferative Disease in a SCID Mouse Model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Tsang, C.M.; Deng, W.; Yip, Y.L.; Lui, V.W.-Y.; Wong, S.C.C.; Cheung, A.L.-M.; Hau, P.M.; Zeng, M.; Lung, M.L.; et al. Enhanced IL-6/IL-6R Signaling Promotes Growth and Malignant Properties in EBV-Infected Premalignant and Cancerous Nasopharyngeal Epithelial Cells. PLoS ONE 2013, 8, e62284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.-Y.; Chang, F.-H.; Tsai, J.-H.; Chang, Y. Epstein-Barr virus Rta promotes invasion of bystander tumor cells through paracrine of matrix metalloproteinase 9. Biochem. Biophys. Res. Commun. 2018, 503, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- Lyngaa, R.; Nørregaard, K.; Kristensen, M.; Kubale, V.; Rosenkilde, M.M.; Kledal, T.N. Cell transformation mediated by the Epstein–Barr virus G protein-coupled receptor BILF1 is dependent on constitutive signaling. Oncogene 2010, 29, 4388–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.; Gao, J.; Cheng, L.; Yang, X.; Li, F.; Jiang, G. The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 upregulates ICAM-1 through a mechanism involving the NF-қB pathway. Biosci. Biotechnol. Biochem. 2020, 84, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, E.K.; Le Large, T.Y.S.; Greijer, A.E.; Middeldorp, J.M. BamHI-A rightward frame 1, an Epstein–Barr virus-encoded oncogene and immune modulator. Rev. Med. Virol. 2013, 23, 367–383. [Google Scholar] [CrossRef] [Green Version]
- Hsu, W.-L.; Chung, P.-J.; Tsai, M.-H.; Chang, C.L.-T.; Liang, C.-L. A role for Epstein–Barr viral BALF1 in facilitating tumor formation and metastasis potential. Virus Res. 2012, 163, 617–627. [Google Scholar] [CrossRef]
- Chiu, S.-H.; Wu, C.-C.; Fang, C.-Y.; Yu, S.-L.; Hsu, H.-Y.; Chow, Y.-H.; Chen, J.-Y. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget 2014, 5, 8583–8601. [Google Scholar] [CrossRef] [Green Version]
- Murayama, K.; Nakayama, S.; Kato-Murayama, M.; Akasaka, R.; Ohbayashi, N.; Kamewari-Hayami, Y.; Terada, T.; Shirouzu, M.; Tsurumi, T.; Yokoyama, S. Crystal Structure of Epstein-Barr Virus DNA Polymerase Processivity Factor BMRF1. J. Biol. Chem. 2009, 284, 35896–35905. [Google Scholar] [CrossRef] [Green Version]
- Zhanga, Q.; Guthriea, E.H.; Gea, J.Q.; Dorskyb, D.; Kenney, S. The Epstein–Barr Virus (EBV) DNA Polymerase Accessory Protein, BMRF1, Activates the Essential Downstream Component of the EBV oriLyt. Virology 1997, 230, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Su, M.-T.; Wang, Y.-T.; Chen, Y.-J.; Lin, S.-F.; Tsai, C.-H.; Chen, M.-R. The SWI/SNF Chromatin Regulator BRG1 Modulates the Transcriptional Regulatory Activity of the Epstein-Barr Virus DNA Polymerase Processivity Factor BMRF1. J. Virol. 2017, 91, e02114-16. [Google Scholar] [CrossRef] [Green Version]
- Salamun, S.G.; Sitz, J.; De La Cruz-Herrera, C.F.; Yockteng-Melgar, J.; Marcon, E.; Greenblatt, J.; Fradet-Turcotte, A.; Frappier, L. The Epstein-Barr Virus BMRF1 Protein Activates Transcription and Inhibits the DNA Damage Response by Binding NuRD. J. Virol. 2019, 93, e01070-19. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.-H.; Sitz, J.; Shen, Q.; Leblanc-Lacroix, A.; Campos, E.I.; Borozan, I.; Marcon, E.; Greenblatt, J.; Fradet-Turcotte, A.; Jin, D.-Y.; et al. A Screen for Epstein-Barr Virus Proteins That Inhibit the DNA Damage Response Reveals a Novel Histone Binding Protein. J. Virol. 2018, 92, e00262-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.-H.; Lee, C.-P.; Su, M.-T.; Wang, J.-T.; Chen, J.-Y.; Lin, S.-F.; Tsai, C.-H.; Hsieh, M.-J.; Takada, K.; Chen, M.-R. Epstein-Barr Virus BGLF4 Kinase Retards Cellular S-Phase Progression and Induces Chromosomal Abnormality. PLoS ONE 2012, 7, e39217. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Liu, M.-T.; Chang, Y.-T.; Fang, C.-Y.; Chou, S.-P.; Liao, H.-W.; Kuo, K.-L.; Hsu, S.-L.; Chen, Y.-R.; Wang, P.-W.; et al. Epstein–Barr Virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2009, 38, 1932–1949. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Wu, C.-C.; Cheng, Y.-J.; Chou, S.-P.; Jiang, Y.-J.; Chu, K.-C.; Tsai, C.-H.; Lin, S.-F.; Chen, J.-Y. Epstein-Barr virus BRLF1 induces genomic instability and progressive malignancy in nasopharyngeal carcinoma cells. Oncotarget 2017, 8, 78948–78964. [Google Scholar] [CrossRef] [Green Version]
- Shumilov, A.; Tsai, M.-H.; Schlosser, Y.T.; Kratz, A.-S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein–Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [Green Version]
- Yiu, S.P.T.; Guo, R.; Zerbe, C.; Weekes, M.P.; Gewurz, B.E. Epstein-Barr virus BNRF1 destabilizes SMC5/6 cohesin complexes to evade its restriction of replication compartments. Cell Rep. 2022, 38, 110411. [Google Scholar] [CrossRef]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.-J.; Lieberman, P.M. EBV Tegument Protein BNRF1 Disrupts DAXX-ATRX to Activate Viral Early Gene Transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Decaussin, G.; Sumner, S.; Ooka, T. N-terminal domain of BARF1 gene encoded by Epstein-Barr virus is essential for malignant transformation of rodent fibroblasts and activation of BCL-2. Oncogene 2001, 20, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Tsao, S.; Ooka, T.; Nicholls, J.M.; Cheung, H.W.; Fu, S.; Wong, Y.; Wang, X. Anti-apoptotic role of BARF1 in gastric cancer cells. Cancer Lett. 2006, 238, 90–103. [Google Scholar] [CrossRef]
- Dawson, C.W.; Eliopoulos, A.G.; Dawson, J.; Young, L.S. BHRF1, a viral homologue of the Bcl-2 oncogene, disturbs epithelial cell differentiation. Oncogene 1995, 10, 69–77. [Google Scholar]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein-Barr Virus Encodes a Novel Homolog of the bcl-2 Oncogene That Inhibits Apoptosis and Associates with Bax and Bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar] [CrossRef] [Green Version]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr Virus Provides a New Paradigm: A Requirement for the Immediate Inhibition of Apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, L.; Cartlidge, R.; Chang, C.; Sejic, N.; Galbraith, L.C.A.; Suraweera, C.; Croom-Carter, D.; Dewson, G.; Tierney, R.J.; Bell, A.; et al. EBV BCL-2 homologue BHRF1 drives chemoresistance and lymphomagenesis by inhibiting multiple cellular pro-apoptotic proteins. Cell Death Differ. 2020, 27, 1554–1568. [Google Scholar] [CrossRef] [PubMed]
- Bellows, D.S.; Howell, M.; Pearson, C.; Hazlewood, S.A.; Hardwick, J.M. Epstein-Barr Virus BALF1 Is a BCL-2-Like Antagonist of the Herpesvirus Antiapoptotic BCL-2 Proteins. J. Virol. 2002, 76, 2469–2479. [Google Scholar] [CrossRef] [Green Version]
- Morrison, T.E.; Mauser, A.; Klingelhutz, A.; Kenney, S.C. Epstein-Barr Virus Immediate-Early Protein BZLF1 Inhibits Tumor Necrosis Factor Alpha-Induced Signaling and Apoptosis by Downregulating Tumor Necrosis Factor Receptor 1. J. Virol. 2004, 78, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Yiu, S.P.T.; Hui, K.F.; Münz, C.; Lo, K.-W.; Tsao, S.W.; Kao, R.Y.T.; Yang, D.; Chiang, A.K.S. Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma. Cancers 2019, 11, 1871. [Google Scholar] [CrossRef] [Green Version]
- Ramasubramanyan, S.; Osborn, K.; Al-Mohammad, R.; Perez-Fernandez, I.B.N.; Zuo, J.; Balan, N.; Godfrey, A.; Patel, H.; Peters, G.; Rowe, M.; et al. Epstein–Barr virus transcription factor Zta acts through distal regulatory elements to directly control cellular gene expression. Nucleic Acids Res. 2015, 43, 3563–3577. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; McBride, J.; Fewell, C.; Lacey, M.; Wang, X.; Lin, Z.; Cameron, J.; Flemington, E.K. MicroRNA-155 Is an Epstein-Barr Virus-Induced Gene That Modulates Epstein-Barr Virus-Regulated Gene Expression Pathways. J. Virol. 2008, 82, 5295–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnstaedt, S.D.; Gottwein, E.; Skalsky, R.L.; Luftig, M.A.; Cullen, B.R. Virally Induced Cellular MicroRNA miR-155 Plays a Key Role in B-Cell Immortalization by Epstein-Barr Virus. J. Virol. 2010, 84, 11670–11678. [Google Scholar] [CrossRef] [Green Version]
- Glaser, R.; Litsky, M.L.; Padgett, D.A.; Baiocchi, R.A.; Yang, E.V.; Chen, M.; Yeh, P.-E.; Green-Church, K.B.; Caligiuri, M.A.; Williams, M.V. EBV-encoded dUTPase induces immune dysregulation: Implications for the pathophysiology of EBV-associated disease. Virology 2006, 346, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Waldman, W.J.; Williams, M.V.; Lemeshow, S.; Binkley, P.; Guttridge, D.; Kiecolt-Glaser, J.K.; Knight, D.A.; Ladner, K.J.; Glaser, R. Epstein-Barr virus-encoded dUTPase enhances proinflammatory cytokine production by macrophages in contact with endothelial cells: Evidence for depression-induced atherosclerotic risk. Brain, Behav. Immun. 2008, 22, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.-E.; Glaser, R.; Kaumaya, P.T.P.; Jones, C.; Williams, M.V. The EBV-Encoded dUTPase Activates NF-κB through the TLR2 and MyD88-Dependent Signaling Pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr Virus Encoded dUTPase Containing Exosomes Modulate Innate and Adaptive Immune Responses in Human Dendritic Cells and Peripheral Blood Mononuclear Cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Stürzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi’s Sarcoma Associated Herpesvirus Tegument Protein ORF75 Is Essential for Viral Lytic Replication and Plays a Critical Role in the Antagonization of ND10-Instituted Intrinsic Immunity. PLoS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Zhang, D.; Gui, C.; Huang, L.; Chang, S.; Dong, L.; Bai, L.; Wu, S.; Lan, K. KSHV RTA antagonizes SMC5/6 complex-induced viral chromatin compaction by hijacking the ubiquitin-proteasome system. PLoS Pathog. 2022, 18, e1010744. [Google Scholar] [CrossRef]
- Decorsière, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Murphy, C.M.; Xu, Y.; Li, F.; Nio, K.; Reszka-Blanco, N.; Li, X.; Wu, Y.; Yu, Y.; Xiong, Y.; Su, L. Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 2016, 16, 2846–2854. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 Complex Restricts HBV when Localized to ND10 without Inducing an Innate Immune Response and Is Counteracted by the HBV X Protein Shortly after Infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef] [Green Version]
- Gibson, R.T.; Androphy, E.J. The SMC5/6 Complex Represses the Replicative Program of High-Risk Human Papillomavirus Type 31. Pathogens 2020, 9, 786. [Google Scholar] [CrossRef]
- Bentley, P.; Tan, M.J.A.; McBride, A.A.; White, E.A.; Howley, P.M. The SMC5/6 Complex Interacts with the Papillomavirus E2 Protein and Influences Maintenance of Viral Episomal DNA. J. Virol. 2018, 92, e00356-18. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kim, J.; Chung, W.-C.; Han, J.H.; Song, M.J. Epstein-Barr Virus Viral Processivity Factor EA-D Facilitates Virus Lytic Replication by Inducing Poly(ADP-Ribose) Polymerase 1 Degradation. J. Virol. 2022, 96, e00371-22. [Google Scholar] [CrossRef] [PubMed]
- Cheong, W.-C.; Park, J.-H.; Kang, H.-R.; Song, M.J. Downregulation of Poly(ADP-Ribose) Polymerase 1 by a Viral Processivity Factor Facilitates Lytic Replication of Gammaherpesvirus. J. Virol. 2015, 89, 9676–9682. [Google Scholar] [CrossRef] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Chung, W.-C.; Lee, S.; Kim, Y.; Seo, J.B.; Song, M.J. Kaposi’s sarcoma-associated herpesvirus processivity factor (PF-8) recruits cellular E3 ubiquitin ligase CHFR to promote PARP1 degradation and lytic replication. PLoS Pathog. 2021, 17, e1009261. [Google Scholar] [CrossRef] [PubMed]
- Strahan, R.C.; McDowell-Sargent, M.; Uppal, T.; Purushothaman, P.; Verma, S.C. KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation. PLoS Pathog. 2017, 13, e1006482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, W.; Chen, X.; Liu, L.; Shu, Y.; Zhang, M.; Zhong, Y. Role of protein arginine methyltransferase 5 in human cancers. Biomed. Pharmacother. 2019, 114, 108790. [Google Scholar] [CrossRef]
- Hiura, K.; Strahan, R.; Uppal, T.; Prince, B.; Rossetto, C.C.; Verma, S.C. KSHV ORF59 and PAN RNA Recruit Histone Demethylases to the Viral Chromatin during Lytic Reactivation. Viruses 2020, 12, 420. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.; Khoufaf, F.Z.H.; Idrissou, M.; Penault-Llorca, F.; Bignon, Y.-J.; Guy, L.; Bernard-Gallon, D. The Functions of the Demethylase JMJD3 in Cancer. Int. J. Mol. Sci. 2021, 22, 968. [Google Scholar] [CrossRef]
- Farzaneh, M.; Kuchaki, Z.; Sheykhahmad, F.R.; Meybodi, S.M.; Abbasi, Y.; Gholami, E.; Ghaedrahmati, F.; Anbiyaee, O. Emerging roles of JMJD3 in cancer. Clin. Transl. Oncol. 2022, 24, 1238–1249. [Google Scholar] [CrossRef]
- McLaughlin-Drubin, M.E.; Crum, C.P.; Munger, K. Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B histone demethylase expression and causes epigenetic reprogramming. Proc. Natl. Acad. Sci. USA 2011, 108, 2130–2135. [Google Scholar] [CrossRef] [Green Version]
- Hyland, P.L.; McDade, S.S.; McCloskey, R.; Dickson, G.J.; Arthur, K.; McCance, D.J.; Patel, D. Evidence for Alteration of EZH2, BMI1, and KDM6A and Epigenetic Reprogramming in Human Papillomavirus Type 16 E6/E7-Expressing Keratinocytes. J. Virol. 2011, 85, 10999–11006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaye, D.; Iqbal, J.; Fujita, N.; Geigerman, C.; Li, S.; Karanam, S.; Fu, K.; Weisenburger, D.; Chan, W.; Moreno, C.; et al. The BCL6-associated transcriptional co-repressor, MTA3, is selectively expressed by germinal centre B cells and lymphomas of putative germinal centre derivation. J. Pathol. 2007, 213, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Kusam, S.; Dent, A. Common mechanisms for the regulation of B cell differentiation and transformation by the transcriptional repressor protein BCL-6. Immunol. Res. 2007, 37, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, C.; Nakagawa, K.; Sancho, R.; Chakraborty, A.; Hendrich, B.; Behrens, A. c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature 2011, 469, 231–235. [Google Scholar] [CrossRef]
- Batisse, J.; Manet, E.; Middeldorp, J.; Sergeant, A.; Gruffat, H. Epstein-Barr Virus mRNA Export Factor EB2 Is Essential for Intranuclear Capsid Assembly and Production of gp350. J. Virol. 2005, 79, 14102–14111. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Marendy, E.; Wang, Y.-D.; Yuan, J.; Sample, J.T.; Swaminathan, S. Multiple Roles of Epstein-Barr Virus SM Protein in Lytic Replication. J. Virol. 2007, 81, 4058–4069. [Google Scholar] [CrossRef] [Green Version]
- Boyer, J.L.; Swaminathan, S.; Silverstein, S.J. The Epstein-Barr Virus SM Protein Is Functionally Similar to ICP27 from Herpes Simplex Virus in Viral Infections. J. Virol. 2002, 76, 9420–9433. [Google Scholar] [CrossRef] [Green Version]
- Winkler, M.; A Rice, S.; Stamminger, T. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 1994, 68, 3943–3954. [Google Scholar] [CrossRef] [Green Version]
- Bello, L.J.; Davison, A.J.; Glenn, M.A.; Whitehouse, A.; Rethmeier, N.; Schulz, T.F.; Clements, J.B. The human herpesvirus-8 ORF 57 gene and its properties. J. Gen. Virol. 1999, 80, 3207–3215. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, A.; Cooper, M.; Meredith, D.M. The Immediate-Early Gene Product Encoded by Open Reading Frame 57 of Herpesvirus Saimiri Modulates Gene Expression at a Posttranscriptional Level. J. Virol. 1998, 72, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Majerciak, V.; Zheng, Z.-M. KSHV ORF57, a Protein of Many Faces. Viruses 2015, 7, 604–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholas, J.; A Gompels, U.; A Craxton, M.; Honess, R.W. Conservation of sequence and function between the product of the 52-kilodalton immediate-early gene of herpesvirus saimiri and the BMLF1-encoded transcriptional effector (EB2) of Epstein-Barr virus. J. Virol. 1988, 62, 3250–3257. [Google Scholar] [CrossRef] [Green Version]
- Majerciak, V.; Yamanegi, K.; Allemand, E.; Kruhlak, M.; Krainer, A.; Zheng, Z.-M. Kaposi’s Sarcoma-Associated Herpesvirus ORF57 Functions as a Viral Splicing Factor and Promotes Expression of Intron-Containing Viral Lytic Genes in Spliceosome-Mediated RNA Splicing. J. Virol. 2008, 82, 2792–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Patel, A.; Krause, P.R. Hidden regulation of herpes simplex virus 1 pre-mRNA splicing and polyadenylation by virally encoded immediate early gene ICP27. PLoS Pathog. 2019, 15, e1007884. [Google Scholar] [CrossRef] [Green Version]
- Hardy, W.R.; Sandri-Goldin, R.M. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J. Virol. 1994, 68, 7790–7799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindberg, A.; Kreivi, J.-P. Splicing Inhibition at the Level of Spliceosome Assembly in the Presence of Herpes Simplex Virus Protein ICP27. Virology 2002, 294, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Semmes, O.J.; Chen, L.; Sarisky, R.T.; Gao, Z.; Zhong, L.; Hayward, S.D. Mta Has Properties of an RNA Export Protein and Increases Cytoplasmic Accumulation of Epstein-Barr Virus Replication Gene mRNA. J. Virol. 1998, 72, 9526–9534. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, V.; Gupta, A.K.; Swaminathan, S. Epstein-Barr Virus SM Protein Interacts with mRNA In Vivo and Mediates a Gene-Specific Increase in Cytoplasmic mRNA. J. Virol. 2001, 75, 6033–6041. [Google Scholar] [CrossRef] [Green Version]
- Gruffat, H.; Batisse, J.; Pich, D.; Neuhierl, B.; Manet, E.; Hammerschmidt, W.; Sergeant, A. Epstein-Barr Virus mRNA Export Factor EB2 Is Essential for Production of Infectious Virus. J. Virol. 2002, 76, 9635–9644. [Google Scholar] [CrossRef] [Green Version]
- Hiriart, E.; Farjot, G.; Gruffat, H.; Nguyen, M.V.C.; Sergeant, A.; Manet, E. A Novel Nuclear Export Signal and a REF Interaction Domain Both Promote mRNA Export by the Epstein-Barr Virus EB2 Protein. J. Biol. Chem. 2003, 278, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, V.; Wang, E.; Boyle, S.; Swaminathan, S. The Epstein–Barr virus nuclear protein SM is both a post-transcriptional inhibitor and activator of gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 8852–8857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buisson, M.; Hans, F.; Kusters, I.; Duran, N.; Sergeant, A. The C-Terminal Region but Not the Arg-X-Pro Repeat of Epstein-Barr Virus Protein EB2 Is Required for Its Effect on RNA Splicing and Transport. J. Virol. 1999, 73, 4090–4100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, V.; Sun, L.; Howard, K.; Sung, S.; Delecluse, H.-J.; Hammerschmidt, W.; Swaminathan, S. Functional Analysis of Epstein-Barr Virus SM Protein: Identification of Amino Acids Essential for Structure, Transactivation, Splicing Inhibition, and Virion Production. J. Virol. 2004, 78, 340–352. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.; Swaminathan, S. Epstein-Barr Virus SM Protein Functions as an Alternative Splicing Factor. J. Virol. 2008, 82, 7180–7188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, D.; Bais, S.; Gaillard, M.; Swaminathan, S. Epstein-Barr Virus SM Protein Utilizes Cellular Splicing Factor SRp20 To Mediate Alternative Splicing. J. Virol. 2010, 84, 11781–11789. [Google Scholar] [CrossRef] [Green Version]
- Juillard, F.; Bazot, Q.; Mure, F.; Tafforeau, L.; Macri, C.; Rabourdin-Combe, C.; Lotteau, V.; Manet, E.; Gruffat, H. Epstein–Barr virus protein EB2 stimulates cytoplasmic mRNA accumulation by counteracting the deleterious effects of SRp20 on viral mRNAs. Nucleic Acids Res. 2012, 40, 6834–6849. [Google Scholar] [CrossRef]
- Gupta, A.K.; Ruvolo, V.; Patterson, C.; Swaminathan, S. The Human Herpesvirus 8 Homolog of Epstein-Barr Virus SM Protein (KS-SM) Is a Posttranscriptional Activator of Gene Expression. J. Virol. 2000, 74, 1038–1044. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hennig, T.; Whisnant, A.W.; Erhard, F.; Prusty, B.K.; Friedel, C.C.; Forouzmand, E.; Hu, W.; Erber, L.; Chen, Y.; et al. Herpes simplex virus blocks host transcription termination via the bimodal activities of ICP27. Nat. Commun. 2020, 11, 293. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, V.; Navarro, L.; Sample, C.E.; David, M.; Sung, S.; Swaminathan, S. The Epstein-Barr Virus SM Protein Induces STAT1 and Interferon-Stimulated Gene Expression. J. Virol. 2003, 77, 3690–3701. [Google Scholar] [CrossRef] [Green Version]
- Brenner, E.; Schörg, B.F.; Ahmetlić, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 2020, 11, 1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braumüller, H.; Wieder, T.; Brenner, E.; Aßmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandhaya-Pillai, R.; Miro-Mur, F.; Alijotas-Reig, J.; Tchkonia, T.; Kirkland, J.L.; Schwartz, S. TNFα-senescence initiates a STAT-dependent positive feedback loop, leading to a sustained interferon signature, DNA damage, and cytokine secretion. Aging 2017, 9, 2411–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, C.F.D.L.C.; Shire, K.; Siddiqi, U.Z.; Frappier, L. A genome-wide screen of Epstein-Barr virus proteins that modulate host SUMOylation identifies a SUMO E3 ligase conserved in herpesviruses. PLoS Pathog. 2018, 14, e1007176. [Google Scholar] [CrossRef] [Green Version]
- Mattoscio, D.; Casadio, C.; Miccolo, C.; Maffini, F.; Raimondi, A.; Tacchetti, C.; Gheit, T.; Tagliabue, M.; Galimberti, V.E.; De Lorenzi, F.; et al. Autophagy regulates UBC9 levels during viral-mediated tumorigenesis. PLoS Pathog. 2017, 13, e1006262. [Google Scholar] [CrossRef] [Green Version]
- Heaton, P.R.; Deyrieux, A.F.; Bian, X.-L.; Wilson, V.G. HPV E6 proteins target Ubc9, the SUMO conjugating enzyme. Virus Res. 2011, 158, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, N.; John, R.; Chand, V.; Nag, A. Oncogenic Human Papillomavirus 16E7 modulates SUMOylation of FoxM1b. Int. J. Biochem. Cell Biol. 2015, 58, 28–36. [Google Scholar] [CrossRef]
- Muller, S.; Dobner, T. The adenovirus E1B-55K oncoprotein induces SUMO modification of p53. Cell Cycle 2008, 7, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Pennella, M.A.; Liu, Y.; Woo, J.L.; Kim, C.A.; Berk, A.J. Adenovirus E1B 55-Kilodalton Protein Is a p53-SUMO1 E3 Ligase That Represses p53 and Stimulates Its Nuclear Export through Interactions with Promyelocytic Leukemia Nuclear Bodies. J. Virol. 2010, 84, 12210–12225. [Google Scholar] [CrossRef] [Green Version]
- Sohn, S.-Y.; Hearing, P. The adenovirus E4-ORF3 protein functions as a SUMO E3 ligase for TIF-1γ sumoylation and poly-SUMO chain elongation. Proc. Natl. Acad. Sci. USA 2016, 113, 6725–6730. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.-C.; Izumiya, Y.; Wu, C.-Y.; Fitzgerald, L.D.; Campbell, M.; Ellison, T.; Lam, K.S.; Luciw, P.A.; Kung, H.-J. Kaposi’s Sarcoma-associated Herpesvirus (KSHV) Encodes a SUMO E3 ligase That Is SIM-dependent and SUMO-2/3-specific. J. Biol. Chem. 2010, 285, 5266–5273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroonen, J.S.; Vertegaal, A.C. Targeting SUMO Signaling to Wrestle Cancer. Trends Cancer 2021, 7, 496–510. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2000 [32] | 2011 [33] | 2022 [35] |
|---|---|---|
| Evading apoptosis | Deregulating cellular energetics | Non-mutational epigenetic reprogramming |
| Self-sufficiency in growth signals | Avoiding immune destruction | Unlocking phenotypic plasticity |
| Insensitivity to anti-growth signals | Genomic instability and mutation | Polymorphic microbiome |
| Sustained angiogenesis | Tumor-promoting inflammation | Senescent cells |
| Limitless replicative potential | ||
| Tissue invasion and metastasis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dorothea, M.; Xie, J.; Yiu, S.P.T.; Chiang, A.K.S. Contribution of Epstein–Barr Virus Lytic Proteins to Cancer Hallmarks and Implications from Other Oncoviruses. Cancers 2023, 15, 2120. https://doi.org/10.3390/cancers15072120
Dorothea M, Xie J, Yiu SPT, Chiang AKS. Contribution of Epstein–Barr Virus Lytic Proteins to Cancer Hallmarks and Implications from Other Oncoviruses. Cancers. 2023; 15(7):2120. https://doi.org/10.3390/cancers15072120
Chicago/Turabian StyleDorothea, Mike, Jia Xie, Stephanie Pei Tung Yiu, and Alan Kwok Shing Chiang. 2023. "Contribution of Epstein–Barr Virus Lytic Proteins to Cancer Hallmarks and Implications from Other Oncoviruses" Cancers 15, no. 7: 2120. https://doi.org/10.3390/cancers15072120
APA StyleDorothea, M., Xie, J., Yiu, S. P. T., & Chiang, A. K. S. (2023). Contribution of Epstein–Barr Virus Lytic Proteins to Cancer Hallmarks and Implications from Other Oncoviruses. Cancers, 15(7), 2120. https://doi.org/10.3390/cancers15072120