
Strategic Decoy Peptides Interfere with MSI1/AGO2 Interaction to Elicit Tumor Suppression Effects

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
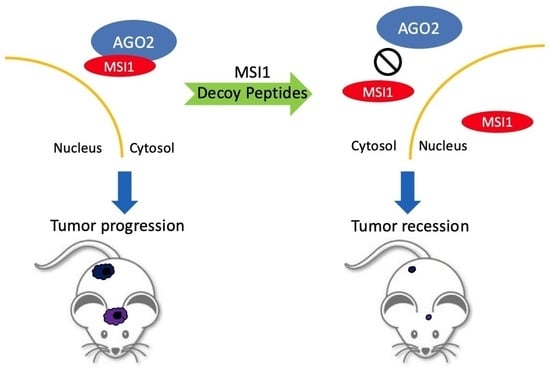
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
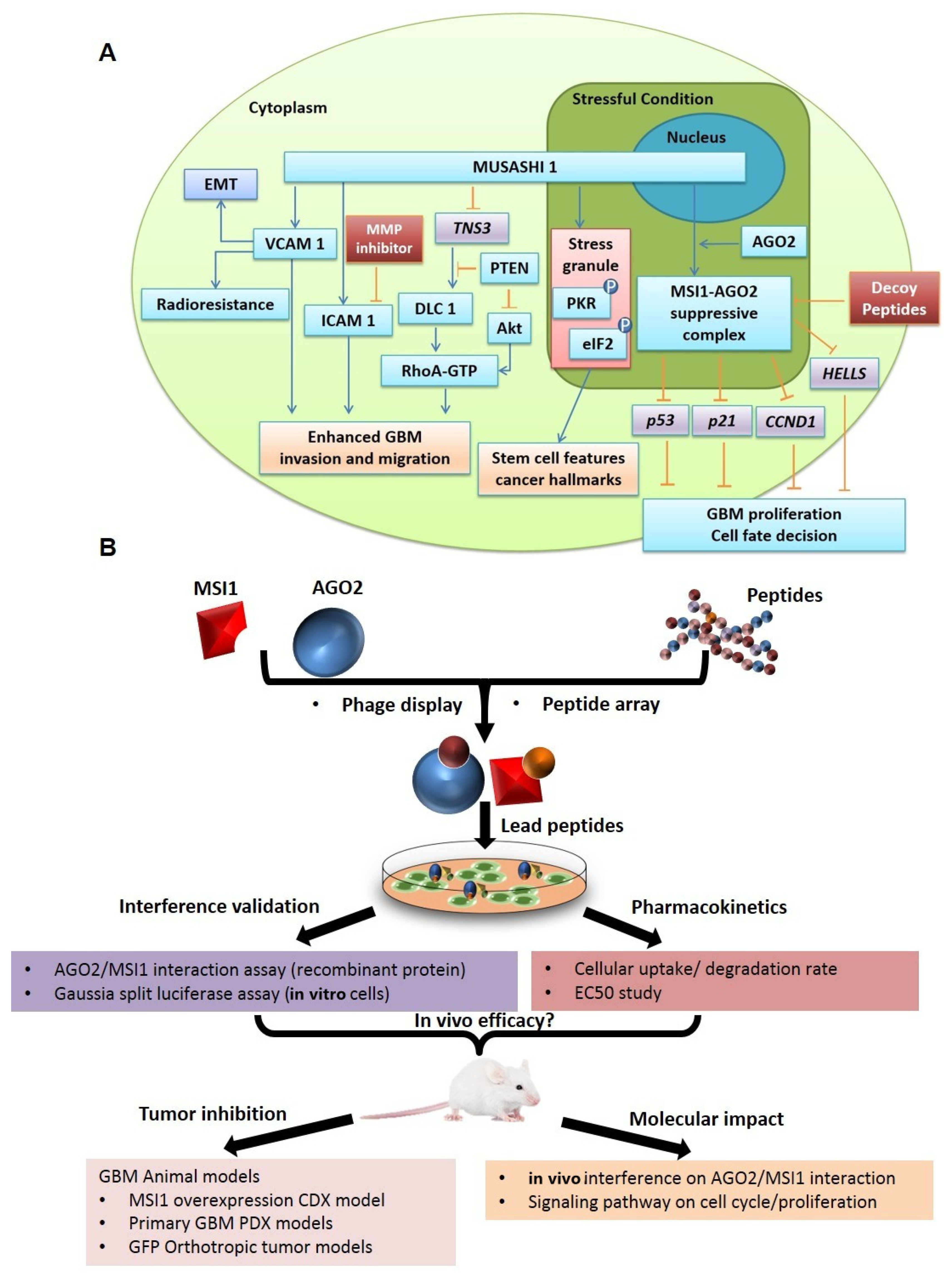
1. Introduction
2. Results
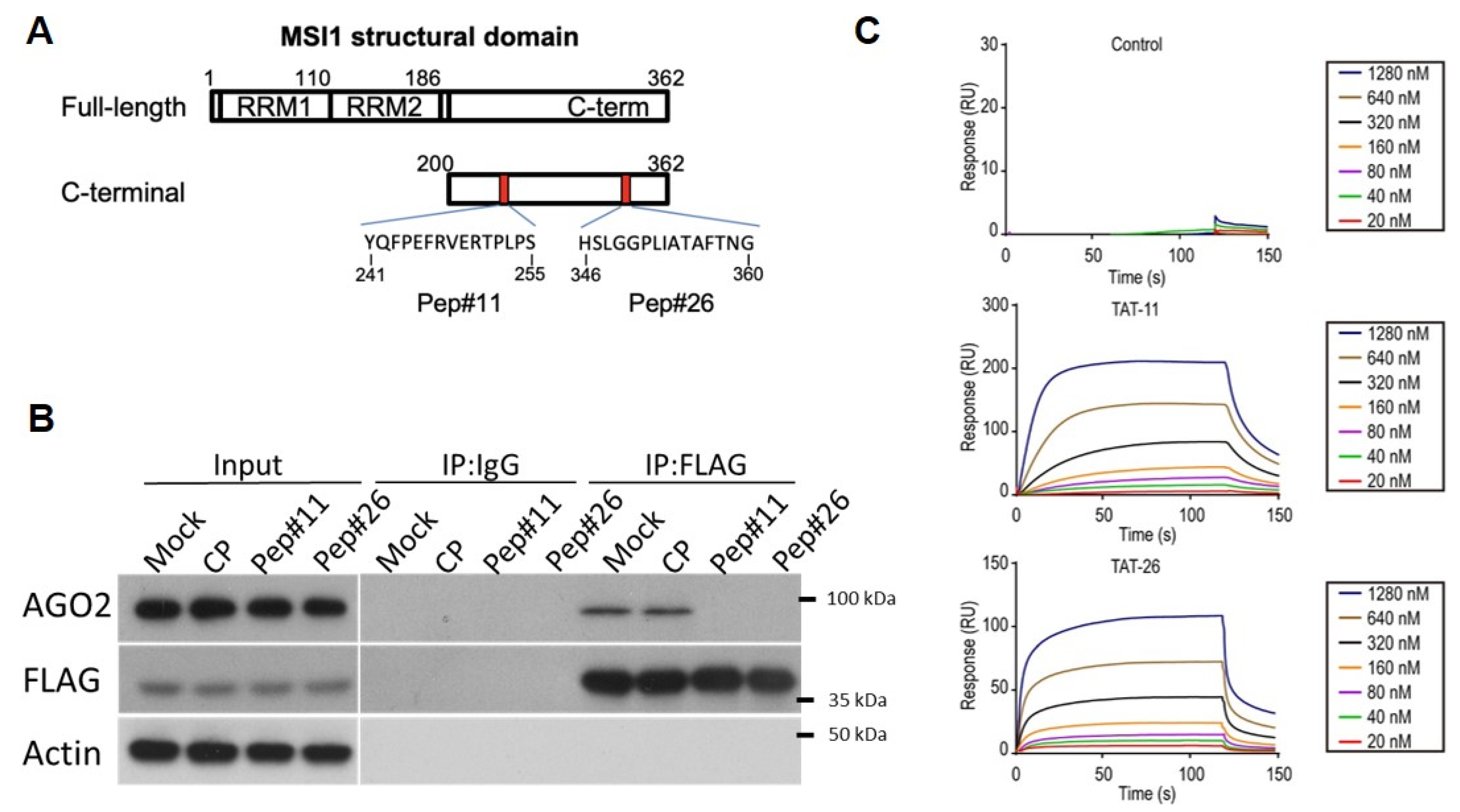
2.1. Identifying C-Terminal Motifs Responsible for MSI1/AGO2 Interaction
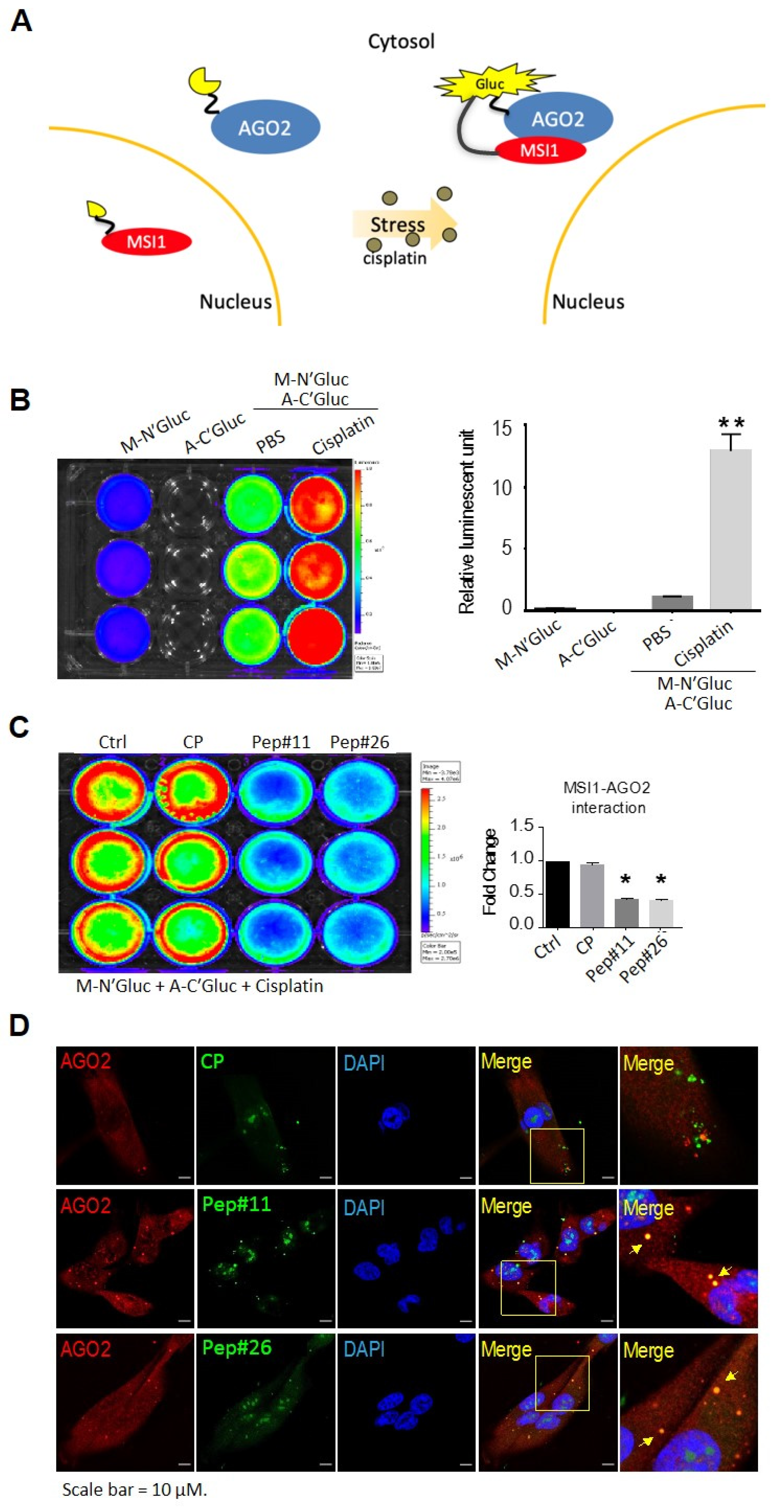
2.2. Stress-Induced MSI1/AGO2 Binding Complex by Decoy Peptides
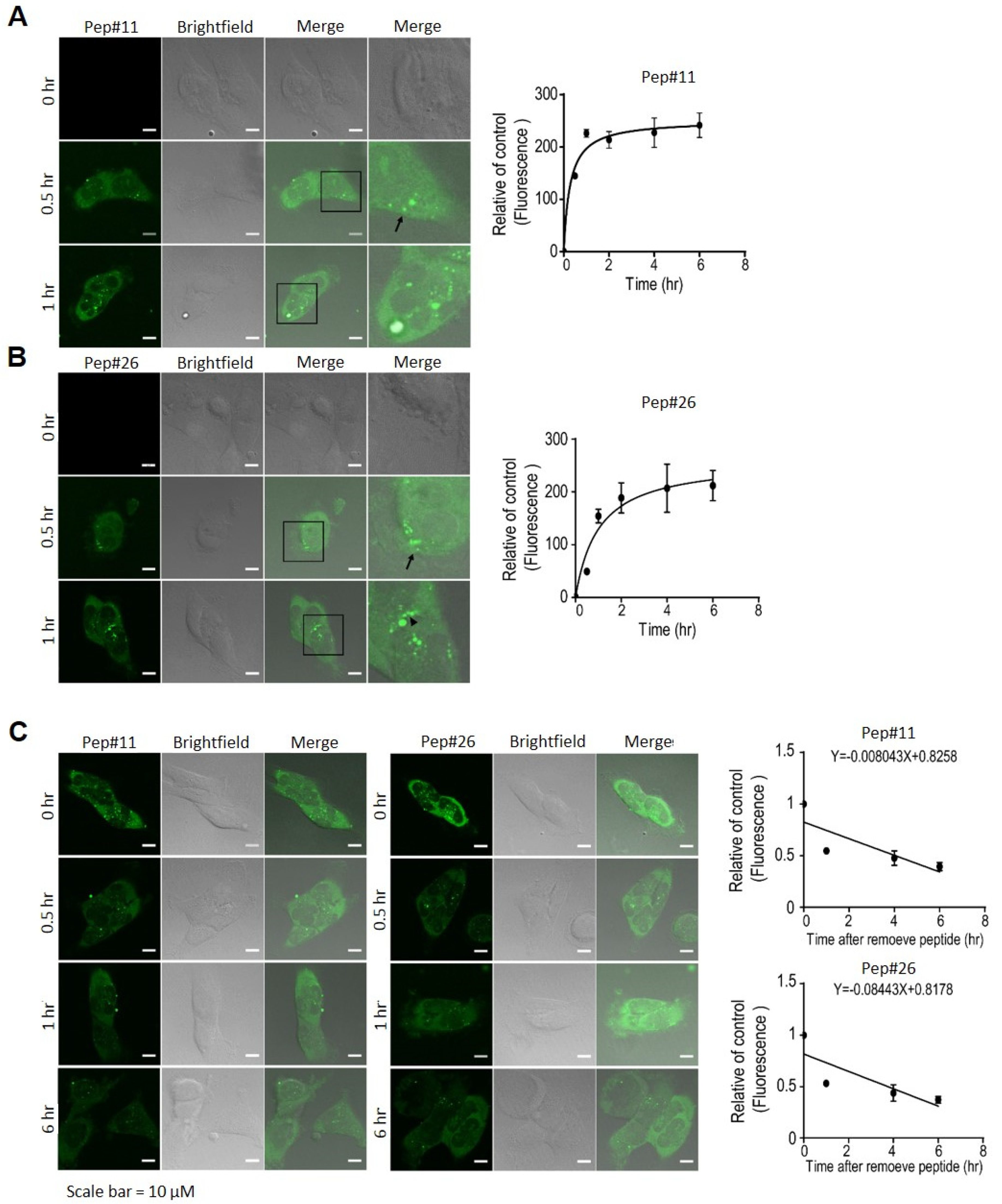
2.3. Uptake and Degradation of Decoy Peptides Pep#11 and Pep#26
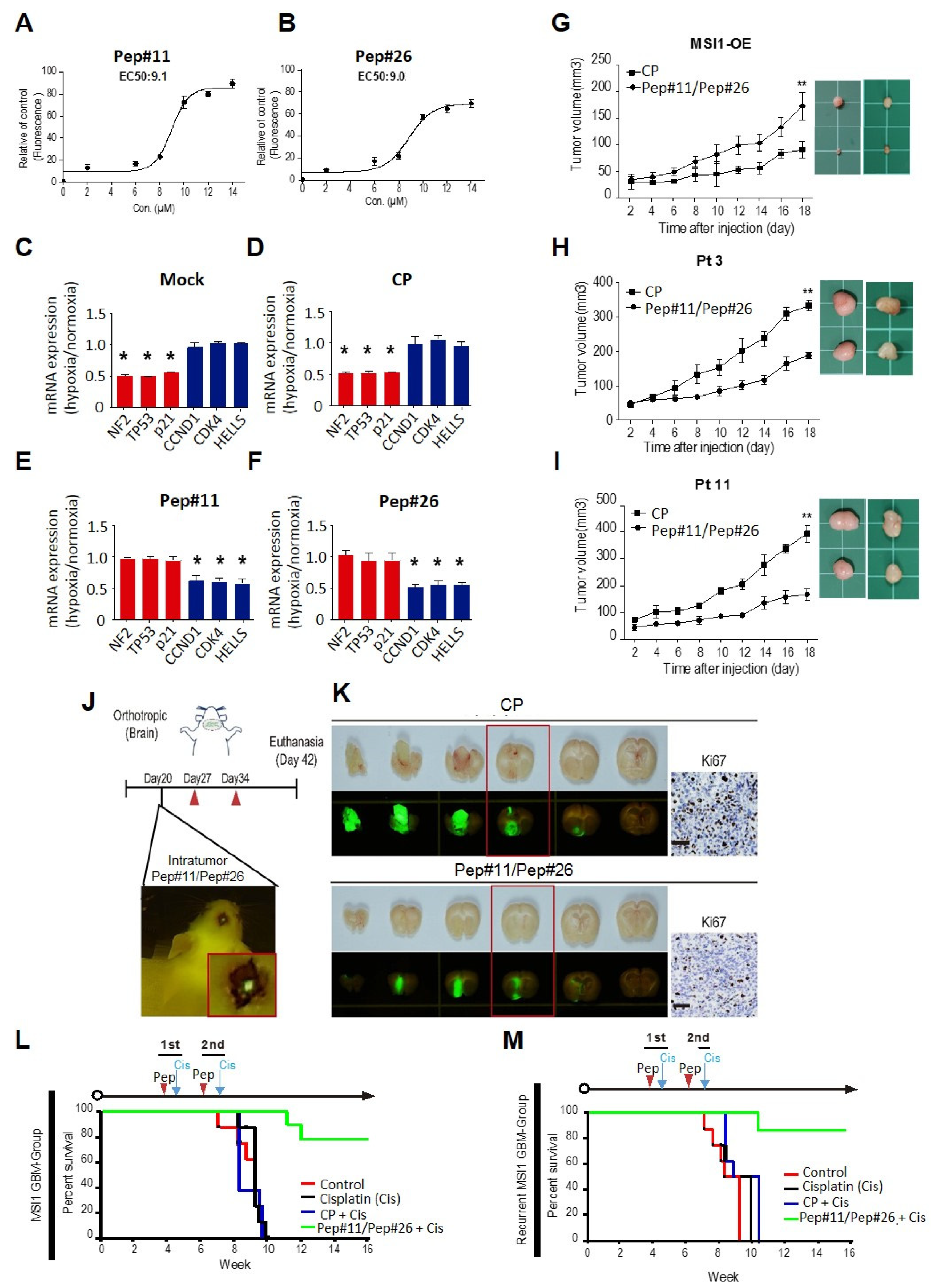
2.4. MSI1 Decoy Peptides Elicit Superior Therapeutic Effects in Tumor Suppression and Prolonging Survival
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animal Care, Tumor Cell Transplantation, and Non-Invasive Imaging
4.3. PepSpot High-Throughput Peptide Tiling Array and Peptide Phage Display
4.4. Split Luciferase Reconstitution Reporter Assay
4.5. Biotinylated Peptide Synthesis and Cell-Penetrating Assay with TAT-Tagged Peptides
4.6. Binding Affinity Measurements between Decoy Peptides and AGO2
4.7. Other General Cellular Molecular Biology Methods
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahjoubin-Tehran, M.; Rezaei, S.; Jalili, A.; Aghaee-Bakhtiari, S.H.; Orafai, H.M.; Jamialahmadi, T.; Sahebkar, A. Peptide decoys: A new technology offering therapeutic opportunities for breast cancer. Drug Discov. Today 2020, 25, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Rahal, R.; Stransky, N.; Lengauer, C.; Hoeflich, K.P. Targeting cancer with kinase inhibitors. J. Clin. Investig. 2015, 125, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Lee, A.C.; Chen, I.J.; Chang, N.C.; Wu, H.C.; Yu, H.M.; Chang, Y.J.; Lee, T.W.; Yu, J.C.; Yu, A.L.; et al. Structure-based optimization of GRP78-binding peptides that enhances efficacy in cancer imaging and therapy. Biomaterials 2016, 94, 31–44. [Google Scholar] [CrossRef]
- Yazdi, M.H.; Faramarzi, M.A.; Nikfar, S.; Abdollahi, M. A Comprehensive Review of Clinical Trials on EGFR Inhibitors Such as Cetuximab and Panitumumab as Monotherapy and in Combination for Treatment of Metastatic Colorectal Cancer. Avicenna J. Med. Biotechnol. 2015, 7, 134–144. [Google Scholar] [PubMed]
- Kumler, I.; Tuxen, M.K.; Nielsen, D.L. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat. Rev. 2014, 40, 259–270. [Google Scholar] [CrossRef]
- Guardiola, S.; Diaz-Lobo, M.; Seco, J.; Garcia, J.; Nevola, L.; Giralt, E. Peptides Targeting EGF Block the EGF-EGFR Interaction. Chembiochem 2016, 17, 702–711. [Google Scholar] [CrossRef]
- Sudhakar, D.R.; Kalaiarasan, P.; Subbarao, N. Docking and molecular dynamics simulation study of EGFR1 with EGF-like peptides to understand molecular interactions. Mol. BioSyst. 2016, 12, 1987–1995. [Google Scholar] [CrossRef]
- Cha, N.; Han, X.; Jia, B.; Liu, Y.; Wang, X.; Gao, Y.; Ren, J. Structure-based design of peptides against HER2 with cytotoxicity on colon cancer. Artif. Cells Nanomed. Biotechnol. 2017, 45, 649–654. [Google Scholar] [CrossRef] [Green Version]
- Geng, L.; Wang, Z.; Yang, X.; Li, D.; Lian, W.; Xiang, Z.; Wang, W.; Bu, X.; Lai, W.; Hu, Z.; et al. Structure-based Design of Peptides with High Affinity and Specificity to HER2 Positive Tumors. Theranostics 2015, 5, 1154–1165. [Google Scholar] [CrossRef] [Green Version]
- Kanemura, Y.; Mori, K.; Sakakibara, S.; Fujikawa, H.; Hayashi, H.; Nakano, A.; Matsumoto, T.; Tamura, K.; Imai, T.; Ohnishi, T.; et al. Musashi1, an evolutionarily conserved neural RNA-binding protein, is a versatile marker of human glioma cells in determining their cellular origin, malignancy, and proliferative activity. Differ. Res. Biol. Divers. 2001, 68, 141–152. [Google Scholar] [CrossRef]
- Okano, H.; Imai, T.; Okabe, M. Musashi: A translational regulator of cell fate. J. Cell Sci. 2002, 115, 1355–1359. [Google Scholar] [CrossRef]
- Lin, J.C.; Tsai, J.T.; Chao, T.Y.; Ma, H.I.; Chien, C.S.; Liu, W.H. MSI1 associates glioblastoma radioresistance via homologous recombination repair, tumor invasion and cancer stem-like cell properties. Radiother. Oncol. 2018, 129, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Lin, L.T.; Wang, M.L.; Lee, S.H.; Tsai, M.L.; Tsai, C.C.; Liu, W.H.; Chen, T.C.; Yang, Y.P.; Lee, Y.Y.; et al. Musashi-1 regulates AKT-derived IL-6 autocrinal/paracrinal malignancy and chemoresistance in glioblastoma. Oncotarget 2016, 7, 42485–42501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.Y.; Lin, L.T.; Wang, M.L.; Laurent, B.; Hsu, C.H.; Pan, C.M.; Jiang, W.R.; Chen, P.Y.; Ma, H.I.; Chen, Y.W.; et al. Musashi-1 Enhances Glioblastoma Cell Migration and Cytoskeletal Dynamics through Translational Inhibition of Tensin3. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, M.; Imai, T.; Kurusu, M.; Hiromi, Y.; Okano, H. Translational repression determines a neuronal potential in Drosophila asymmetric cell division. Nature 2001, 411, 94–98. [Google Scholar] [CrossRef]
- Imai, T.; Tokunaga, A.; Yoshida, T.; Hashimoto, M.; Mikoshiba, K.; Weinmaster, G.; Nakafuku, M.; Okano, H. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol. Cell. Biol. 2001, 21, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Lin, L.T.; Wang, M.L.; Tsai, K.L.; Huang, P.I.; Yang, Y.P.; Lee, Y.Y.; Chen, Y.W.; Lo, W.L.; Lan, Y.T.; et al. Musashi-1 promotes chemoresistant granule formation by PKR/eIF2 alpha signalling cascade in refractory glioblastoma. Mol. Basis Dis. 2018, 1864, 1850–1861. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; George, R.J.; Dieckgraefe, B.K.; McLeod, H.L.; Ramalingam, S.; Bishnupuri, K.S.; Natarajan, G.; Anant, S.; Houchen, C.W. Knockdown of RNA binding protein musashi-1 leads to tumor regression in vivo. Gastroenterology 2008, 134, 1448–1458. [Google Scholar] [CrossRef]
- Lagadec, C.; Vlashi, E.; Frohnen, P.; Alhiyari, Y.; Chan, M.; Pajonk, F. The RNA-binding protein Musashi-1 regulates proteasome subunit expression in breast cancer- and glioma-initiating cells. Stem. Cells 2014, 32, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Dahlrot, R.H.; Hansen, S.; Herrstedt, J.; Schroder, H.D.; Hjelmborg, J.; Kristensen, B.W. Prognostic value of Musashi-1 in gliomas. J. Neuro-Oncol. 2013, 115, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Huntzinger, E.; Izaurralde, E. Getting to the root of miRNA-mediated gene silencing. Cell 2008, 132, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karginov, F.V.; Hannon, G.J. Remodeling of Ago2-mRNA interactions upon cellular stress reflects miRNA complementarity and correlates with altered translation rates. Genes Dev. 2013, 27, 1624–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Gen. 2013, 14, 447–459. [Google Scholar] [CrossRef]
- Shen, J.; Xia, W.; Khotskaya, Y.B.; Huo, L.; Nakanishi, K.; Lim, S.O.; Du, Y.; Wang, Y.; Chang, W.C.; Chen, C.H.; et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 2013, 497, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Golden, R.J.; Chen, B.; Li, T.; Braun, J.; Manjunath, H.; Chen, X.; Wu, J.; Schmid, V.; Chang, T.C.; Kopp, F.; et al. An Argonaute phosphorylation cycle promotes microRNA-mediated silencing. Nature 2017, 542, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Shankar, S.; Tien, J.C.; Siebenaler, R.F.; Chugh, S.; Dommeti, V.L.; Zelenka-Wang, S.; Wang, X.M.; Apel, I.J.; Waninger, J.; Eyunni, S.; et al. An essential role for Argonaute 2 in EGFR-KRAS signaling in pancreatic cancer development. Nat. Commun. 2020, 11, 2817. [Google Scholar] [CrossRef]
- Casey, M.C.; Prakash, A.; Holian, E.; McGuire, A.; Kalinina, O.; Shalaby, A.; Curran, C.; Webber, M.; Callagy, G.; Bourke, E.; et al. Quantifying Argonaute 2 (Ago2) expression to stratify breast cancer. BMC Cancer 2019, 19, 712. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Dou, J.; Guo, Y.; He, J.; Li, L.; Liu, X.; Chen, R.; Deng, R.; Huang, J.; et al. Acetylation of AGO2 promotes cancer progression by increasing oncogenic miR-19b biogenesis. Oncogene 2019, 38, 1410–1431. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Wang, M.L.; Laurent, B.; Hsu, C.H.; Chen, M.T.; Lin, L.T.; Shen, J.; Chang, W.C.; Hsu, J.; Hung, M.C.; et al. Musashi-1 promotes stress-induced tumor progression through recruitment of AGO2. Theranostics 2020, 10, 201–217. [Google Scholar] [CrossRef]
- Futaki, S. Membrane-permeable arginine-rich peptides and the translocation mechanisms. Adv. Drug Deliv. Rev. 2005, 57, 547–558. [Google Scholar] [CrossRef]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef]
- Jackson, C.M.; Choi, J.; Lim, M. Mechanisms of immunotherapy resistance: Lessons from glioblastoma. Nat. Immunol. 2019, 20, 1100–1109. [Google Scholar] [CrossRef]
- Fox, R.G.; Lytle, N.K.; Jaquish, D.V.; Park, F.D.; Ito, T.; Bajaj, J.; Koechlein, C.S.; Zimdahl, B.; Yano, M.; Kopp, J.L.; et al. Image-based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature 2016, 534, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Fox, R.G.; Park, F.D.; Koechlein, C.S.; Kritzik, M.; Reya, T. Musashi signaling in stem cells and cancer. Ann. Rev. Cell Dev. Biol. 2015, 31, 249–267. [Google Scholar] [CrossRef] [Green Version]
- Hunt, E.A.; Moutsiopoulou, A.; Ioannou, S.; Ahern, K.; Woodward, K.; Dikici, E.; Daunert, S.; Deo, S.K. Truncated Variants of Gaussia Luciferase with Tyrosine Linker for Site-Specific Bioconjugate Applications. Sci. Rep. 2016, 6, 26814. [Google Scholar] [CrossRef] [Green Version]
- Tannous, B.A. Gaussia luciferase reporter assay for monitoring biological processes in culture and in vivo. Nat. Protoc. 2009, 4, 582–591. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Chien, C.S.; Wang, M.L.; Chen, M.T.; Yang, Y.P.; Yu, Y.L.; Chien, Y.; Chang, Y.C.; Shen, C.C.; Chio, C.C.; et al. Epigenetic regulation of the miR142-3p/interleukin-6 circuit in glioblastoma. Mol. Cell 2013, 52, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Wang-Gillam, A.; Li, C.P.; Bodoky, G.; Dean, A.; Shan, Y.S.; Jameson, G.; Macarulla, T.; Lee, K.H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet (Lond. Engl.) 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Tahbaz, N.; Kolb, F.A.; Zhang, H.; Jaronczyk, K.; Filipowicz, W.; Hobman, T.C. Characterization of the interactions between mammalian PAZ PIWI domain proteins and Dicer. EMBO Rep. 2004, 5, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelin, J.; Cardo-Vila, M.; Driessen, W.H.; Mathew, P.; Navone, N.M.; Lin, S.H.; Logothetis, C.J.; Rietz, A.C.; Dobroff, A.S.; Proneth, B.; et al. Selection and identification of ligand peptides targeting a model of castrate-resistant osteogenic prostate cancer and their receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 3776–3781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Cambuli, F.M.; Correa, B.R.; Rezza, A.; Burns, S.C.; Qiao, M.; Uren, P.J.; Kress, E.; Boussouar, A.; Galante, P.A.; Penalva, L.O.; et al. A Mouse Model of Targeted Musashi1 Expression in Whole Intestinal Epithelium Suggests Regulatory Roles in Cell Cycle and Stemness. Stem Cells 2015, 33, 3621–3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minuesa, G.; Albanese, S.K.; Xie, W.; Kazansky, Y.; Worroll, D.; Chow, A.; Schurer, A.; Park, S.M.; Rotsides, C.Z.; Taggart, J.; et al. Small-molecule targeting of MUSASHI RNA-binding activity in acute myeloid leukemia. Nat. Commun. 2019, 10, 2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.; von Richter, O.; Kovar, A.; Scheible, H.; van Lier, J.J.; Johne, A. Metabolism and disposition of the alphav-integrin ss3/ss5 receptor antagonist cilengitide, a cyclic polypeptide, in humans. J. Clin. Pharmacol. 2015, 55, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, L.; Dong, Y.; Tan, Y.; Li, Y.; Zhao, M.; Xie, H.; Ju, H.; Wang, H.; Zhao, Y.; et al. EGFRvIII/integrin beta3 interaction in hypoxic and vitronectinenriching microenvironment promote GBM progression and metastasis. Oncotarget 2016, 7, 4680–4694. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-P.; Chien, C.-S.; Yarmishyn, A.A.; Chan, M.-S.; Lee, A.C.-L.; Chen, Y.-W.; Huang, P.-I.; Ma, H.-I.; Lo, W.-L.; Chien, Y.; et al. Musashi-1 Regulates MIF1-Mediated M2 Macrophage Polarization in Promoting Glioblastoma Progression. Cancers 2021, 13, 1799. [Google Scholar] [CrossRef]
- Xue, V.W.; Chung, J.Y.; Tang, P.C.; Chan, A.S.; To, T.H.; Chung, J.S.; Mussal, F.; Lam, E.W.; Li, C.; To, K.F.; et al. USMB-shMincle: A virus-free gene therapy for blocking M1/M2 polarization of tumor-associated macrophages. Mol. Ther. Oncol. 2021, 23, 26–37. [Google Scholar] [CrossRef]
- Li, R.; Liu, T.; Wong, B.S.; Pan, T.; Morillas, M.; Swietnicki, W.; O′Rourke, K.; Gambetti, P.; Surewicz, W.K.; Sy, M.S. Identification of an epitope in the C terminus of normal prion protein whose expression is modulated by binding events in the N terminus. J. Mol. Biol. 2000, 301, 567–573. [Google Scholar] [CrossRef]
- Kato, N.; Jones, J. The split luciferase complementation assay. Methods Mol. Biol. 2010, 655, 359–376. [Google Scholar] [CrossRef]
- Paulmurugan, R.; Umezawa, Y.; Gambhir, S.S. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc. Natl. Acad. Sci. USA. 2002, 99, 15608–15613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, R.; Gurbaxani, B.; Morrison, S.L.; Seyfzadeh, M. Optimization of codon pair use within the (GGGGS)3 linker sequence results in enhanced protein expression. Mol. Immunol. 2004, 40, 717–722. [Google Scholar] [CrossRef]
- Newton, D.L.; Xue, Y.; Olson, K.A.; Fett, J.W.; Rybak, S.M. Angiogenin single-chain immunofusions: Influence of peptide linkers and spacers between fusion protein domains. Biochemistry 1996, 35, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Chiou, S.H.; Lee, T.W.; Liu, R.S.; Hwang, J.J.; Chang, C.H.; Ma, H.I.; Lee, Y.J. A comparative study of primary and recurrent human glioblastoma multiforme using the small animal imaging and molecular expressive profiles. Mol. Imaging Biol. 2013, 15, 262–272. [Google Scholar] [CrossRef]
- Abes, R.; Arzumanov, A.; Moulton, H.; Abes, S.; Ivanova, G.; Gait, M.J.; Iversen, P.; Lebleu, B. Arginine-rich cell penetrating peptides: Design, structure-activity, and applications to alter pre-mRNA splicing by steric-block oligonucleotides. J. Pept. Sci. 2008, 14, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharmacol. 2005, 145, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Thoren, P.E.; Persson, D.; Isakson, P.; Goksor, M.; Onfelt, A.; Norden, B. Uptake of analogs of penetratin, Tat(48-60) and oligoarginine in live cells. Biochem. Biophys. Res. Commun. 2003, 307, 100–107. [Google Scholar] [CrossRef]
- Kawahara, H.; Okada, Y.; Imai, T.; Iwanami, A.; Mischel, P.S.; Okano, H. Musashi1 cooperates in abnormal cell lineage protein 28 (Lin28)-mediated let-7 family microRNA biogenesis in early neural differentiation. J. Biol. Chem. 2011, 286, 16121–16130. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.; Pachter, L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat. Methods 2013, 10, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.S.; Wang, M.L.; Chu, P.Y.; Chang, Y.L.; Liu, W.H.; Yu, C.C.; Lan, Y.T.; Huang, P.I.; Lee, Y.Y.; Chen, Y.W.; et al. Lin28B/Let-7 Regulates Expression of Oct4 and Sox2 and Reprograms Oral Squamous Cell Carcinoma Cells to a Stem-like State. Cancer Res. 2015, 75, 2553–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.-P.; Lee, A.C.-L.; Lin, L.-T.; Chen, Y.-W.; Huang, P.-I.; Ma, H.-I.; Chen, Y.-C.; Lo, W.-L.; Lan, Y.-T.; Fang, W.-L.; et al. Strategic Decoy Peptides Interfere with MSI1/AGO2 Interaction to Elicit Tumor Suppression Effects. Cancers 2022, 14, 505. https://doi.org/10.3390/cancers14030505
Yang Y-P, Lee AC-L, Lin L-T, Chen Y-W, Huang P-I, Ma H-I, Chen Y-C, Lo W-L, Lan Y-T, Fang W-L, et al. Strategic Decoy Peptides Interfere with MSI1/AGO2 Interaction to Elicit Tumor Suppression Effects. Cancers. 2022; 14(3):505. https://doi.org/10.3390/cancers14030505
Chicago/Turabian StyleYang, Yi-Ping, Andy Chi-Lung Lee, Liang-Ting Lin, Yi-Wei Chen, Pin-I Huang, Hsin-I Ma, Yi-Chen Chen, Wen-Liang Lo, Yuan-Tzu Lan, Wen-Liang Fang, and et al. 2022. "Strategic Decoy Peptides Interfere with MSI1/AGO2 Interaction to Elicit Tumor Suppression Effects" Cancers 14, no. 3: 505. https://doi.org/10.3390/cancers14030505
APA StyleYang, Y. -P., Lee, A. C. -L., Lin, L. -T., Chen, Y. -W., Huang, P. -I., Ma, H. -I., Chen, Y. -C., Lo, W. -L., Lan, Y. -T., Fang, W. -L., Wang, C. -Y., Liu, Y. -Y., Hsu, P. -K., Lin, W. -C., Li, C. -P., Chen, M. -T., Chien, C. -S., & Wang, M. -L. (2022). Strategic Decoy Peptides Interfere with MSI1/AGO2 Interaction to Elicit Tumor Suppression Effects. Cancers, 14(3), 505. https://doi.org/10.3390/cancers14030505