
Targeted Drug Delivery and Theranostic Strategies in Malignant Lymphomas

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Malignant Lymphomas
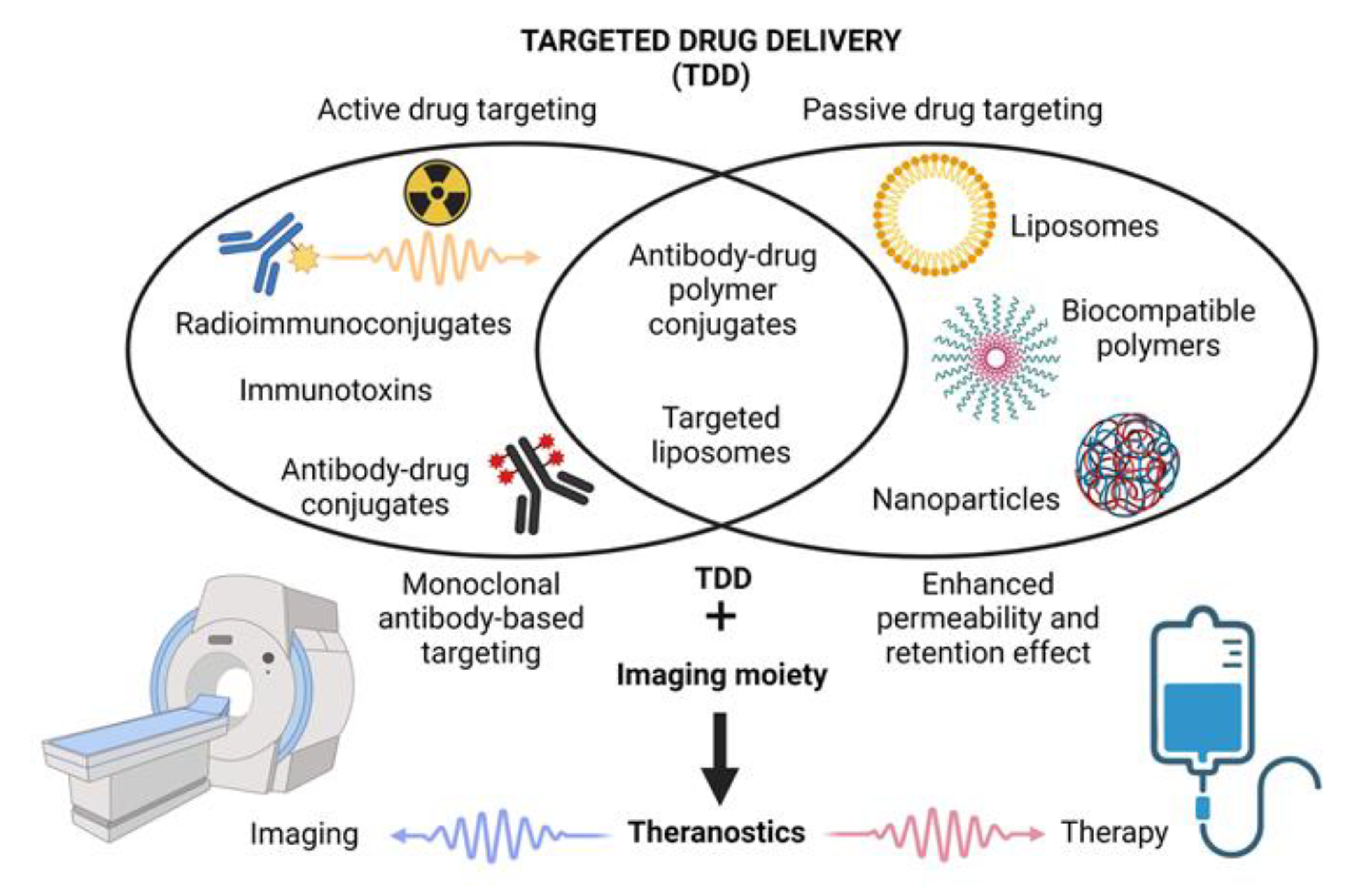
1.2. The Concept of Targeted Drug Delivery and Controlled Drug Release
2. Antibody–Drug Conjugates (ADCs) and Immunotoxins: MAb-Mediated TDD
2.1. The Structure of ADCs
2.2. Gemtuzumab Ozogamicin (GO): The First Global Approval
2.3. Brentuximab Vedotin (BV): The First Clinical Approval for the Therapy of Lymphomas
2.4. Inotuzumab Ozogamicin: Targeting B-Cell Acute Lymphoblastic Leukemia/Lymphoma
2.5. Polatuzumab Vedotin (PV): The Game Changer in DLBCL?
2.6. Loncastuximab Tesirine (LT): Another Player in CD19-Directed Strategies
2.7. Next-Generation ADCs
2.8. Immunotoxins: From Denileukin Diftitox to Moxetumomab Pasudotox and Beyond
3. Liposomes, Polymeric Nanocarriers and Other Types of Nanomedicines
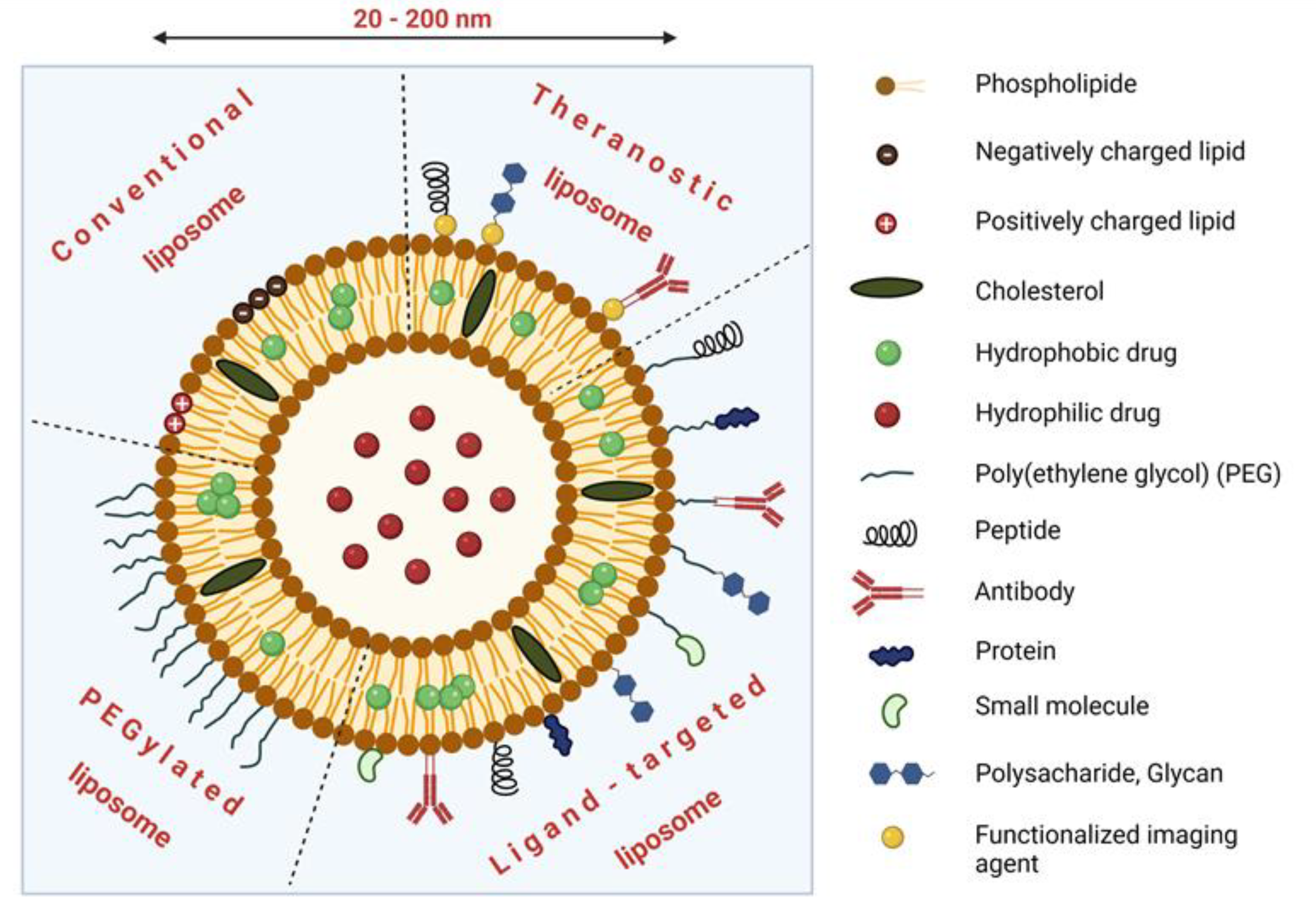
3.1. Nanosystems for TDD: Structure, Passive and Active Targeting
3.2. Liposomal Formulations of Cytostatics
3.3. Liposomal Formulations of Anthracyclines
3.3.1. Liposomal Formulations of Doxorubicin—The First Nanomedicines Approved for the Treatment of Cancer
3.3.2. Liposomal Formulations of Doxorubicin in the Therapy of Aggressive Lymphomas
3.4. Liposomal Formulations of Vinca Alkaloids and Cytarabine
3.5. Targeted Liposomes
4. Polymer-Based Nanomedicines
4.1. PEG-Based Polymers
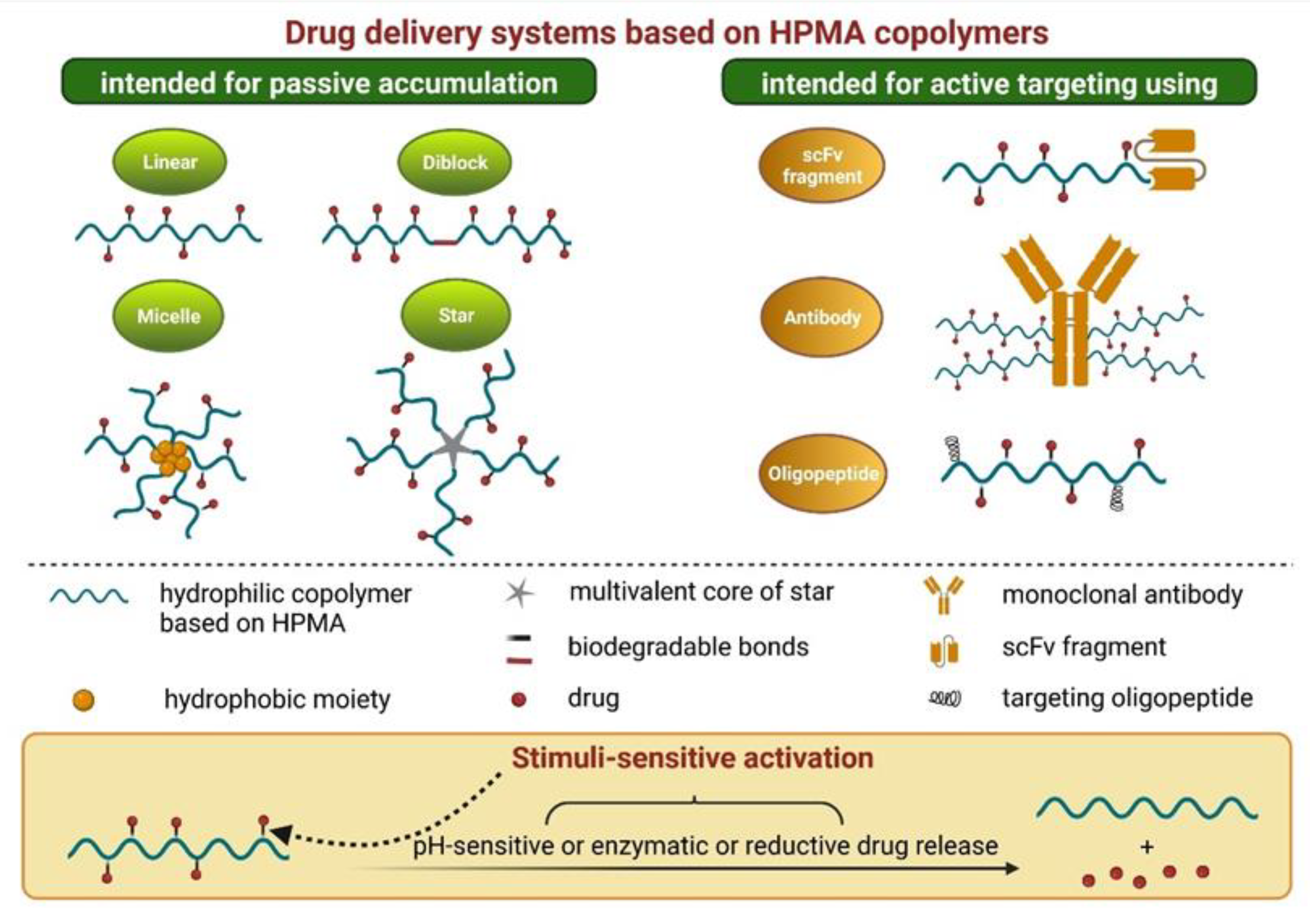
4.2. HPMA-Based Polymers
5. Theranostics
5.1. The Origins and Evolution of the Theranostic Concept
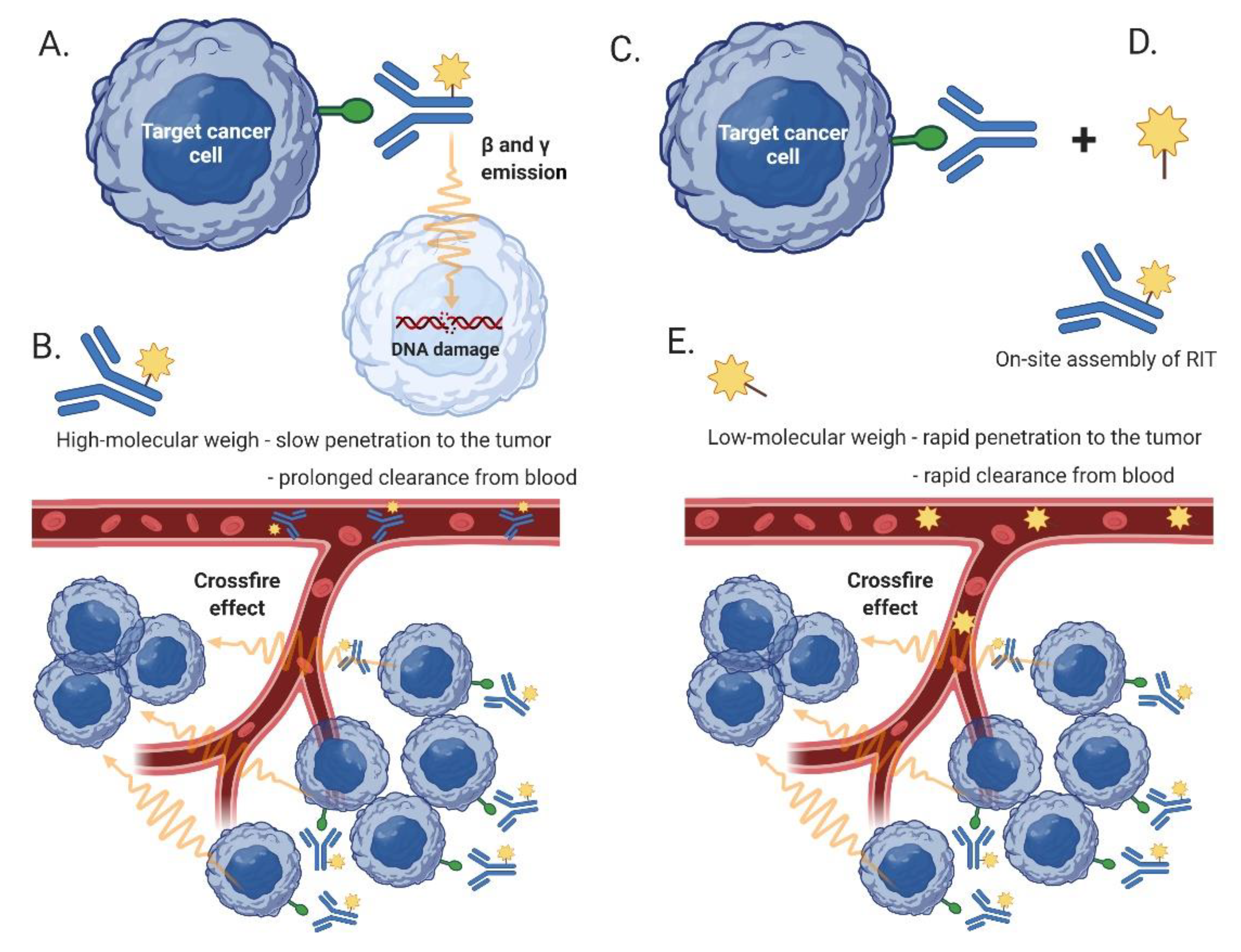
5.2. Radioimmunotherapy (RIT)
5.3. Pretargeted RIT
5.4. Immuno-PET
5.5. CXCR4-Targeted Therapy and Other Theranostic Concepts
6. Theranostic Strategies Based on Magnetic Resonance Imaging
6.1. Gadolinium-Based Tracers and Manganese Oxide Nanoparticles
6.2. Magnetic Nanoparticles
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADC | ntibody drug conjugate |
| ADCC | antibody-dependent cell-mediated cytotoxicity |
| ADPC | antibody-dependent cell-mediated phagocytosis |
| ALL | acute lymphoblastic leukemia |
| AML | acute myeloid leukemia |
| BCMA | B-cell maturation antigen |
| BM | belantamab mafodotin |
| B-NHL | B-cell NHL |
| BV | brentuximab vedotin |
| CAR | chimeric antigen receptor |
| CAR19 | CAR against CD19-positive cells |
| CDC | complement-dependent cytotoxicity |
| DAR | drug to antibody ration |
| DDS | drug delivery systems |
| DLBCL | diffuse large B-cell lymphoma |
| EPR | enhanced permeability and retention |
| FDG | 2-[F18] fluoro-2-deoxy-D-glucose |
| FL | follicular lymphoma |
| Fv | fragment variable (of the monoclonal antibody) |
| Gd | gadolinium |
| GFLG | glycylphenylalanylleucylglycine |
| GO | gemtuzumab ozogamicin |
| HL | Hodgkin lymphoma |
| HPMA | N-(2-hydroxypropyl)methacrylamide |
| InO | inotuzumab ozogamicin |
| IT | immunotoxin |
| LT | loncastuximab tesirine |
| MCL | mantle cell lymphoma |
| MDR-1 | multi-drug resistance protein 1 |
| MM | multiple myeloma |
| MMAE | monomethyl auristatin E |
| MoA | mode of action |
| MRI | magnetic resonance imaging |
| NHL | non-Hodgkin lymphoma |
| NPLD | non-pegylated liposomal doxorubicin |
| MON | manganese oxide nanoparticles |
| MZL | marginal zone lymphoma |
| Mab | monoclonal antibody |
| NK | natural killer |
| PEG | polyethylene glycol |
| PGA | poly(L-glutamic acid) |
| PLD | pegylated liposomal doxorubicin |
| PET | positron emission tomography |
| PBDD | pyrrolobenzodiazepine dimer |
| Pox | poly(2-oxazoline) polymers |
| PRIT | pretargeted RIT |
| PSMA | prostate specific membrane antigen |
| PV | polatuzumab vedotin |
| R-CHOP | rituximab + cyclophosphamide + doxorubicin + vincristine + prednisone |
| RIT | radioimmunotherapy |
| R/R | relapsed/refractory |
| scFc | single-chain fragment variable |
| SPECT | single-photon emission computed tomography |
| SPION | superparamagnetic iron oxide nanoparticles |
| TDD | targeted drug delivery |
| T-NHL | T-cell NHL |
| VSLI | vincristine sulphate liposome injection |
References
- Nogai, H.; Dorken, B.; Lenz, G. Pathogenesis of non-Hodgkin’s lymphoma. J. Clin. Oncol. 2011, 29, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Cook, J.R. As the world turns, evolving lymphoma classifications-past, present and future. Hum. Pathol. 2019, 95, 55–77. [Google Scholar] [CrossRef]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, M.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Crump, M.; Neelapu, S.S.; Farooq, U.; Van Den Neste, E.; Kuruvilla, J.; Westin, J.; Link, B.K.; Hay, A.; Cerhan, J.R.; Zhu, L.; et al. Outcomes in refractory diffuse large B-cell lymphoma: Results from the international SCHOLAR-1 study. Blood 2017, 130, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Tafasitamab: First Approval. Drugs 2020, 80, 1731–1737. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; González Barca, E.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Wang, M.L.; Rule, S.; Martin, P.; Goy, A.; Auer, R.; Kahl, B.S.; Jurczak, W.; Advani, R.H.; Romaguera, J.E.; Williams, M.E.; et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2013, 369, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Dreyling, M.; Jurczak, W.; Jerkeman, M.; Silva, R.S.; Rusconi, C.; Trneny, M.; Offner, F.; Caballero, D.; Joao, C.; Witzens-Harig, M.; et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: An international, randomised, open-label, phase 3 study. Lancet 2016, 387, 770–778. [Google Scholar] [CrossRef] [Green Version]
- Trneny, M.; Lamy, T.; Walewski, J.; Belada, D.; Mayer, J.; Radford, J.; Jurczak, W.; Morschhauser, F.; Alexeeva, J.; Rule, S.; et al. Lenalidomide versus investigator’s choice in relapsed or refractory mantle cell lymphoma (MCL-002; SPRINT): A phase 2, randomised, multicentre trial. Lancet Oncol. 2016, 17, 319–331. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, J.; Ramchandren, R.; Santoro, A.; Paszkiewicz-Kozik, E.; Gasiorowski, R.; Johnson, N.A.; Fogliatto, L.M.; Goncalves, I.; de Oliveira, J.S.R.; Buccheri, V.; et al. Pembrolizumab versus brentuximab vedotin in relapsed or refractory classical Hodgkin lymphoma (KEYNOTE-204): An interim analysis of a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 2021, 22, 512–524. [Google Scholar] [CrossRef]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, R.S. Paul Ehrlich’s magic bullets. N. Engl. J. Med. 2004, 350, 1079–1080. [Google Scholar] [CrossRef]
- Maloney, D.G.; Grillo-Lopez, A.J.; Bodkin, D.J.; White, C.A.; Liles, T.M.; Royston, I.; Varns, C.; Rosenberg, J.; Levy, R. IDEC-C2B8: Results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J. Clin. Oncol. 1997, 15, 3266–3274. [Google Scholar] [CrossRef] [PubMed]
- Maloney, D.G.; Grillo-Lopez, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.; Davies, A.; Ando, K.; Klapper, W.; Opat, S.; Owen, C.; Phillips, E.; Sangha, R.; Schlag, R.; Seymour, J.F.; et al. Obinutuzumab for the First-Line Treatment of Follicular Lymphoma. N. Engl. J. Med. 2017, 377, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Dhodapkar, M.V.; Ferrone, S. Monoclonal antibodies for cancer immunotherapy. Lancet 2009, 373, 1033–1040. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Jin, Y.; Schladetsch, M.A.; Huang, X.; Balunas, M.J.; Wiemer, A.J. Stepping forward in antibody-drug conjugate development. Pharmacol. Ther. 2021, 229, 107917. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Maderna, A.; Leverett, C.A. Recent advances in the development of new auristatins: Structural modifications and application in antibody drug conjugates. Mol. Pharm. 2015, 12, 1798–1812. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Kim, Y.H.; Horwitz, S.M.; Dummer, R.; Scarisbrick, J.; Quaglino, P.; Zinzani, P.L.; Wolter, P.; Sanches, J.A.; Ortiz-Romero, P.L.; et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): An international, open-label, randomised, phase 3, multicentre trial. Lancet 2017, 390, 555–566. [Google Scholar] [CrossRef]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Forero-Torres, A.; Leonard, J.P.; Younes, A.; Rosenblatt, J.D.; Brice, P.; Bartlett, N.L.; Bosly, A.; Pinter-Brown, L.; Kennedy, D.; Sievers, E.L.; et al. A Phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br. J. Haematol. 2009, 146, 171–179. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Advani, A.S.; Marks, D.I.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; Jabbour, E.; Merchant, A.; Wang, T.; et al. Inotuzumab ozogamicin for relapsed/refractory acute lymphoblastic leukemia: Outcomes by disease burden. Blood Cancer J. 2020, 10, 81. [Google Scholar] [CrossRef]
- Dang, N.H.; Ogura, M.; Castaigne, S.; Fayad, L.E.; Jerkeman, M.; Radford, J.; Pezzutto, A.; Bondarenko, I.; Stewart, D.A.; Shnaidman, M.; et al. Randomized, phase 3 trial of inotuzumab ozogamicin plus rituximab versus chemotherapy plus rituximab for relapsed/refractory aggressive B-cell non-Hodgkin lymphoma. Br. J. Haematol. 2018, 182, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients with Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, K. The era of CD19-directed therapy in diffuse large B-cell lymphoma. Lancet Oncol. 2021, 22, 741–742. [Google Scholar] [CrossRef]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef]
- Tolcher, A.W. The Evolution of Antibody-Drug Conjugates: A Positive Inflexion Point. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Tiefenthaler, G.; Georges, G. Proteases as activators for cytotoxic prodrugs in antitumor therapy. Cancer Genom. Proteom. 2014, 11, 67–79. [Google Scholar]
- Hinrichs, M.J.M.; Ryan, P.M.; Zheng, B.; Afif-Rider, S.; Yu, X.Q.; Gunsior, M.; Zhong, H.; Harper, J.; Bezabeh, B.; Vashisht, K.; et al. Fractionated Dosing Improves Preclinical Therapeutic Index of Pyrrolobenzodiazepine-Containing Antibody Drug Conjugates. Clin. Cancer Res. 2017, 23, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Ghia, E.M.; Chen, L.; Rassenti, L.Z.; DeBoever, C.; Widhopf, G.F., 2nd; Yu, J.; Neuberg, D.S.; Wierda, W.G.; Rai, K.R.; et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood 2016, 128, 2931–2940. [Google Scholar] [CrossRef] [Green Version]
- Vaisitti, T.; Arruga, F.; Vitale, N.; Lee, T.T.; Ko, M.; Chadburn, A.; Braggio, E.; Di Napoli, A.; Iannello, A.; Allan, J.N.; et al. ROR1 targeting with the antibody-drug conjugate VLS-101 is effective in Richter syndrome patient-derived xenograft mouse models. Blood 2021, 137, 3365–3377. [Google Scholar] [CrossRef]
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci. Transl. Med. 2013, 5, 207ra144. [Google Scholar] [CrossRef]
- Bodyak, N.D.; Mosher, R.; Yurkovetskiy, A.V.; Yin, M.; Bu, C.; Conlon, P.R.; Demady, D.R.; DeVit, M.J.; Gumerov, D.R.; Gurijala, V.R.; et al. The Dolaflexin-based Antibody-Drug Conjugate XMT-1536 Targets the Solid Tumor Lineage Antigen SLC34A2/NaPi2b. Mol. Cancer Ther. 2021, 20, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, A.V.; Bodyak, N.D.; Yin, M.; Thomas, J.D.; Clardy, S.M.; Conlon, P.R.; Stevenson, C.A.; Uttard, A.; Qin, L.; Gumerov, D.R.; et al. Dolaflexin: A Novel Antibody-Drug Conjugate Platform Featuring High Drug Loading and a Controlled Bystander Effect. Mol. Cancer Ther. 2021, 20, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.Y.; Do, P.; Goswami, S.; Nunes, J.; Chiang, C.L.; Elgamal, S.; Ventura, A.M.; Cheney, C.; Zapolnik, K.; Williams, E.; et al. The ROR1 antibody-drug conjugate huXBR1-402-G5-PNU effectively targets ROR1+ leukemia. Blood Adv. 2021, 5, 3152–3162. [Google Scholar] [CrossRef]
- Prince, H.M.; Duvic, M.; Martin, A.; Sterry, W.; Assaf, C.; Sun, Y.; Straus, D.; Acosta, M.; Negro-Vilar, A. Phase III placebo-controlled trial of denileukin diftitox for patients with cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 1870–1877. [Google Scholar] [CrossRef]
- Kawai, H.; Ando, K.; Maruyama, D.; Yamamoto, K.; Kiyohara, E.; Terui, Y.; Fukuhara, N.; Miyagaki, T.; Tokura, Y.; Sakata-Yanagimoto, M.; et al. Phase II study of E7777 in Japanese patients with relapsed/refractory peripheral and cutaneous T-cell lymphoma. Cancer Sci. 2021, 112, 2426–2435. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Karlin, L.; Robak, T.; Gladstone, D.E.; le Coutre, P.; Dietrich, S.; Gotic, M.; et al. Moxetumomab pasudotox in relapsed/refractory hairy cell leukemia. Leukemia 2018, 32, 1768–1777. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Elbayoumi, T.A.; Torchilin, V.P. Current trends in liposome research. Methods Mol. Biol. 2010, 605, 1–27. [Google Scholar] [CrossRef]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control. Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshantri, A.K.; Varela Moreira, A.; Ecker, V.; Mandhane, S.N.; Schiffelers, R.M.; Buchner, M.; Fens, M. Nanomedicines for the treatment of hematological malignancies. J. Control. Release 2018, 287, 194–215. [Google Scholar] [CrossRef] [PubMed]
- Monfardini, C.; Veronese, F.M. Stabilization of substances in circulation. Bioconjug. Chem. 1998, 9, 418–450. [Google Scholar] [CrossRef]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visani, G.; Isidori, A. Doxorubicin variants for hematological malignancies. Nanomedicine 2011, 6, 303–306. [Google Scholar] [CrossRef]
- Houshmand, M.; Garello, F.; Circosta, P.; Stefania, R.; Aime, S.; Saglio, G.; Giachino, C. Nanocarriers as Magic Bullets in the Treatment of Leukemia. Nanomaterials 2020, 10, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef] [Green Version]
- Briuglia, M.L.; Rotella, C.; McFarlane, A.; Lamprou, D.A. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv. Transl. Res. 2015, 5, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef] [Green Version]
- Mu, L.M.; Ju, R.J.; Liu, R.; Bu, Y.Z.; Zhang, J.Y.; Li, X.Q.; Zeng, F.; Lu, W.L. Dual-functional drug liposomes in treatment of resistant cancers. Adv. Drug Deliv. Rev. 2017, 115, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Avilés, A.; Neri, N.; Castañeda, C.; Talavera, A.; Huerta-Guzmán, J.; González, M. Pegylated liposomal doxorubicin in combination chemotherapy in the treatment of previously untreated aggressive diffuse large-B-cell lymphoma. Med. Oncol. 2002, 19, 55–58. [Google Scholar] [CrossRef]
- Tahover, E.; Patil, Y.P.; Gabizon, A.A. Emerging delivery systems to reduce doxorubicin cardiotoxicity and improve therapeutic index: Focus on liposomes. Anticancer Drugs 2015, 26, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines. J. Control. Release 2014, 187, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Barenholz, Y. Doxil®--the first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; Bixby, D.L.; et al. CPX-351 versus 7+3 cytarabine and daunorubicin chemotherapy in older adults with newly diagnosed high-risk or secondary acute myeloid leukaemia: 5-year results of a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2021, 8, e481–e491. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients with Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Blair, H.A. Daunorubicin/Cytarabine Liposome: A Review in Acute Myeloid Leukaemia. Drugs 2018, 78, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Mayer, L.D.; Tardi, P.; Louie, A.C. CPX-351: A nanoscale liposomal co-formulation of daunorubicin and cytarabine with unique biodistribution and tumor cell uptake properties. Int. J. Nanomed. 2019, 14, 3819–3830. [Google Scholar] [CrossRef] [Green Version]
- Visco, C.; Pregnolato, F.; Ferrarini, I.; De Marco, B.; Bonuomo, V.; Sbisà, E.; Fraenza, C.; Bernardelli, A.; Tanasi, I.; Quaglia, F.M.; et al. Efficacy of R-COMP in comparison to R-CHOP in patients with DLBCL: A systematic review and single-arm metanalysis. Crit. Rev. Oncol. Hematol. 2021, 163, 103377. [Google Scholar] [CrossRef]
- Sancho, J.M.; Fernández-Alvarez, R.; Gual-Capllonch, F.; González-García, E.; Grande, C.; Gutiérrez, N.; Peñarrubia, M.J.; Batlle-López, A.; González-Barca, E.; Guinea, J.M.; et al. R-COMP versus R-CHOP as first-line therapy for diffuse large B-cell lymphoma in patients ≥60 years: Results of a randomized phase 2 study from the Spanish GELTAMO group. Cancer Med. 2021, 10, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Fridrik, M.A.; Jaeger, U.; Petzer, A.; Willenbacher, W.; Keil, F.; Lang, A.; Andel, J.; Burgstaller, S.; Krieger, O.; Oberaigner, W.; et al. Cardiotoxicity with rituximab, cyclophosphamide, non-pegylated liposomal doxorubicin, vincristine and prednisolone compared to rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone in frontline treatment of patients with diffuse large B-cell lymphoma: A randomised phase-III study from the Austrian Cancer Drug Therapy Working Group [Arbeitsgemeinschaft Medikamentöse Tumortherapie AGMT](NHL-14). Eur. J. Cancer 2016, 58, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, A.J.; Shah, G.; Schöder, H.; Ganesan, N.; Drill, E.; Hancock, H.; Davey, T.; Perez, L.; Ryu, S.; Sohail, S.; et al. Phase II Trial of Pembrolizumab Plus Gemcitabine, Vinorelbine, and Liposomal Doxorubicin as Second-Line Therapy for Relapsed or Refractory Classical Hodgkin Lymphoma. J. Clin. Oncol. 2021, 39, 3109–3117. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Zhang, W.; Liu, Y.; Yang, Q.; Rasko, J.E.J.; Nie, J.; Liu, J.; Li, X.; Dong, L.; Chen, M.; et al. Camrelizumab Plus Gemcitabine, Vinorelbine, and Pegylated Liposomal Doxorubicin in Relapsed/Refractory Primary Mediastinal B-Cell Lymphoma: A Single-Arm, Open-Label, Phase II Trial. Clin. Cancer Res. 2020, 26, 4521–4530. [Google Scholar] [CrossRef] [PubMed]
- Vu, K.; Wu, C.H.; Yang, C.Y.; Zhan, A.; Cavallone, E.; Berry, W.; Heeter, P.; Pincus, L.; Wieduwilt, M.J.; William, B.M.; et al. Romidepsin Plus Liposomal Doxorubicin Is Safe and Effective in Patients with Relapsed or Refractory T-Cell Lymphoma: Results of a Phase I Dose-Escalation Study. Clin. Cancer Res. 2020, 26, 1000–1008. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, X.Q.; Xia, Z.J. New tricks for old drugs: Combination of rituximab and two nanoparticle-delivered chemotherapy drugs, albumin-bound paclitaxel and pegylated liposomal doxorubicin, in the treatment of relapsed/refractory diffuse large B cell lymphoma. Leuk. Lymphoma 2020, 61, 2502–2506. [Google Scholar] [CrossRef]
- Kaplan, L.D.; Deitcher, S.R.; Silverman, J.A.; Morgan, G. Phase II study of vincristine sulfate liposome injection (Marqibo) and rituximab for patients with relapsed and refractory diffuse large B-Cell lymphoma or mantle cell lymphoma in need of palliative therapy. Clin. Lymphoma Myeloma Leuk. 2014, 14, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Hagemeister, F.; Rodriguez, M.A.; Deitcher, S.R.; Younes, A.; Fayad, L.; Goy, A.; Dang, N.H.; Forman, A.; McLaughlin, P.; Medeiros, L.J.; et al. Long term results of a phase 2 study of vincristine sulfate liposome injection (Marqibo(®) ) substituted for non-liposomal vincristine in cyclophosphamide, doxorubicin, vincristine, prednisone with or without rituximab for patients with untreated aggressive non-Hodgkin lymphomas. Br. J. Haematol. 2013, 162, 631–638. [Google Scholar] [CrossRef]
- Wang, D.; Sun, Y.; Liu, Y.; Meng, F.; Lee, R.J. Clinical translation of immunoliposomes for cancer therapy: Recent perspectives. Expert Opin. Drug Deliv. 2018, 15, 893–903. [Google Scholar] [CrossRef]
- Merino, M.; Zalba, S.; Garrido, M.J. Immunoliposomes in clinical oncology: State of the art and future perspectives. J. Control. Release 2018, 275, 162–176. [Google Scholar] [CrossRef]
- Sun, H.; Zhu, X.; Lu, P.Y.; Rosato, R.R.; Tan, W.; Zu, Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, e182. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. PEG conjugates in clinical development or use as anticancer agents: An overview. Adv. Drug Deliv. Rev. 2009, 61, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Pasut, G.; Veronese, F.M. State of the art in PEGylation: The great versatility achieved after forty years of research. J. Control. Release 2012, 161, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Viegas, T.X.; Bentley, M.D.; Harris, J.M.; Fang, Z.; Yoon, K.; Dizman, B.; Weimer, R.; Mero, A.; Pasut, G.; Veronese, F.M. Polyoxazoline: Chemistry, properties, and applications in drug delivery. Bioconjug. Chem. 2011, 22, 976–986. [Google Scholar] [CrossRef]
- Hoelzer, D.; Leiske, M.N.; Hartlieb, M.; Bus, T.; Pretzel, D.; Hoeppener, S.; Kempe, K.; Thierbach, R.; Schubert, U.S. Tumor targeting with pH-responsive poly(2-oxazoline)-based nanogels for metronomic doxorubicin treatment. Oncotarget 2018, 9, 22316–22331. [Google Scholar] [CrossRef] [Green Version]
- Zahoranová, A.; Luxenhofer, R. Poly(2-oxazoline)- and Poly(2-oxazine)-Based Self-Assemblies, Polyplexes, and Drug Nanoformulations-An Update. Adv. Healthc. Mater. 2021, 10, e2001382. [Google Scholar] [CrossRef]
- Kopeček, J.; Yang, J. Polymer nanomedicines. Adv. Drug Deliv. Rev. 2020, 156, 40–64. [Google Scholar] [CrossRef]
- Chytil, P.; Kostka, L.; Etrych, T. HPMA Copolymer-Based Nanomedicines in Controlled Drug Delivery. J. Pers. Med. 2021, 11, 115. [Google Scholar] [CrossRef]
- Lidický, O.; Klener, P.; Machová, D.; Vočková, P.; Pokorná, E.; Helman, K.; Mavis, C.; Janoušková, O.; Etrych, T. Overcoming resistance to rituximab in relapsed non-Hodgkin lymphomas by antibody-polymer drug conjugates actively targeted by anti-CD38 daratumumab. J. Control. Release 2020, 328, 160–170. [Google Scholar] [CrossRef]
- Etrych, T.; Daumová, L.; Pokorná, E.; Tušková, D.; Lidický, O.; Kolářová, V.; Pankrác, J.; Šefc, L.; Chytil, P.; Klener, P. Effective doxorubicin-based nano-therapeutics for simultaneous malignant lymphoma treatment and lymphoma growth imaging. J. Control. Release 2018, 289, 44–55. [Google Scholar] [CrossRef]
- Wiesing, U. Theranostics: Is it really a revolution? Evaluating a new term in medicine. Med. Health Care Philos. 2019, 22, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, A.; Eppard, E.; Kürpig, S.; Bundschuh, R.A.; Schönberger, S.; Gonzalez-Carmona, M.; Feldmann, G.; Ahmadzadehfar, H.; Essler, M. Theranostics in nuclear medicine practice. Onco Targets Ther. 2017, 10, 4821–4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidlin, S.M.; Rossman, I. Radioiodine therapy of metastases from carcinoma of the thyroid; a 6-year progress report. J. Clin. Endocrinol. Metab. 1949, 9, 1122–1137. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.C., Jr.; Libby, R.L.; Cassen, B. The scintillation counter in clinical studies of human thyroid physiology using I131. J. Clin. Endocrinol. Metab. 1951, 11, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Bono, J.S.D.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Phase III study of lutetium-177-PSMA-617 in patients with metastatic castration-resistant prostate cancer (VISION). J. Clin. Oncol. 2021, 39, LBA4. [Google Scholar] [CrossRef]
- Czernin, J.; Calais, J. (177)Lu-PSMA617 and the VISION Trial: One of the Greatest Success Stories in the History of Nuclear Medicine. J. Nucl. Med. 2021, 62, 1025–1026. [Google Scholar] [CrossRef]
- Kang, L.; Li, C.; Rosenkrans, Z.T.; Huo, N.; Chen, Z.; Ehlerding, E.B.; Huo, Y.; Ferreira, C.A.; Barnhart, T.E.; Engle, J.W.; et al. CD38-Targeted Theranostics of Lymphoma with (89)Zr/(177)Lu-Labeled Daratumumab. Adv. Sci. 2021, 8, 2001879. [Google Scholar] [CrossRef]
- Skarbnik, A.P.; Smith, M.R. Radioimmunotherapy in mantle cell lymphoma. Best Pract. Res. Clin. Haematol. 2012, 25, 201–210. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, M.S.; Tuck, M.; Estes, J.; Kolstad, A.; Ross, C.W.; Zasadny, K.; Regan, D.; Kison, P.; Fisher, S.; Kroll, S.; et al. 131I-tositumomab therapy as initial treatment for follicular lymphoma. N. Engl. J. Med. 2005, 352, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Buchsbaum, D.J. CD38 pretargeted RIT of B-cell tumors. Blood 2018, 131, 589–590. [Google Scholar] [CrossRef]
- Green, D.J.; O’Steen, S.; Lin, Y.; Comstock, M.L.; Kenoyer, A.L.; Hamlin, D.K.; Wilbur, D.S.; Fisher, D.R.; Nartea, M.; Hylarides, M.D.; et al. CD38-bispecific antibody pretargeted radioimmunotherapy for multiple myeloma and other B-cell malignancies. Blood 2018, 131, 611–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dun, Y.; Huang, G.; Liu, J.; Wei, W. ImmunoPET imaging of hematological malignancies: From preclinical promise to clinical reality. Drug Discov. Today 2021. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Rosenkrans, Z.T.; Liu, J.; Huang, G.; Luo, Q.Y.; Cai, W. ImmunoPET: Concept, Design, and Applications. Chem. Rev. 2020, 120, 3787–3851. [Google Scholar] [CrossRef] [PubMed]
- Kahle, X.U.; Montes de Jesus, F.M.; Glaudemans, A.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Plattel, W.J.; van Meerten, T.; Diepstra, A.; van den Berg, A.; Kwee, T.C.; et al. Molecular imaging in lymphoma beyond (18)F-FDG-PET: Understanding the biology and its implications for diagnostics and therapy. Lancet Haematol. 2020, 7, e479–e489. [Google Scholar] [CrossRef]
- Muylle, K.; Flamen, P.; Vugts, D.J.; Guiot, T.; Ghanem, G.; Meuleman, N.; Bourgeois, P.; Vanderlinden, B.; van Dongen, G.A.; Everaert, H.; et al. Tumour targeting and radiation dose of radioimmunotherapy with (90)Y-rituximab in CD20+ B-cell lymphoma as predicted by (89)Zr-rituximab immuno-PET: Impact of preloading with unlabelled rituximab. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1304–1314. [Google Scholar] [CrossRef]
- Schottelius, M.; Osl, T.; Poschenrieder, A.; Hoffmann, F.; Beykan, S.; Hänscheid, H.; Schirbel, A.; Buck, A.K.; Kropf, S.; Schwaiger, M.; et al. [(177)Lu]pentixather: Comprehensive Preclinical Characterization of a First CXCR4-directed Endoradiotherapeutic Agent. Theranostics 2017, 7, 2350–2362. [Google Scholar] [CrossRef]
- Lapa, C.; Hänscheid, H.; Kircher, M.; Schirbel, A.; Wunderlich, G.; Werner, R.A.; Samnick, S.; Kotzerke, J.; Einsele, H.; Buck, A.K.; et al. Feasibility of CXCR4-Directed Radioligand Therapy in Advanced Diffuse Large B-Cell Lymphoma. J. Nucl. Med. 2019, 60, 60–64. [Google Scholar] [CrossRef]
- Paul, D.M.; Ghiuzeli, C.M.; Rini, J.; Palestro, C.J.; Fung, E.K.; Ghali, M.; Ben-Levi, E.; Prideaux, A.; Vallabhajosula, S.; Popa, E.C. A pilot study treatment of malignant tumors using low-dose (18)F-fluorodeoxyglucose ((18)F-FDG). Am. J. Nucl. Med. Mol. Imaging 2020, 10, 334–341. [Google Scholar]
- Anani, T.; Rahmati, S.; Sultana, N.; David, A.E. MRI-traceable theranostic nanoparticles for targeted cancer treatment. Theranostics 2021, 11, 579–601. [Google Scholar] [CrossRef]
- Li, F.; Liang, Z.; Liu, J.; Sun, J.; Hu, X.; Zhao, M.; Liu, J.; Bai, R.; Kim, D.; Sun, X.; et al. Dynamically Reversible Iron Oxide Nanoparticle Assemblies for Targeted Amplification of T1-Weighted Magnetic Resonance Imaging of Tumors. Nano Lett. 2019, 19, 4213–4220. [Google Scholar] [CrossRef]
- Yang, L.; Wang, L.; Huang, G.; Zhang, X.; Chen, L.; Li, A.; Gao, J.; Zhou, Z.; Su, L.; Yang, H.; et al. Improving the sensitivity of T (1) contrast-enhanced MRI and sensitive diagnosing tumors with ultralow doses of MnO octahedrons. Theranostics 2021, 11, 6966–6982. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhu, Q.; Zeng, Y.; Zeng, Q.; Chen, X.; Zhan, Y. Manganese Oxide Nanoparticles As MRI Contrast Agents In Tumor Multimodal Imaging And Therapy. Int. J. Nanomed. 2019, 14, 8321–8344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, M.; Mallela, J.; Wang, C.; Ravi, S.; Dixit, S.; Garapati, U.; Mohapatra, S. Manganese-loaded lipid-micellar theranostics for simultaneous drug and gene delivery to lungs. J. Control. Release 2013, 167, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Lin, H.; Zhang, G.; Si, Y.; Yang, H.; Bai, G.; Yang, C.; Zhong, K.; Cai, D.; Wu, Z.; et al. Effective pH-Activated Theranostic Platform for Synchronous Magnetic Resonance Imaging Diagnosis and Chemotherapy. ACS Appl. Mater. Interfaces 2018, 10, 31114–31123. [Google Scholar] [CrossRef]
- Klenk, C.; Gawande, R.; Uslu, L.; Khurana, A.; Qiu, D.; Quon, A.; Donig, J.; Rosenberg, J.; Luna-Fineman, S.; Moseley, M.; et al. Ionising radiation-free whole-body MRI versus (18)F-fluorodeoxyglucose PET/CT scans for children and young adults with cancer: A prospective, non-randomised, single-centre study. Lancet Oncol. 2014, 15, 275–285. [Google Scholar] [CrossRef]
- Zhi, D.; Yang, T.; Yang, J.; Fu, S.; Zhang, S. Targeting strategies for superparamagnetic iron oxide nanoparticles in cancer therapy. Acta Biomater. 2020, 102, 13–34. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generic Name | Trade Name | Target Antigen | Linker | Toxic Payload | Target Patient Population | Approval Date |
|---|---|---|---|---|---|---|
| Brentuximab vedotin | Adcetris® | CD30 | Enzyme cleavable | Auristatin | R/R HL, CD30+ T-NHL, MF | 2017 |
| Inotuzumab ozogamicin | Besponsa® | CD22 | pH cleavable | Calicheamicin | R/R B-ALL | 2017 |
| Moxetumomab pasudotox | Lumoxiti® | CD22 | Enzyme cleavable | Pseudomonas exotoxin | R/R HCL | 2018 |
| Polatuzumab vedotin | Polivy® | CD79B | Enzyme cleavable | Auristatin | R/R DLBCL | 2019 |
| Loncastuximab tesirine | Zynlonta® | CD19 | Enzyme cleavable | PBD dimer | R/R DLBCL | 2021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etrych, T.; Braunova, A.; Zogala, D.; Lambert, L.; Renesova, N.; Klener, P. Targeted Drug Delivery and Theranostic Strategies in Malignant Lymphomas. Cancers 2022, 14, 626. https://doi.org/10.3390/cancers14030626
Etrych T, Braunova A, Zogala D, Lambert L, Renesova N, Klener P. Targeted Drug Delivery and Theranostic Strategies in Malignant Lymphomas. Cancers. 2022; 14(3):626. https://doi.org/10.3390/cancers14030626
Chicago/Turabian StyleEtrych, Tomas, Alena Braunova, David Zogala, Lukas Lambert, Nicol Renesova, and Pavel Klener. 2022. "Targeted Drug Delivery and Theranostic Strategies in Malignant Lymphomas" Cancers 14, no. 3: 626. https://doi.org/10.3390/cancers14030626
APA StyleEtrych, T., Braunova, A., Zogala, D., Lambert, L., Renesova, N., & Klener, P. (2022). Targeted Drug Delivery and Theranostic Strategies in Malignant Lymphomas. Cancers, 14(3), 626. https://doi.org/10.3390/cancers14030626