
Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives


Abstract
:Simple Summary
Abstract
1. Introduction
2. Glycosphingolipids
2.1. GD2
2.2. GD3
2.3. Fucosyl-GM1
2.4. GM3
2.5. Globo-Series
3. Simple Mucin-Type O-Glycan Antigens
3.1. Tn Antigen
3.2. Sialyl-Tn Antigen
3.3. TF Antigen
3.4. Parasite Glycans and Cancer Immunotherapy
4. Lewis Antigens
5. Polysialic Acid
6. Polyvalent TACA Vaccines
7. CAR-T Cells
7.1. CAR-T Cells Targeting GD2
7.2. CAR-T Cells Targeting MUC1/MUC1-Tn
7.3. CAR-T Cells Targeting STn
8. Concluding Remarks and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Finck, A.; Gill, S.I.; June, C.H. Cancer immunotherapy comes of age and looks for maturity. Nat. Commun. 2020, 11, 3325. [Google Scholar] [CrossRef] [PubMed]
- Goydel, R.S.; Rader, C. Antibody-based cancer therapy. Oncogene 2021, 40, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; O'Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; Guérardel, Y.; Delannoy, P. Gangliosides: Structures, Biosynthesis, Analysis, and Roles in Cancer. ChemBioChem 2017, 18, 1146–1154. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Biochim Biophys Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Acta Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef]
- Croci, D.O.; Cerliani, J.P.; Pinto, N.A.; Morosi, L.G.; Rabinovich, G.A. Regulatory role of glycans in the control of hypoxia-driven angiogenesis and sensitivity to anti-angiogenic treatment. Glycobiology 2014, 24, 1283–1290. [Google Scholar] [CrossRef] [Green Version]
- Mereiter, S.; Balmaña, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Dusoswa, S.A.; Verhoeff, J.; Abels, E.; Méndez-Huergo, S.P.; Croci, D.O.; Kuijper, L.H.; de Miguel, E.; Wouters, V.M.C.J.; Best, M.G.; Rodriguez, E.; et al. Glioblastomas exploit truncated O-linked glycans for local and distant immune modulation via the macrophage galactose-type lectin. Proc. Natl. Acad. Sci. USA 2020, 117, 3693–3703. [Google Scholar] [CrossRef] [PubMed]
- Fuster, M.M.; Esko, J.D. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 2005, 5, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Vankemmelbeke, M.; Chua, J.X.; Durrant, L.G. Cancer cell associated glycans as targets for immunotherapy. Oncoimmunology 2015, 5, e1061177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, E.; Schetters, S.T.T.; van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, M.; Berti, F.; Bzducha-Wróbel, A.; Chiodo, F.; Colombo, C.; Compostella, F.; Durlik, K.; Ferhati, X.; Holmdahl, R.; Jovanovic, D.; et al. Emerging glyco-based strategies to steer immune responses. FEBS J. 2021, 288, 4746–4772. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S.; Zhang, Y. Glycosphingolipid antigens and cancer therapy. Chem. Biol. 1997, 4, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.T.; Lan, H.R.; Chen, X.Y.; Wang, S.B.; Ying, X.J.; Lin, Y.; Mou, X.Z. Recent advances in carbohydrate-based cancer vaccines. Biotechnol. Lett. 2019, 41, 641–650. [Google Scholar] [CrossRef]
- Ragupathi, G.; Liu, N.X.; Musselli, C.; Powell, S.; Lloyd, K.; Livingston, P.O. Antibodies against tumor cell glycolipids and proteins, but not mucins, mediate complement-dependent cytotoxicity. J. Immunol. 2005, 174, 5706–5712. [Google Scholar] [CrossRef] [Green Version]
- Hubert, P.; Heitzmann, A.; Viel, S.; Nicolas, A.; Sastre-Garau, X.; Oppezzo, P.; Pritsch, O.; Osinaga, E.; Amigorena, S. Antibody-dependent cell cytotoxicity synapses form in mice during tumor-specific antibody immunotherapy. Cancer Res. 2011, 71, 5134–5143. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Dinutuximab: First global approval. Drugs 2015, 75, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S.I. Structure and function of glycosphingolipids and sphingolipids: Recollections and future trends. Biochim. Biophys. Acta 2008, 1780, 325–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D'Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef] [PubMed]
- Hakomori, S.I. Glycosynaptic microdomains controlling tumor cell phenotype through alteration of cell growth, adhesion, and motility. FEBS Lett. 2010, 584, 1901–1906. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hung, J.T.; Wang, S.H.; Cheng, J.Y.; Yu, A.L. Targeting glycosphingolipids for cancer immunotherapy. FEBS Lett. 2020, 594, 3602–3618. [Google Scholar] [CrossRef]
- Yu, A.L.; Hung, J.T.; Ho, M.Y.; Yu, J. Alterations of Glycosphingolipids in Embryonic Stem Cell Differentiation and Development of Glycan-Targeting Cancer Immunotherapy. Stem Cells Dev. 2016, 25, 1532–1548. [Google Scholar] [CrossRef]
- Regina Todeschini, A.; Hakomori, S.I. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta 2008, 1780, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Cumin, C.; Huang, Y.L.; Everest-Dass, A.; Jacob, F. Deciphering the Importance of Glycosphingolipids on Cellular and Molecular Mechanisms Associated with Epithelial-to-Mesenchymal Transition in Cancer. Biomolecules 2021, 11, 62. [Google Scholar] [CrossRef]
- Giussani, P.; Tringali, C.; Riboni, L.; Viani, P.; Venerando, B. Sphingolipids: Key regulators of apoptosis and pivotal players in cancer drug resistance. Int. J. Mol. Sci. 2014, 15, 4356–4392. [Google Scholar] [CrossRef] [Green Version]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Yuki, N.; Yamada, M.; Tagawa, Y.; Takahashi, H.; Handa, S. Pathogenesis of the neurotoxicity caused by anti-GD2 antibody therapy. J. Neurol. Sci. 1997, 149, 127–130. [Google Scholar] [CrossRef]
- Cavdarli, S.; Groux-Degroote, S.; Delannoy, P. Gangliosides: The Double-Edge Sword of Neuro-Ectodermal Derived Tumors. Biomolecules 2019, 9, 311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Kawaguchi, H.; Sato, S.; Ueda, R.; Furukawa, K. An anti-GD2 monoclonal antibody enhances apoptotic effects of anti-cancer drugs against small cell lung cancer cells via JNK (c-Jun terminal kinase) activation. Jpn. J. Cancer Res. 2002, 93, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Doronin, I.I.; Vishnyakova, P.A.; Kholodenko, I.V.; Ponomarev, E.D.; Ryazantsev, D.Y.; Molotkovskaya, I.M.; Kholodenko, R.V. Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer 2014, 14, 295. [Google Scholar] [CrossRef] [Green Version]
- Shurin, G.V.; Shurin, M.R.; Bykovskaia, S.; Shogan, J.; Lotze, M.T.; Barksdale, E.M. Jr. Neuroblastoma-derived gangliosides inhibit dendritic cell generation and function. Cancer Res. 2001, 61, 363–369. [Google Scholar] [PubMed]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albertini, M.R.; Hank, J.A.; Gadbaw, B.; Kostlevy, J.; Haldeman, J.; Schalch, H.; Gan, J.; Kim, K.; Eickhoff, J.; Gillies, S.D.; et al. Phase II trial of hu14.18-IL2 for patients with metastatic melanoma. Cancer Immunol. Immunother. 2012, 61, 2261–2271. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.K.; Guo, H.; Hu, J.; Tassev, D.V.; Cheung, I.Y. Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology 2012, 1, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.K.; Cheung, I.Y.; Kushner, B.H.; Ostrovnaya, I.; Chamberlain, E.; Kramer, K.; Modak, S. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J. Clin. Oncol. 2012, 30, 3264–3270. [Google Scholar] [CrossRef] [Green Version]
- Mueller, B.M.; Romerdahl, C.A.; Gillies, S.D.; Reisfeld, R.A. Enhancement of antibody-dependent cytotoxicity with a chimeric anti-GD2 antibody. J. Immunol. 1990, 144, 1382–1386. [Google Scholar]
- Barker, E.; Mueller, B.M.; Handgretinger, R.; Herter, M.; Yu, A.L.; Reisfeld, R.A. Effect of a chimeric anti-ganglioside GD2 antibody on cell-mediated lysis of human neuroblastoma cells. Cancer Res. 1991, 51, 144–149. [Google Scholar] [PubMed]
- Seeger, R.C. Immunology and immunotherapy of neuroblastoma. Semin. Cancer Biol. 2011, 21, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.K. Humanized 3F8 Anti-GD2 Monoclonal Antibody Dosing with Granulocyte-Macrophage Colony-Stimulating Factor in Patients with Resistant Neuroblastoma: A Phase 1 Clinical Trial. JAMA Oncol. 2018, 4, 1729–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, I.Y.; Kushner, B.H.; Modak, S.; Basu, E.M.; Roberts, S.S.; Cheung, N.V. Phase I trial of anti-GD2 monoclonal antibody hu3F8 plus GM-CSF: Impact of body weight, immunogenicity and anti-GD2 response on pharmacokinetics and survival. Oncoimmunology 2017, 6, e1358331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. . An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert. Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Albertini, M.R.; Yang, R.K.; Ranheim, E.A.; Hank, J.A.; Zuleger, C.L.; Weber, S.; Neuman, H.; Hartig, G.; Weigel, T.; Mahvi, D.; et al. Pilot trial of the hu14.18-IL2 immunocytokine in patients with completely resectable recurrent stage III or stage IV melanoma. Cancer Immunol. Immunother. 2018, 67, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.K.; Kuznetsov, I.B.; Ranheim, E.A.; Wei, J.S.; Sindiri, S.; Gryder, B.E.; Gangalapudi, V.; Song, Y.K.; Patel, V.; Hank, J.A.; et al. Outcome-Related Signatures Identified by Whole Transcriptome Sequencing of Resectable Stage III/IV Melanoma Evaluated after Starting Hu14.18-IL2. Clin. Cancer Res. 2020, 26, 3296–3306. [Google Scholar] [CrossRef] [Green Version]
- Park, J.A.; Cheung, N.V. Targets and Antibody Formats for Immunotherapy of Neuroblastoma. J. Clin. Oncol. 2020, 38, 1836–1848. [Google Scholar] [CrossRef]
- Forero, A.; Shah, J.; Carlisle, R.; Triozzi, P.L.; LoBuglio, A.F.; Wang, W.Q.; Fujimori, M.; Conry, R.M. A phase I study of an anti-GD3 monoclonal antibody, KW-2871, in patients with metastatic melanoma. Cancer Biother. Radiopharm. 2006, 21, 561–568. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Moschos, S.J.; Lin, Y.; Lin, H.M.; Sander, C.; Yin, Y.; Venhaus, R.; Gajewski, T.F.; Kirkwood, J.M. Safety and efficacy of the antiganglioside GD3 antibody ecromeximab (KW2871) combined with high-dose interferon-α2b in patients with metastatic melanoma. Melanoma Res. 2017, 27, 342–350. [Google Scholar] [CrossRef]
- Chu, Q.; Leighl, N.; Surmont, V.; Van Herpen, C.; Sibille, A.; Markman, B.; Clarke, S.; Juergens, R.; Acosta Rivera, M.; Andelkovic, V.; et al. Clinical Activity of BMS-986012, an AntieFucosyl-GM1 Monoclonal Antibody, Plus Nivolumab in Small Cell Lung Cancer. J. Thorac. Oncol. 2021, 16, S195. [Google Scholar] [CrossRef]
- Fiedler, W.; DeDosso, S.; Cresta, S.; Weidmann, J.; Tessari, A.; Salzberg, M.; Dietrich, B.; Baumeister, H.; Goletz, S.; Gianni, L.; et al. A phase I study of PankoMab-GEX, a humanised glyco-optimised monoclonal antibody to a novel tumour-specific MUC1 glycopeptide epitope in patients with advanced carcinomas. Eur. J. Cancer 2016, 63, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.; Sehouli, J.; Zurawski, B.; Raspagliesi, F.; De Giorgi, U.; Banerjee, S.; Arranz Arija, J.; Romeo Marin, M.; Lisyanskaya, A.; Póka, R. LBA41A double-blind, placebo-controlled, randomized, phase 2 study to evaluate the efficacy and safety of switch maintenance therapy with the anti-TA-MUC1 antibody PankoMab-GEX after chemotherapy in patients with recurrent epithelial ovarian carcinoma. Ann. Oncol. 2017, 28 (Suppl. 5), v626. [Google Scholar] [CrossRef] [Green Version]
- Garralda, E.; Del Conte, G.; Macchini, M.; Matos, I.; Klinghammer, K.F.; Saavedra, O.; Fiedler, W.M.; Rolling, C.C.; Kebenko, M.; Raspagliesi, F.; et al. Activity results of the GATTO study, a phase Ib study combining the anti-TA-MUC1 antibody gatipotuzumab with the anti-EGFR tomuzotuximab or panitumumab in patients with refractory solid tumors. J. Clin. Oncol. 2021, 39, 2522. [Google Scholar] [CrossRef]
- McQuarrie, S.A.; MacLean, G.D.; Boniface, G.R.; Golberg, K.; McEwan, A.J. Radioimmunoscintigraphy in patients with breast adenocarcinoma using technetium-99m labelled monoclonal antibody 170H.82: Report of a phase II study. Eur J. Nucl. Med. 1997, 24, 381–389. [Google Scholar] [CrossRef]
- Gupta, S.; McDonald, J.D.; Ayabe, R.I.; Khan, T.M.; Gamble, L.A.; Sinha, S.; Hannah, C.; Blakely, A.M.; Davis, J.L.; Hernandez, J.M. Targeting CA 19-9 with a humanized monoclonal antibody at the time of surgery may decrease recurrence rates for patients undergoing resections for pancreatic cancer, cholangiocarcinoma and metastatic colorectal cancer. J. Gastrointest. Oncol. 2020, 11, 231–235. [Google Scholar] [CrossRef]
- Krug, L.M.; Milton, D.T.; Jungbluth, A.A.; Chen, L.C.; Quaia, E.; Pandit-Taskar, N.; Nagel, A.; Jones, J.; Kris, M.G.; Finn, R.; et al. Targeting Lewis Y (Le(y)) in small cell lung cancer with a humanized monoclonal antibody, hu3S193: A pilot trial testing two dose levels. J. Thorac. Oncol. 2007, 2, 947–952. [Google Scholar] [CrossRef]
- Smaletz, O.; Diz, M.D.; do Carmo, C.C.; Sabbaga, J.; Cunha-Junior, G.F.; Azevedo, S.J.; Maluf, F.C.; Barrios, C.H.; Costa, R.L.; Fontana, A.G.; et al. A phase II trial with anti-Lewis-Y monoclonal antibody (hu3S193) for the treatment of platinum resistant/refractory ovarian, fallopian tube and primary peritoneal carcinoma. Gynecol. Oncol. 2015, 138, 272–277. [Google Scholar] [CrossRef]
- Ross, H.J.; Hart, L.L.; Swanson, P.M.; Rarick, M.U.; Figlin, R.A.; Jacobs, A.D.; McCune, D.E.; Rosenberg, A.H.; Baron, A.D.; Grove, L.E.; et al. A randomized, multicenter study to determine the safety and efficacy of the immunoconjugate SGN-15 plus docetaxel for the treatment of non-small cell lung carcinoma. Lung Cancer. 2006, 54, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Rader, C. Bispecific antibodies in cancer immunotherapy. Curr. Opin. Biotechnol. 2020, 65, 9–16. [Google Scholar] [CrossRef]
- Rashidijahanabad, Z.; Huang, X. Recent advances in tumor associated carbohydrate antigen based chimeric antigen receptor T cells and bispecific antibodies for anti-cancer immunotherapy. Semin. Immunol. 2020, 47, 101390. [Google Scholar] [CrossRef]
- Michon, J.; Perdereau, B.; Brixy, F.; Moutel, S.; Fridman, W.H.; Teillaud, J.L. In vivo targeting of human neuroblastoma xenograft by anti-GD2/anti-Fc gamma RI (CD64) bispecific antibody. Eur. J. Cancer 1995, 31A, 631–636. [Google Scholar] [CrossRef]
- Cheng, M.; Ahmed, M.; Xu, H.; Cheung, N.K. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int. J. Cancer 2015, 136, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Santich, B.H.; Xu, H.; Ahmed, M.; Huse, M.; Cheung, N.K. Successful engineering of a highly potent single-chain variable-fragment (scFv) bispecific antibody to target disialoganglioside (GD2) positive tumors. Oncoimmunology 2016, 5, e1168557. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Cheng, M.; Guo, H.; Chen, Y.; Huse, M.; Cheung, N.K. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol. Res. 2015, 3, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zirngibl, F.; Ivasko, S.M.; Grunewald, L.; Klaus, A.; Schwiebert, S.; Ruf, P.; Lindhofer, H.; Astrahantseff, K.; Andersch, L.; Schulte, J.H.; et al. GD2-directed bispecific trifunctional antibody outperforms dinutuximab beta in a murine model for aggressive metastasized neuroblastoma. J. Immunother. Cancer 2021, 9, e002923. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.H.; Cheung, I.Y.; Modak, S.; Kramer, K.; Ragupathi, G.; Cheung, N.K. Phase I trial of a bivalent gangliosides vaccine in combination with β-glucan for high-risk neuroblastoma in second or later remission. Clin. Cancer Res. 2014, 20, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Cheung, I.Y.; Cheung, N.V.; Modak, S.; Mauguen, A.; Feng, Y.; Basu, E.; Roberts, S.S.; Ragupathi, G.; Kushner, B.H. Survival Impact of Anti-GD2 Antibody Response in a Phase II Ganglioside Vaccine Trial Among Patients With High-Risk Neuroblastoma With Prior Disease Progression. J. Clin. Oncol. 2021, 39, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.C.; Kris, M.G.; Houghton, A.N.; Chapman, P.B. Long survival of patients with small cell lung cancer after adjuvant treatment with the anti-idiotypic antibody BEC2 plus Bacillus Calmette-Guérin. Clin. Cancer Res. 1999, 5, 1319–1323. [Google Scholar]
- Giaccone, G.; Debruyne, C.; Felip, E.; Chapman, P.B.; Grant, S.C.; Millward, M.; Thiberville, L.; D'addario, G.; Coens, C.; Rome, L.S.; et al. Phase III study of adjuvant vaccination with Bec2/bacille Calmette-Guerin in responding patients with limited-disease small-cell lung cancer (European Organisation for Research and Treatment of Cancer 08971-08971B; Silva Study). J. Clin. Oncol. 2005, 23, 6854–6864. [Google Scholar] [CrossRef]
- Bottomley, A.; Debruyne, C.; Felip, E.; Millward, M.; Thiberville, L.; D'Addario, G.; Rome, L.; Zatloukal, P.; Coens, C.; Giaccone, G. Symptom and quality of life results of an international randomised phase III study of adjuvant vaccination with Bec2/BCG in responding patients with limited disease small-cell lung cancer. Eur. J. Cancer 2008, 44, 2178–2184. [Google Scholar] [CrossRef] [PubMed]
- Cacciavillano, W.; Sampor, C.; Venier, C.; Gabri, M.R.; de Dávila, M.T.; Galluzzo, M.L.; Guthmann, M.D.; Fainboim, L.; Alonso, D.F.; Chantada, G.L. A Phase I Study of the Anti-Idiotype Vaccine Racotumomab in Neuroblastoma and Other Pediatric Refractory Malignancies. Pediatr. Blood Cancer. 2015, 62, 2120–2124. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Yu, A.L.; Tseng, L.M.; Chow, L.W.C.; Hou, M.F.; Hurvitz, S.A.; Schwab, R.B.; Murray, J.; Chang, H.K.; Chang, H.T.; et al. Globo H-KLH vaccine adagloxad simolenin (OBI-822)/OBI-821 in patients with metastatic breast cancer: Phase II randomized, placebo-controlled study. J. Immunother. Cancer 2020, 8, e000342. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, P.; Artaud, C.; Bay, S.; Ganneau, C.; Campone, M.; Delaloge, S.; Gourmelon, C.; Loirat, D.; Medioni, J.; Pein, F.; et al. The fully synthetic glycopeptide MAG-Tn3 therapeutic vaccine induces tumor-specific cytotoxic antibodies in breast cancer patients. Cancer Immunol. Immunother. 2020, 69, 703–716. [Google Scholar] [CrossRef]
- Ibrahim, N.K.; Murray, J.L.; Zhou, D.; Mittendorf, E.A.; Sample, D.; Tautchin, M.; Miles, D. Survival Advantage in Patients with Metastatic Breast Cancer Receiving Endocrine Therapy plus Sialyl Tn-KLH Vaccine: Post Hoc Analysis of a Large Randomized Trial. J. Cancer 2013, 4, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Miles, D.; Roché, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III multicenter clinical trial of the sialyl-TN (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–2100. [Google Scholar] [CrossRef] [Green Version]
- Slovin, S.F.; Ragupathi, G.; Musselli, C.; Fernandez, C.; Diani, M.; Verbel, D.; Danishefsky, S.; Livingston, P.; Scher, H.I. Thomsen-Friedenreich (TF) antigen as a target for prostate cancer vaccine: Clinical trial results with TF cluster (c)-KLH plus QS21 conjugate vaccine in patients with biochemically relapsed prostate cancer. Cancer Immunol. Immunother. 2005, 54, 694–702. [Google Scholar] [CrossRef]
- Diab, A.; Ragupathi, G.; Scholz, W.W.; Panageas, K.; Hudis, C.; Livingston, P.O.; Gilewski, T. A pilot study of vaccination with sialyl Lewisa (sLea)–keyhole limpet hemocyanin (KLH) conjugate plus the immunologic adjuvant QS-21 in metastatic breast cancer patients (pts). J. Clin. Oncol. 2011, 29, 2599. [Google Scholar] [CrossRef]
- Krug, L.M.; Ragupathi, G.; Hood, C.; George, C.; Hong, F.; Shen, R.; Abrey, L.; Jennings, H.J.; Kris, M.G.; Livingston, P.O. Immunization with N-propionyl polysialic acid-KLH conjugate in patients with small cell lung cancer is safe and induces IgM antibodies reactive with SCLC cells and bactericidal against group B meningococci. Cancer Immunol. Immunother. 2012, 61, 9–18. [Google Scholar] [CrossRef] [Green Version]
- O'Cearbhaill, R.E.; Ragupathi, G.; Zhu, J.; Wan, Q.; Mironov, S.; Yang, G.; Spassova, M.K.; Iasonos, A.; Kravetz, S.; Ouerfelli, O.; et al. A Phase I Study of Unimolecular Pentavalent (Globo-H-GM2-sTn-TF-Tn) Immunization of Patients with Epithelial Ovarian, Fallopian Tube, or Peritoneal Cancer in First Remission. Cancers 2016, 8, 46. [Google Scholar] [CrossRef] [Green Version]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front. Neurosci. 2020, 14, 572965. [Google Scholar] [CrossRef] [PubMed]
- Kohla, G.; Stockfleth, E.; Schauer, R. Gangliosides with O-acetylated sialic acids in tumors of neuroectodermal origin. Neurochem. Res. 2002, 27, 583–592. [Google Scholar] [CrossRef]
- Itokazu, Y.; Yu, R. Glycolipid Antigens in Neural Stem Cells. In Neural Surface Antigens; Pruszak, J., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 91–102. [Google Scholar] [CrossRef]
- Yeh, S.C.; Wang, P.Y.; Lou, Y.W.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Glycolipid GD3 and GD3 synthase are key drivers for glioblastoma stem cells and tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zheng, X.; Pang, X.; Li, L.; Wang, J.; Yang, C.; Du, G. Ganglioside GD3 synthase (GD3S), a novel cancer drug target. Acta Pharm. Sin. B 2018, 8, 713–720. [Google Scholar] [CrossRef]
- Nakakuma, H.; Horikawa, K.; Kawaguchi, T.; Hidaka, M.; Nagakura, S.; Hirai, S.; Kageshita, T.; Ono, T.; Kagimoto, T.; Iwamori, M.; et al. Common phenotypic expression of gangliosides GM3 and GD3 in normal human tissues and neoplastic skin lesions. Jpn. J. Clin. Oncol. 1992, 22, 308–312. [Google Scholar] [PubMed]
- Ohkawa, Y.; Miyazaki, S.; Hamamura, K.; Kambe, M.; Miyata, M.; Tajima, O.; Ohmi, Y.; Yamauchi, Y.; Furukawa, K.; Furukawa, K. Ganglioside GD3 enhances adhesion signals and augments malignant properties of melanoma cells by recruiting integrins to glycolipid-enriched microdomains. J. Biol. Chem. 2010, 285, 27213–27223. [Google Scholar] [CrossRef] [Green Version]
- Ramos, R.I.; Bustos, M.A.; Wu, J.; Jones, P.; Chang, S.C.; Kiyohara, E.; Tran, K.; Zhang, X.; Stern, S.L.; Izraely, S.; et al. Upregulation of cell surface GD3 ganglioside phenotype is associated with human melanoma brain metastasis. Mol. Oncol. 2020, 14, 1760–1778. [Google Scholar] [CrossRef]
- Dippold, W.G.; Lloyd, K.O.; Li, L.T.; Ikeda, H.; Oettgen, H.F.; Old, L.J. Cell surface antigens of human malignant melanoma: Definition of six antigenic systems with mouse monoclonal antibodies. Proc. Natl. Acad. Sci. USA 1980, 77, 6114–6118. [Google Scholar] [CrossRef] [Green Version]
- Hellström, I.; Brankovan, V.; Hellström, K.E. Strong antitumor activities of IgG3 antibodies to a human melanoma-associated ganglioside. Proc. Natl. Acad. Sci. USA 1985, 82, 1499–1502. [Google Scholar] [CrossRef] [Green Version]
- Ohta, S.; Honda, A.; Tokutake, Y.; Yoshida, H.; Hanai, N. Antitumor effects of a novel monoclonal antibody with high binding affinity to ganglioside GD3. Cancer Immunol. Immunother. 1993, 36, 260–266. [Google Scholar] [CrossRef]
- Real, F.X.; Houghton, A.N.; Albino, A.P.; Cordon-Cardo, C.; Melamed, M.R.; Oettgen, H.F.; Old, L.J. Surface antigens of melanomas and melanocytes defined by mouse monoclonal antibodies: Specificity analysis and comparison of antigen expression in cultured cells and tissues. Cancer Res. 1985, 45, 4401–4411. [Google Scholar]
- Nasi, M.L.; Meyers, M.; Livingston, P.O.; Houghton, A.N.; Chapman, P.B. Anti-melanoma effects of R24, a monoclonal antibody against GD3 ganglioside. Melanoma Res. 1997, 7 (Suppl. 2), S155–S162. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Gillies, S.D.; Houghton, A.N.; Reilly, R.M. Mapping effector functions of a monoclonal antibody to GD3 by characterization of a mouse-human chimeric antibody. Cancer Immunol. Immunother. 1994, 39, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Lee, F.T.; Hopkins, W.; Cebon, J.S.; Wheatley, J.M.; Liu, Z.; Smyth, F.E.; Murone, C.; Sturrock, S.; MacGregor, D.; et al. Specific targeting, biodistribution, and lack of immunogenicity of chimeric anti-GD3 monoclonal antibody KM871 in patients with metastatic melanoma: Results of a phase I trial. J. Clin. Oncol. 2001, 19, 3976–3987. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Houghton, A.N. Induction of IgG antibodies against GD3 ganglioside in rabbits by an anti-idiotypic monoclonal antibody. J. Clin. Investig. 1991, 88, 186–192. [Google Scholar] [CrossRef] [Green Version]
- McCaffery, M.; Yao, T.J.; Williams, L.; Livingston, P.O.; Houghton, A.N.; Chapman, P.B. Immunization of melanoma patients with BEC2 anti-idiotypic monoclonal antibody that mimics GD3 ganglioside: Enhanced immunogenicity when combined with adjuvant. Clin. Cancer Res. 1996, 2, 679–686. [Google Scholar]
- Brezicka, F.T.; Olling, S.; Nilsson, O.; Bergh, J.; Holmgren, J.; Sörenson, S.; Yngvason, F.; Lindholm, L. Immunohistological detection of fucosyl-GM1 ganglioside in human lung cancer and normal tissues with monoclonal antibodies. Cancer Res. 1989, 49, 1300–1305. [Google Scholar]
- Zhang, S.; Cordon-Cardo, C.; Zhang, H.S.; Reuter, V.E.; Adluri, S.; Hamilton, W.B.; Lloyd, K.O.; Livingston, P.O. Selection of tumor antigens as targets for immune attack using immunohistochemistry: I. Focus on gangliosides. Int. J. Cancer 1997, 73, 42–49. [Google Scholar] [CrossRef]
- Groux-Degroote, S.; Delannoy, P. Cancer-Associated Glycosphingolipids as Tumor Markers and Targets for Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 6145. [Google Scholar] [CrossRef]
- Dickler, M.N.; Ragupathi, G.; Liu, N.X.; Musselli, C.; Martino, D.J.; Miller, V.A.; Kris, M.G.; Brezicka, F.T.; Livingston, P.O.; Grant, S.C. Immunogenicity of a fucosyl-GM1-keyhole limpet hemocyanin conjugate vaccine in patients with small cell lung cancer. Clin. Cancer Res. 1999, 5, 2773–2779. [Google Scholar]
- Krug, L.M.; Ragupathi, G.; Hood, C.; Kris, M.G.; Miller, V.A.; Allen, J.R.; Keding, S.J.; Danishefsky, S.J.; Gomez, J.; Tyson, L.; et al. Vaccination of patients with small-cell lung cancer with synthetic fucosyl GM-1 conjugated to keyhole limpet hemocyanin. Clin. Cancer Res. 2004, 10, 6094–6100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brezicka, F.T.; Holmgren, J.; Kalies, I.; Lindholm, L. Tumor-cell killing by MAbs against fucosyl GM1, a ganglioside antigen associated with small-cell lung carcinoma. Int. J. Cancer 1991, 49, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Brezicka, F.; Einbeigi, Z.; Bergman, B. Functional assessment in vitro of human-complement-dependent antibody-induced cytotoxicity of neoplastic cells. Cancer Immunol. Immunother. 2000, 49, 235–242. [Google Scholar] [CrossRef]
- Ponath, P.; Menezes, D.; Pan, C.; Chen, B.; Oyasu, M.; Strachan, D.; LeBlanc, H.; Sun, H.; Wang, X.T.; Rangan, V.S.; et al. A Novel, Fully Human Anti-fucosyl-GM1 Antibody Demonstrates Potent In Vitro and In Vivo Antitumor Activity in Preclinical Models of Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 5178–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Saxton, R.E.; Morton, D.L.; Irie, R.F. Gangliosides of human melanoma. J. Natl. Cancer Inst. 1987, 78, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Terreni, M.; Sollogoub, M.; Zhang, Y. Ganglioside GM3 and Its Role in Cancer. Curr. Med. Chem. 2019, 26, 2933–2947. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Zhang, J.; Mi, W.; Yang, J.; Han, F.; Lu, X.; Yu, W. Silencing of GM3 synthase suppresses lung metastasis of murine breast cancer cells. Breast Cancer Res. 2008, 10, R1. [Google Scholar] [CrossRef] [Green Version]
- Tringali, C.; Silvestri, I.; Testa, F.; Baldassari, P.; Anastasia, L.; Mortarini, R.; Anichini, A.; López-Requena, A.; Tettamanti, G.; Venerando, B. Molecular subtyping of metastatic melanoma based on cell ganglioside metabolism profiles. BMC Cancer 2014, 14, 560. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, Y.; Hamaoka, A.; Matsumoto, M.; Matsubara, T.; Tagawa, M.; Wakabayashi, S.; Taniguchi, M. Syngeneic monoclonal antibody against melanoma antigen with interspecies cross-reactivity recognizes GM3, a prominent ganglioside of B16 melanoma. J. Biol. Chem. 1985, 260, 13328–13333. [Google Scholar] [CrossRef]
- Nores, G.A.; Dohi, T.; Taniguchi, M.; Hakomori, S. Density-dependent recognition of cell surface GM3 by a certain anti-melanoma antibody, and GM3 lactone as a possible immunogen: Requirements for tumor-associated antigen and immunogen. J. Immunol. 1987, 139, 3171–3176. [Google Scholar]
- Liu, J.W.; Sun, P.; Yan, Q.; Paller, A.S.; Gerami, P.; Ho, N.; Vashi, N.; Le Poole, I.C.; Wang, X.Q. De-N-acetyl GM3 promotes melanoma cell migration and invasion through urokinase plasminogen activator receptor signaling-dependent MMP-2 activation. Cancer Res. 2009, 69, 8662–8669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, L.E.; Gabri, M.R.; Guthmann, M.D.; Gomez, R.E.; Gold, S.; Fainboim, L.; Gomez, D.E.; Alonso, D.F. NGcGM3 ganglioside: A privileged target for cancer vaccines. Clin. Dev. Immunol. 2010, 2010, 814397. [Google Scholar] [CrossRef] [PubMed]
- Irie, A.; Koyama, S.; Kozutsumi, Y.; Kawasaki, T.; Suzuki, A. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J. Biol. Chem. 1998, 273, 15866–15871. [Google Scholar] [CrossRef] [Green Version]
- Blanco, R.; Quintana, Y.; Blanco, D.; Cedeño, M.; Rengifo, C.E.; Frómeta, M.; Ríos, M.; Rengifo, E.; Carr, A. Tissue Reactivity of the 14F7 Mab Raised against N-Glycolyl GM3 Ganglioside in Tumors of Neuroectodermal, Mesodermal, and Epithelial Origin. J. Biomark. 2013, 2013, 02417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samraj, A.N.; Läubli, H.; Varki, N.; Varki, A. Involvement of a non-human sialic Acid in human cancer. Front. Oncol. 2014, 4, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, J.P.; Valdés, Z.; Casacó, A.; Pimentel, G.; González, J.; Alvarez, I.; Osorio, M.; Velazco, M.; Figueroa, M.; Ortiz, R.; et al. Clinical evidences of GM3 (NeuGc) ganglioside expression in human breast cancer using the 14F7 monoclonal antibody labelled with (99m)Tc. Breast Cancer Res Treat. 2006, 96, 115–121. [Google Scholar] [CrossRef]
- Blanco, R.; Domínguez, E.; Morales, O.; Blanco, D.; Martínez, D.; Rengifo, C.E.; Viada, C.; Cedeño, M.; Rengifo, E.; Carr, A. Prognostic Significance of N-Glycolyl GM3 Ganglioside Expression in Non-Small Cell Lung Carcinoma Patients: New Evidences. Patholog. Res. Int. 2015, 2015, 132326. [Google Scholar] [CrossRef] [Green Version]
- Labrada, M.; Dorvignit, D.; Hevia, G.; Rodríguez-Zhurbenko, N.; Hernández, A.M.; Vázquez, A.M.; Fernández, L.E. GM3(Neu5Gc) ganglioside: An evolution fixed neoantigen for cancer immunotherapy. Semin. Oncol. 2018, 45, 41–51. [Google Scholar] [CrossRef]
- Carr, A.; Mullet, A.; Mazorra, Z.; Vázquez, A.M.; Alfonso, M.; Mesa, C.; Rengifo, E.; Pérez, R.; Fernández, L.E. A mouse IgG1 monoclonal antibody specific for N-glycolyl GM3 ganglioside recognized breast and melanoma tumors. Hybridoma 2000, 19, 241–247. [Google Scholar] [CrossRef]
- Fernández-Marrero, Y.; Roque-Navarro, L.; Hernández, T.; Dorvignit, D.; Molina-Pérez, M.; González, A.; Sosa, K.; López-Requena, A.; Pérez, R.; de Acosta, C.M. A cytotoxic humanized anti-ganglioside antibody produced in a murine cell line defective of N-glycolylated-glycoconjugates. Immunobiology 2011, 216, 1239–1247. [Google Scholar] [CrossRef]
- Dorvignit, D.; García-Martínez, L.; Rossin, A.; Sosa, K.; Viera, J.; Hernández, T.; Mateo, C.; Hueber, A.O.; Mesa, C.; López-Requena, A. Antitumor and cytotoxic properties of a humanized antibody specific for the GM3(Neu5Gc) ganglioside. Immunobiology 2015, 220, 1343–1350. [Google Scholar] [CrossRef]
- Dorvignit, D.; Boligan, K.F.; Relova-Hernández, E.; Clavell, M.; López, A.; Labrada, M.; Simon, H.U.; López-Requena, A.; Mesa, C.; von Gunten, S. Antitumor effects of the GM3(Neu5Gc) ganglioside-specific humanized antibody 14F7hT against Cmah-transfected cancer cells. Sci. Rep. 2019, 9, 9921. [Google Scholar] [CrossRef] [PubMed]
- Estevez, F.; Carr, A.; Solorzano, L.; Valiente, O.; Mesa, C.; Barroso, O.; Sierra, G.V.; Fernandez, L.E. Enhancement of the immune response to poorly immunogenic gangliosides after incorporation into very small size proteoliposomes (VSSP). Vaccine 1999, 18, 190–197. [Google Scholar] [CrossRef]
- Osorio, M.; Gracia, E.; Reigosa, E.; Hernandez, J.; de la Torre, A.; Saurez, G.; Perez, K.; Viada, C.; Cepeda, M.; Carr, A.; et al. Effect of vaccination with N-glycolyl GM3/VSSP vaccine by subcutaneous injection in patients with advanced cutaneous melanoma. Cancer Manag. Res. 2012, 4, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, A.; Rodríguez, E.; Arango, M. del C.; Camacho, R.; Osorio, M.; Gabri, M.; Carrillo, G.; Valdés, Z.; Bebelagua, Y.; Pérez, R.; et al. Immunotherapy of advanced breast cancer with a heterophilic ganglioside (NeuGcGM3) cancer vaccine. J. Clin. Oncol. 2003, 21, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Mulens, V.; de la Torre, A.; Marinello, P.; Rodríguez, R.; Cardoso, J.; Díaz, R.; O'Farrill, M.; Macias, A.; Viada, C.; Saurez, G.; et al. Immunogenicity and safety of a NeuGcGM3 based cancer vaccine: Results from a controlled study in metastatic breast cancer patients. Hum. Vaccines 2010, 6, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Pérez, K.; Osorio, M.; Hernández, J.; Carr, A.; Fernández, L.E. NGcGM3/VSSP vaccine as treatment for melanoma patients. Hum Vaccines Immunother. 2013, 9, 1237–1240. [Google Scholar] [CrossRef] [Green Version]
- de la Torre, A.; Pérez, K.; Vega, A.M.; Santiesteban, E.; Ruiz, R.; Hernández, L.; Durrutí, D.; Viada, C.E.; Sánchez, L.; Álvarez, M.; et al. Superior Efficacy and Safety of a Nonemulsive Variant of the NGcGM3/VSSP Vaccine in Advanced Breast Cancer Patients. Breast Cancer 2016, 10, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Jerne, N.K. Towards a network theory of the immune system. Ann. Immunol. 1974, 125C, 373–389. [Google Scholar]
- Foon, K.A.; Lutzky, J.; Baral, R.N.; Yannelli, J.R.; Hutchins, L.; Teitelbaum, A.; Kashala, O.L.; Das, R.; Garrison, J.; Reisfeld, R.A.; et al. Clinical and immune responses in advanced melanoma patients immunized with an anti-idiotype antibody mimicking disialoganglioside GD2. J. Clin. Oncol. 2000, 18, 376–384. [Google Scholar] [CrossRef]
- Kohler, H.; Pashov, A.; Kieber-Emmons, T. The Promise of Anti-idiotype Revisited. Front. Immunol. 2019, 10, 808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez, A.M.; Pérez, A.; Hernández, A.M.; Macías, A.; Alfonso, M.; Bombino, G.; Pérez, R. Syngeneic anti-idiotypic monoclonal antibodies to an anti-NeuGc-containing ganglioside monoclonal antibody. Hybridoma 1998, 17, 527–534. [Google Scholar] [CrossRef]
- Segatori, V.I.; Cuello, H.A.; Gulino, C.A.; Albertó, M.; Venier, C.; Guthmann, M.D.; Demarco, I.A.; Alonso, D.F.; Gabri, M.R. Antibody-dependent cell-mediated cytotoxicity induced by active immunotherapy based on racotumomab in non-small cell lung cancer patients. Cancer Immunol. Immunother. 2018, 67, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Guthmann, M.D.; Castro, M.A.; Cinat, G.; Venier, C.; Koliren, L.; Bitton, R.J.; Vázquez, A.M.; Fainboim, L. Cellular and humoral immune response to N-Glycolyl-GM3 elicited by prolonged immunotherapy with an anti-idiotypic vaccine in high-risk and metastatic breast cancer patients. J. Immunother. 2006, 29, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Säljö, K.; Barone, A.; Mölne, J.; Rydberg, L.; Teneberg, S.; Breimer, M.E. HLA and Histo-Blood Group Antigen Expression in Human Pluripotent Stem Cells and their Derivatives. Sci. Rep. 2017, 7, 13072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, H.S.; Reuter, V.E.; Slovin, S.F.; Scher, H.I.; Livingston, P.O. Expression of potential target antigens for immunotherapy on primary and metastatic prostate cancers. Clin. Cancer Res. 1998, 4, 295–302. [Google Scholar]
- Cheng, J.Y.; Wang, S.H.; Lin, J.; Tsai, Y.C.; Yu, J.; Wu, J.C.; Hung, J.T.; Lin, J.J.; Wu, Y.Y.; Yeh, K.T.; et al. Globo-H ceramide shed from cancer cells triggers translin-associated factor X-dependent angiogenesis. Cancer Res. 2014, 74, 6856–6866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slovin, S.F.; Ragupathi, G.; Adluri, S.; Ungers, G.; Terry, K.; Kim, S.; Spassova, M.; Bornmann, W.G.; Fazzari, M.; Dantis, L.; et al. Carbohydrate vaccines in cancer: Immunogenicity of a fully synthetic globo H hexasaccharide conjugate in man. Proc. Natl. Acad. Sci. USA 1999, 96, 5710–5715. [Google Scholar] [CrossRef] [Green Version]
- Gilewski, T.; Ragupathi, G.; Bhuta, S.; Williams, L.J.; Musselli, C.; Zhang, X.F.; Bornmann, W.G.; Spassova, M.; Bencsath, K.P.; Panageas, K.S.; et al. Immunization of metastatic breast cancer patients with a fully synthetic globo H conjugate: A phase I trial. Proc. Natl. Acad. Sci. USA 2001, 98, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Chuang, P.K.; Hsiao, M.; Hsu, T.L.; Chang, C.F.; Wu, C.Y.; Chen, B.R.; Huang, H.W.; Liao, K.S.; Chen, C.C.; Chen, C.L.; et al. Signaling pathway of globo-series glycosphingolipids and β1,3-galactosyltransferase V (β3GalT5) in breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3518–3523. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, F.M.; Rodríguez-Walker, M.; Daniotti, J.L. Exploiting the internalization feature of an antibody against the glycosphingolipid SSEA-4 to deliver immunotoxins in breast cancer cells. Immunol. Cell Biol. 2020, 98, 187–202. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.C.; Shia, C.S.; Li, W.F.; Wang, C.C.; Chen, I.J.; Huang, T.Y.; Chen, Y.J.; Chang, H.W.; Lu, C.H.; Wu, Y.C.; et al. Preclinical Studies of OBI-999: A Novel Globo H-Targeting Antibody-Drug Conjugate. Mol. Cancer Ther. 2021, 20, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Cervoni, G.E.; Cheng, J.J.; Stackhouse, K.A.; Heimburg-Molinaro, J.; Cummings, R.D. O-glycan recognition and function in mice and human cancers. Biochem. J. 2020, 477, 1541–1564. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babino, A.; Oppezzo, P.; Bianco, S.; Barrios, E.; Berois, N.; Navarrete, H.; Osinaga, E. Tn antigen is a pre-cancerous biomarker in breast tissue and serum in n-nitrosomethylurea-induced rat mammary carcinogenesis. Int. J. Cancer 2000, 86, 753–759. [Google Scholar] [CrossRef]
- Berriel, E.; Hill, M.; Barcia, J.J.; Ubillos, L.; Gonzalez, M.; Detjen, G.; Rondan, M.; Navarrete, H.; Osinaga, E. Oncol Simple mucin-type cancer associated antigens are pre-cancerous biomarkers during 1,2-dimethylhydrazine-induced rat colon carcinogenesis. Oncol. Rep. 2005, 14, 219–227. [Google Scholar] [PubMed]
- An, G.; Wei, B.; Xia, B.; McDaniel, J.M.; Ju, T.; Cummings, R.D.; Braun, J.; Xia, L. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J. Exp. Med. 2007, 204, 1417–1429. [Google Scholar] [CrossRef]
- Chia, J.; Goh, G.; Bard, F. Short O-GalNAc glycans: Regulation and role in tumor development and clinical perspectives. Biochim. Biophys. Acta 2016, 1860, 1623–1639. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Zhao, H.; Wang, Y.; Cai, H.; Xiao, Y.; Zeng, Y.; Chen, H. Tumor-associated antigens: Tn antigen, sTn antigen, and T antigen. HLA 2016, 88, 275–286. [Google Scholar] [CrossRef]
- Munkley, J. The Role of Sialyl-Tn in Cancer. Int. J. Mol. Sci. 2016, 17, 275. [Google Scholar] [CrossRef] [Green Version]
- Ju, T.; Lanneau, G.S.; Gautam, T.; Wang, Y.; Xia, B.; Stowell, S.R.; Willard, M.T.; Wang, W.; Xia, J.Y.; Zuna, R.E.; et al. Human tumor antigens Tn and sialyl Tn arise from mutations in Cosmc. Cancer Res. 2008, 68, 1636–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, R.; Song, L.; Wang, Y.; Ding, X.; Zeng, J.; Lehoux, S.; Aryal, R.P.; Wang, J.; Crew, V.K.; van Die, I.; et al. Epigenetic silencing of the chaperone Cosmc in human leukocytes expressing Tn antigen. J. Biol. Chem. 2012, 287, 41523–41533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, D.J.; Chia, J.; Senewiratne, J.; Bard, F. Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J. Cell Biol. 2010, 189, 843–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, D.J.; Tham, K.M.; Chia, J.; Wang, S.C.; Steentoft, C.; Clausen, H.; Bard-Chapeau, E.A.; Bard, F.A. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc. Natl. Acad. Sci. USA 2013, 110, E3152–E3161. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.K.; Metoki, R.; Hakomori, S. Immunoglobulin G3 monoclonal antibody directed to Tn antigen (tumor-associated alpha-N-acetylgalactosaminyl epitope) that does not cross-react with blood group A antigen. Cancer Res. 1988, 48, 4361–4367. [Google Scholar]
- Springer, G.F.; Desai, P.R.; Wise, W.; Carlstedt, S.C.; Tegtmeyer, H.; Stein, R.; Scanlon, E.F. Pancarcinoma T and Tn epitopes: Autoimmunogens and diagnostic markers that reveal incipient carcinomas and help establish prognosis. Immunol. Ser. 1990, 53, 587–612. [Google Scholar]
- Numata, Y.; Nakada, H.; Fukui, S.; Kitagawa, H.; Ozaki, K.; Inoue, M.; Kawasaki, T.; Funakoshi, I.; Yamashina, I. A monoclonal antibody directed to Tn antigen. Biochem. Biophys. Res. Commun. 1990, 170, 981–985. [Google Scholar] [CrossRef]
- King, M.J.; Parsons, S.F.; Wu, A.M.; Jones, N. Immunochemical studies on the differential binding properties of two monoclonal antibodies reacting with Tn red cells. Transfusion 1991, 31, 142–149. [Google Scholar] [CrossRef]
- Thurnher, M.; Clausen, H.; Sharon, N.; Berger, E.G. Use of O-glycosylation-defective human lymphoid cell lines and flow cytometry to delineate the specificity of Moluccella laevis lectin and monoclonal antibody 5F4 for the Tn antigen (GalNAc alpha 1-O-Ser/Thr). Immunol. Lett. 1993, 36, 239–243. [Google Scholar] [CrossRef]
- Terasawa, K.; Furumoto, H.; Kamada, M.; Aono, T. Expression of Tn and sialyl-Tn antigens in the neoplastic transformation of uterine cervical epithelial cells. Cancer Res. 1996, 56, 2229–2232. [Google Scholar]
- Reis, C.A.; Sørensen, T.; Mandel, U.; David, L.; Mirgorodskaya, E.; Roepstorff, P.; Kihlberg, J.; Hansen, J.E.; Clausen, H. Development and characterization of an antibody directed to an alpha-N-acetyl-D-galactosamine glycosylated MUC2 peptide. Glycoconj. J. 1998, 15, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Osinaga, E.; Bay, S.; Tello, D.; Babino, A.; Pritsch, O.; Assemat, K.; Cantacuzene, D.; Nakada, H.; Alzari, P. Analysis of the fine specificity of Tn-binding proteins using synthetic glycopeptide epitopes and a biosensor based on surface plasmon resonance spectroscopy. FEBS Lett. 2000, 469, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Möller, H.; Serttas, N.; Paulsen, H.; Burchell, J.M.; Taylor-Papadimitriou, J.; Meyer, B. NMR-based determination of the binding epitope and conformational analysis of MUC-1 glycopeptides and peptides bound to the breast cancer-selective monoclonal antibody SM3. Eur. J. Biochem. 2002, 269, 1444–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schietinger, A.; Philip, M.; Yoshida, B.A.; Azadi, P.; Liu, H.; Meredith, S.C.; Schreiber, H. A mutant chaperone converts a wild-type protein into a tumor-specific antigen. Science 2006, 314, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Danielczyk, A.; Stahn, R.; Faulstich, D.; Löffler, A.; Märten, A.; Karsten, U.; Goletz, S. PankoMab: A potent new generation anti-tumour MUC1 antibody. Cancer Immunol. Immunother. 2006, 55, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Tarp, M.A.; Sørensen, A.L.; Mandel, U.; Paulsen, H.; Burchell, J.; Taylor-Papadimitriou, J.; Clausen, H. Identification of a novel cancer-specific immunodominant glycopeptide epitope in the MUC1 tandem repeat. Glycobiology 2007, 17, 197–209. [Google Scholar] [CrossRef]
- Ando, H.; Matsushita, T.; Wakitani, M.; Sato, T.; Kodama-Nishida, S.; Shibata, K.; Shitara, K.; Ohta, S. Mouse-human chimeric anti-Tn IgG1 induced anti-tumor activity against Jurkat cells in vitro and in vivo. Biol. Pharm. Bull. 2008, 31, 1739–1744. [Google Scholar] [CrossRef] [Green Version]
- Danussi, C.; Coslovi, A.; Campa, C.; Mucignat, M.T.; Spessotto, P.; Uggeri, F.; Paoletti, S.; Colombatti, A. A newly generated functional antibody identifies Tn antigen as a novel determinant in the cancer cell-lymphatic endothelium interaction. Glycobiology 2009, 19, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Welinder, C.; Baldetorp, B.; Borrebaeck, C.; Fredlund, B.M.; Jansson, B. A new murine IgG1 anti-Tn monoclonal antibody with in vivo anti-tumor activity. Glycobiology 2011, 21, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Trabbic, K.R.; Kleski, K.A.; Shi, M.; Bourgault, J.P.; Prendergast, J.M.; Dransfield, D.T.; Andreana, P.R. Production of a mouse monoclonal IgM antibody that targets the carbohydrate Thomsen-nouveau cancer antigen resulting in in vivo and in vitro tumor killing. Cancer Immunol. Immunother. 2018, 67, 1437–1447. [Google Scholar] [CrossRef]
- Steentoft, C.; Fuhrmann, M.; Battisti, F.; Van Coillie, J.; Madsen, T.D.; Campos, D.; Halim, A.; Vakhrushev, S.Y.; Joshi, H.J.; Schreiber, H.; et al. A strategy for generating cancer-specific monoclonal antibodies to aberrant O-glycoproteins: Identification of a novel dysadherin-Tn antibody. Glycobiology 2019, 29, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Kudelka, M.R.; Hanes, M.S.; Lehoux, S.; Dutta, S.; Jones, M.B.; Stackhouse, K.A.; Cervoni, G.E.; Heimburg-Molinaro, J.; Smith, D.F.; et al. Identification of Tn antigen O-GalNAc-expressing glycoproteins in human carcinomas using novel anti-Tn recombinant antibodies. Glycobiology 2020, 30, 282–300. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Takio, K.; Titani, K.; Greene, T.; Levery, S.B.; Salyan, M.E.; Hakomori, S. The oncofetal structure of human fibronectin defined by monoclonal antibody FDC-6. Unique structural requirement for the antigenic specificity provided by a glycosylhexapeptide. J. Biol. Chem. 1988, 263, 3314–3322. [Google Scholar] [CrossRef]
- Grinstead, J.S.; Koganty, R.R.; Krantz, M.J.; Longenecker, B.M.; Campbell, A.P. Effect of glycosylation on MUC1 humoral immune recognition: NMR studies of MUC1 glycopeptide-antibody interactions. Biochemistry 2002, 41, 9946–9961. [Google Scholar] [CrossRef]
- Canals Hernaez, D.; Hughes, M.R.; Dean, P.; Bergqvist, P.; Samudio, I.; Blixt, O.; Wiedemeyer, K.; Li, Y.; Bond, C.; Cruz, E.; et al. PODO447: A novel antibody to a tumor-restricted epitope on the cancer antigen podocalyxin. J. Immunother. Cancer 2020, 8, e001128. [Google Scholar] [CrossRef]
- Medeiros, A.; Bianchi, S.; Calvete, J.J.; Balter, H.; Bay, S.; Robles, A.; Cantacuzène, D.; Nimtz, M.; Alzari, P.M.; Osinaga, E. Biochemical and functional characterization of the Tn-specific lectin from Salvia sclarea seeds. Eur. J. Biochem. 2000, 267, 1434–1440. [Google Scholar] [CrossRef] [Green Version]
- Nakada, H.; Inoue, M.; Numata, Y.; Tanaka, N.; Funakoshi, I.; Fukui, S.; Mellors, A.; Yamashina, I. Epitopic structure of Tn glycophorin A for an anti-Tn antibody (MLS 128). Proc. Natl. Acad. Sci. USA 1993, 90, 2495–2499. [Google Scholar] [CrossRef] [Green Version]
- Mazal, D.; Lo-Man, R.; Bay, S.; Pritsch, O.; Dériaud, E.; Ganneau, C.; Medeiros, A.; Ubillos, L.; Obal, G.; Berois, N. Monoclonal antibodies toward different Tn-amino acid backbones display distinct recognition patterns on human cancer cells. Implications for effective immuno-targeting of cancer. Cancer Immunol. Immunother. 2013, 62, 1107–1122. [Google Scholar] [CrossRef]
- Manimala, J.C.; Li, Z.; Jain, A.; VedBrat, S.; Gildersleeve, J.C. Carbohydrate array analysis of anti-Tn antibodies and lectins reveals unexpected specificities: Implications for diagnostic and vaccine development. ChemBioChem 2005, 6, 2229–2241. [Google Scholar] [CrossRef]
- Blixt, O.; Lavrova, O.I.; Mazurov, D.V.; Cló, E.; Kracun, S.K.; Bovin, N.V.; Filatov, A.V. Analysis of Tn antigenicity with a panel of new IgM and IgG1 monoclonal antibodies raised against leukemic cells. Glycobiology 2012, 22, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Lavrsen, K.; Madsen, C.B.; Rasch, M.G.; Woetmann, A.; Ødum, N.; Mandel, U.; Clausen, H.; Pedersen, A.E.; Wandall, H.H. Aberrantly glycosylated MUC1 is expressed on the surface of breast cancer cells and a target for antibody-dependent cell-mediated cytotoxicity. Glycoconj. J. 2013, 30, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Klein Wolterink, R.G.J.; Gulaia, V.; Cloosen, S.; Ehlers, F.A.I.; Wieten, L.; Graus, Y.F.; Bos, G.M.J.; Germeraad, W.T.V. Defucosylation of Tumor-Specific Humanized Anti-MUC1 Monoclonal Antibody Enhances NK Cell-Mediated Anti-Tumor Cell Cytotoxicity. Cancers 2021, 13, 2579. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Yajima, Y.; Asanuma, H.; Nakada, H.; Fujita-Yamaguchi, Y. Inhibition of cancer cell growth by anti-Tn monoclonal antibody MLS128. Biosci. Trends. 2009, 3, 32–37. [Google Scholar] [PubMed]
- von Mensdorff-Pouilly, S.; Petrakou, E.; Kenemans, P.; van Uffelen, K.; Verstraeten, A.A.; Snijdewint, F.G.; van Kamp, G.J.; Schol, D.J.; Reis, C.A.; Price, M.R.; et al. Reactivity of natural and induced human antibodies to MUC1 mucin with MUC1 peptides and n-acetylgalactosamine (GalNAc) peptides. Int. J. Cancer 2000, 86, 702–712. [Google Scholar] [CrossRef]
- Oppezzo, P.; Osinaga, E.; Tello, D.; Bay, S.; Cantacuzene, D.; Irigoín, F.; Ferreira, A.; Roseto, A.; Cayota, A.; Alzari, P.; et al. Production and functional characterization of two mouse/human chimeric antibodies with specificity for the tumor-associated Tn-antigen. Hybridoma 2000, 19, 229–339. [Google Scholar] [CrossRef]
- Pancino, G.F.; Osinaga, E.; Vorauher, W.; Kakouche, A.; Mistro, D.; Charpin, C.; Roseto, A. Production of a monoclonal antibody as immunohistochemical marker on paraffin embedded tissues using a new immunization method. Hybridoma 1990, 9, 389–395. [Google Scholar] [CrossRef]
- Sedlik, C.; Heitzmann, A.; Viel, S.; Ait Sarkouh, R.; Batisse, C.; Schmidt, F.; De La Rochere, P.; Amzallag, N.; Osinaga, E.; Oppezzo, P.; et al. Effective antitumor therapy based on a novel antibody-drug conjugate targeting the Tn carbohydrate antigen. Oncoimmunology 2016, 5, e1171434. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.; Berois, N.; Malanga, A.; Ortega, C.; Oppezzo, P.; Pristch, O.; Mombrú, A.W.; Osinaga, E.; Pardo, H. Docetaxel in chitosan-based nanocapsules conjugated with an anti-Tn antigen mouse/human chimeric antibody as a promising targeting strategy of lung tumors. Int. J. Biol. Macromol. 2021, 182, 806–814. [Google Scholar] [CrossRef]
- Springer, G.F. Immunoreactive T and Tn epitopes in cancer diagnosis, prognosis, and immunotherapy. J. Mol. Med. 1997, 75, 594–602. [Google Scholar] [CrossRef]
- Slovin, S.F.; Ragupathi, G.; Musselli, C.; Olkiewicz, K.; Verbel, D.; Kuduk, S.D.; Schwarz, J.B.; Sames, D.; Danishefsky, S.; Livingston, P.O.; et al. Fully synthetic carbohydrate-based vaccines in biochemically relapsed prostate cancer: Clinical trial results with alpha-N-acetylgalactosamine-O-serine/threonine conjugate vaccine. J. Clin. Oncol. 2003, 21, 4292–4298. [Google Scholar] [CrossRef]
- Kuduk, S.; Schwarz, J.; Chen, X.T.; Glunz, P.; Sames, D.; Ragupathi, G.; Livingston, P.O.; Danishefsky, S. Synthetic and immunological studies of clustered modes of mucin-related Tn and TF O-linked antigens: The preparation of a glycopeptide-based vaccines for clinical trials against prostate cancer. J. Am. Chem. Soc. 1998, 120, 12474–12485. [Google Scholar] [CrossRef]
- Lo-Man, R.; Vichier-Guerre, S.; Bay, S.; Deriaud, E.; Cantacuzene, D.; Leclerc, C. Anti-tumor immunity provided by a synthetic multiple antigenic glycopeptide displaying a tri-Tn glycotope. J. Immunol. 2001, 166, 2849–2854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubreton, D.; Bay, S.; Sedlik, C.; Artaud, C.; Ganneau, C.; Dériaud, E.; Viel, S.; Puaux, A.L.; Amigorena, S.; Gérard, C.; et al. The fully synthetic MAG-Tn3 therapeutic vaccine containing the tetanus toxoid-derived TT830-844 universal epitope provides anti-tumor immunity. Cancer Immunol. Immunother. 2016, 65, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeland, E.; van Vliet, S.J.; Bäckström, M.; van den Berg, V.C.; Geijtenbeek, T.B.; Meijer, G.A.; van Kooyk, Y. The C-type lectin MGL expressed by dendritic cells detects glycan changes on MUC1 in colon carcinoma. Cancer Immunol. Immunother. 2007, 56, 1225–1236. [Google Scholar] [CrossRef]
- da Costa, V.; van Vliet, S.J.; Carasi, P.; Frigerio, S.; García, P.A.; Croci, D.O.; Festari, M.F.; Costa, M.; Landeira, M.; Rodríguez-Zraquia, S.A.; et al. The Tn antigen promotes lung tumor growth by fostering immunosuppression and angiogenesis via interaction with Macrophage Galactose-type lectin 2 (MGL2). Cancer Lett. 2021, 518, 72–81. [Google Scholar] [CrossRef]
- Cornelissen, L.A.M.; Blanas, A.; Zaal, A.; van der Horst, J.C.; Kruijssen, L.J.W.; O'Toole, T.; van Kooyk, Y.; van Vliet, S.J. Tn Antigen Expression Contributes to an Immune Suppressive Microenvironment and Drives Tumor Growth in Colorectal Cancer. Front. Oncol. 2020, 10, 1622. [Google Scholar] [CrossRef]
- Pinho, S.; Marcos, N.T.; Ferreira, B.; Carvalho, A.S.; Oliveira, M.J.; Santos-Silva, F.; Harduin-Lepers, A.; Reis, C.A. Biological significance of cancer-associated sialyl-Tn antigen: Modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett. 2007, 249, 157–170. [Google Scholar] [CrossRef]
- Julien, S.; Adriaenssens, E.; Ottenberg, K.; Furlan, A.; Courtand, G.; Vercoutter-Edouart, A.S.; Hanisch, F.G.; Delannoy, P.; Le Bourhis, X. ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumourigenicity. Glycobiology 2006, 16, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, H.; Matsuzaki, H.; Ando, H.; Kaji, H.; Nakanishi, H.; Ikehara, Y.; Narimatsu, H. Enhancement of metastatic ability by ectopic expression of ST6GalNAcI on a gastric cancer cell line in a mouse model. Clin. Exp. Metastasis 2012, 29, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Carrascal, M.A.; Severino, P.F.; Guadalupe Cabral, M.; Silva, M.; Ferreira, J.A.; Calais, F.; Quinto, H.; Pen, C.; Ligeiro, D.; Santos, L.L.; et al. Sialyl Tn-expressing bladder cancer cells induce a tolerogenic phenotype in innate and adaptive immune cells. Mol. Oncol. 2014, 8, 753–765. [Google Scholar] [CrossRef]
- Colcher, D.; Hand, P.H.; Nuti, M.; Schlom, J. A spectrum of monoclonal antibodies reactive with human mammary tumor cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3199–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, V.G.; Schlom, J.; Paterson, A.J.; Bennett, J.; Magnani, J.L.; Colcher, D. Analysis of a human tumor-associated glycoprotein (TAG-72) identified by monoclonal antibody B72.3. Cancer Res. 1986, 46, 850–857. [Google Scholar] [PubMed]
- Muraro, R.; Kuroki, M.; Wunderlich, D.; Poole, D.J.; Colcher, D.; Thor, A.; Greiner, J.W.; Simpson, J.F.; Molinolo, A.; Noguchi, P.; et al. Generation and characterization of B72.3 second generation monoclonal antibodies reactive with the tumor-associated glycoprotein 72 antigen. Cancer Res. 1988, 48, 4588–4596. [Google Scholar] [PubMed]
- Kjeldsen, T.; Clausen, H.; Hirohashi, S.; Ogawa, T.; Iijima, H.; Hakomori, S. Preparation and characterization of monoclonal antibodies directed to the tumor-associated O-linked sialosyl-2—6 alpha-N-acetylgalactosaminyl (sialosyl-Tn) epitope. Cancer Res. 1988, 48, 2214–2220. [Google Scholar]
- Zhang, S.; Walberg, L.; Ogata, S.; Itzkowitz, S.H.; Koganty, R.R.; Reddish, M.; Gandhi, S.S.; Longenecker, B.M.; Lloyd, K.O.; Livingston, P.O. Immune sera and monoclonal antibodies define two configurations for the sialyl Tn tumor antigen. Cancer Res. 1995, 55, 3364–3368. [Google Scholar]
- Ogata, S.; Koganty, R.; Reddish, M.; Longenecker, B.M.; Chen, A.; Perez, C.; Itzkowitz, S.H. ; Different modes of sialyl-Tn expression during malignant transformation of human colonic mucosa. Glycoconj. J. 1998, 15, 29–35. [Google Scholar] [CrossRef]
- Reddish, M.A.; Jackson, L.; Koganty, R.R.; Qiu, D.; Hong, W.; Longenecker, B.M. Specificities of anti-sialyl-Tn and anti-Tn monoclonal antibodies generated using novel clustered synthetic glycopeptide epitopes. Glycoconj. J. 1997, 14, 549–560. [Google Scholar] [CrossRef]
- Colcher, D.; Minelli, M.F.; Roselli, M.; Muraro, R.; Simpson-Milenic, D.; Schlom, J. Radioimmunolocalization of human carcinoma xenografts with B72.3 second generation monoclonal antibodies. Cancer Res. 1988, 48, 4597–4603. [Google Scholar]
- Kashmiri, S.V.; Shu, L.; Padlan, E.A.; Milenic, D.E.; Schlom, J.; Hand, P.H. Generation, characterization, and in vivo studies of humanized anticarcinoma antibody CC49. Hybridoma 1995, 14, 461–473. [Google Scholar] [CrossRef]
- Meredith, R.F.; Partridge, E.E.; Alvarez, R.D.; Khazaeli, M.B.; Plott, G.; Russell, C.D.; Wheeler, R.H.; Liu, T.; Grizzle, W.E.; Schlom, J.; et al. Intraperitoneal radioimmunotherapy of ovarian cancer with lutetium-177-CC49. J. Nucl. Med. 1996, 37, 1491–1496. [Google Scholar]
- Alvarez, R.D.; Partridge, E.E.; Khazaeli, M.B.; Plott, G.; Austin, M.; Kilgore, L.; Russell, C.D.; Liu, T.; Grizzle, W.E.; Schlom, J.; et al. Intraperitoneal radioimmunotherapy of ovarian cancer with 177Lu-CC49: A phase I/II study. Gynecol. Oncol. 1997, 65, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.F.; Alvarez, R.D.; Partridge, E.E.; Khazaeli, M.B.; Lin, C.Y.; Macey, D.J.; Austin, J.M., Jr.; Kilgore, L.C.; Grizzle, W.E.; Schlom, J.; et al. Intraperitoneal radioimmunochemotherapy of ovarian cancer: A phase I study. Cancer Biother. Radiopharm. 2001, 16, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.F.; Khazaeli, M.B.; Macey, D.J.; Grizzle, W.E.; Mayo, M.; Schlom, J.; Russell, C.D.; LoBuglio, A.F. Phase II study of interferon-enhanced 131I-labeled high affinity CC49 monoclonal antibody therapy in patients with metastatic prostate cancer. Clin. Cancer Res. 1999, 5, 3254s–3258s. [Google Scholar] [PubMed]
- Rogers, B.E.; Roberson, P.L.; Shen, S.; Khazaeli, M.B.; Carpenter, M.; Yokoyama, S.; Brechbiel, M.W.; LoBuglio, A.F.; Buchsbaum, D.J. Intraperitoneal radioimmunotherapy with a humanized anti-TAG-72 (CC49) antibody with a deleted CH2 region. Cancer Biother. Radiopharm. 2005, 20, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Minnix, M.; Li, L.; Yazaki, P.J.; Miller, A.D.; Chea, J.; Poku, E.; Liu, A.; Wong, J.Y.C.; Rockne, R.C.; Colcher, D.; et al. TAG-72-Targeted alpha-Radionuclide Therapy of Ovarian Cancer Using (225)Ac-Labeled DOTAylated-huCC49 Antibody. J. Nucl. Med. 2021, 62, 55–61. [Google Scholar] [CrossRef]
- Kim, K.S.; Lee, Y.K.; Kim, J.S.; Koo, K.H.; Hong, H.J.; Park, Y.S. Targeted gene therapy of LS174 T human colon carcinoma by anti-TAG-72 immunoliposomes. Cancer Gene Ther. 2008, 15, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Minnix, M.; Li, L.; Yazaki, P.J.; Chea, J.; Poku, E.; Colcher, D.; Shively, J.E. Improved targeting of an anti-TAG-72 antibody drug conjugate for the treatment of ovarian cancer. Cancer Med. 2020, 9, 4756–4767. [Google Scholar] [CrossRef]
- Prendergast, J.M.; Galvao da Silva, A.P.; Eavarone, D.A.; Ghaderi, D.; Zhang, M.; Brady, D.; Wicks, J.; DeSander, J.; Behrens, J.; Rueda, B.R. Novel anti-Sialyl-Tn monoclonal antibodies and antibody-drug conjugates demonstrate tumor specificity and anti-tumor activity. MAbs 2017, 9, 615–627. [Google Scholar] [CrossRef]
- Ragupathi, G.; Howard, L.; Cappello, S.; Koganty, R.R.; Qiu, D.; Longenecker, B.M.; Reddish, M.A.; Lloyd, K.O.; Livingston, P.O. Vaccines prepared with sialyl-Tn and sialyl-Tn trimers using the 4-(4-maleimidomethyl)cyclohexane-1-carboxyl hydrazide linker group result in optimal antibody titers against ovine submaxillary mucin and sialyl-Tn-positive tumor cells. Cancer Immunol. Immunother. 1999, 48, 1–8. [Google Scholar] [CrossRef]
- Julien, S.; Picco, G.; Sewell, R.; Vercoutter-Edouart, A.S.; Tarp, M.; Miles, D.; Clausen, H.; Taylor-Papadimitriou, J.; Burchell, J.M. Sialyl-Tn vaccine induces antibody-mediated tumour protection in a relevant murine model. Br. J. Cancer 2009, 100, 1746–1754. [Google Scholar] [CrossRef] [Green Version]
- Holmberg, L.A.; Sandmaier, B.M. Vaccination with Theratope (STn-KLH) as treatment for breast cancer. Expert. Rev. Vaccines 2004, 3, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Videira, P.A.; Delannoy, P. Sialyl-tn in cancer: (how) did we miss the target? Biomolecules. 2012, 2, 435–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindrewicz, P.; Lian, L.Y.; Yu, L.G. Interaction of the Oncofetal Thomsen-Friedenreich Antigen with Galectins in Cancer Progression and Metastasis. Front. Oncol. 2016, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsten, U.; Goletz, S. What controls the expression of the core-1 (Thomsen-Friedenreich) glycotope on tumor cells? Biochemistry (Mosc). 2015, 80, 801–807. [Google Scholar] [CrossRef]
- Henderson, G.; Ulsemer, P.; Schöber, U.; Löffler, A.; Alpert, C.A.; Zimmermann-Kordmann, M.; Reutter, W.; Karsten, U.; Goletz, S.; Blaut, M. Occurrence of the human tumor-specific antigen structure Galβ1-3GalNAcα- (Thomsen-Friedenreich) and related structures on gut bacteria: Prevalence, immunochemical analysis and structural confirmation. Glycobiology 2011, 21, 1277–1289. [Google Scholar] [CrossRef]
- Kurtenkov, O.; Izotova, J.; Klaamas, K.; Sergeyev, B. Increased sialylation of anti-Thomsen-Friedenreich antigen (CD176) antibodies in patients with gastric cancer: A diagnostic and prognostic potential. Biomed. Res. Int. 2014, 2014, 830847. [Google Scholar] [CrossRef] [Green Version]
- Kurtenkov, O.; Bubina, M.; Klaamas, K. Signatures of anti-Thomsen—Friedenreich antigen antibody diversity in colon cancer patients. Exp Oncol. 2018, 40, 48–58. [Google Scholar] [CrossRef]
- Kurtenkov, O. Profiling of Naturally Occurring Antibodies to the Thomsen-Friedenreich Antigen in Health and Cancer: The Diversity and Clinical Potential. Biomed. Res. Int. 2020, 2020, 9747040. [Google Scholar] [CrossRef]
- Hanisch, F.G.; Baldus, S.E. The Thomsen-Friedenreich (TF) antigen: A critical review on the structural, biosynthetic and histochemical aspects of a pancarcinoma-associated antigen. Histol. Histopathol. 1997, 12, 263–281. [Google Scholar]
- Rittenhouse-Diakun, K.; Xia, Z.; Pickhardt, D.; Morey, S.; Baek, M.G.; Roy, R. Development and characterization of monoclonal antibody to T-antigen: (gal beta1-3GalNAc-alpha-O). Hybridoma 1998, 17, 165–173. [Google Scholar] [CrossRef]
- Heimburg, J.; Yan, J.; Morey, S.; Glinskii, O.V.; Huxley, V.H.; Wild, L.; Klick, R.; Roy, R.; Glinsky, V.V.; Rittenhouse-Olson, K. Inhibition of spontaneous breast cancer metastasis by anti-Thomsen-Friedenreich antigen monoclonal antibody JAA-F11. Neoplasia 2006, 8, 939–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rittenhouse-Olson, K. Jaa-f11: Extending the life of mice with breast cancer. Expert. Opin. Biol. Ther. 2007, 7, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.; Yadav, A.; Morey, S.; Abdullah, J.; Hrysenko, G.; Eng, J.Y.; Sajjad, M.; Koury, S.; Rittenhouse-Olson, K. Preclinical studies with JAA-F11 anti-Thomsen-Friedenreich monoclonal antibody for human breast cancer. Future Oncol. 2014, 10, 385–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tati, S.; Fisk, J.C.; Abdullah, J.; Karacosta, L.; Chrisikos, T.; Philbin, P.; Morey, S.; Ghazal, D.; Zazala, F.; Jessee, J.; et al. Humanization of JAA-F11, a Highly Specific Anti-Thomsen-Friedenreich Pancarcinoma Antibody and InVitro Efficacy Analysis. Neoplasia 2017, 19, 716–733. [Google Scholar] [CrossRef] [PubMed]
- Longenecker, B.M.; Willans, D.J.; MacLean, G.D.; Selvaraj, S.; Suresh, M.R.; Noujaim, A.A. Monoclonal antibodies and synthetic tumor-associated glycoconjugates in the study of the expression of Thomsen-Friedenreich-like and Tn-like antigens on human cancers. J. Natl. Cancer Inst. 1987, 78, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Richman, C.M.; Denardo, S.J.; O'Donnell, R.T.; Yuan, A.; Shen, S.; Goldstein, D.S.; Tuscano, J.M.; Wun, T.; Chew, H.K.; Lara, P.; et al. High-dose radioimmunotherapy combined with fixed, low-dose paclitaxel in metastatic prostate and breast cancer by using a MUC-1 monoclonal antibody, m170, linked to indium-111/yttrium-90 via a cathepsin cleavable linker with cyclosporine to prevent human anti-mouse antibody. Clin. Cancer Res. 2005, 11, 5920–5927. [Google Scholar] [CrossRef] [Green Version]
- Cartwright, O.C.; Beekman, A.M.; Cominetti, M.M.D.; Russell, D.A.; Searcey, M. A Peptide-Duocarmycin Conjugate Targeting the Thomsen-Friedenreich Antigen Has Potent and Selective Antitumor Activity. Bioconjug. Chem. 2020, 31, 1745–1749. [Google Scholar] [CrossRef]
- Osinaga, E. Expression of cancer-associated simple mucin-type O-glycosylated antigens in parasites. IUBMB Life 2007, 59, 269–273. [Google Scholar] [CrossRef]
- Alvarez Errico, D.; Medeiros, A.; Míguez, M.; Casaravilla, C.; Malgor, R.; Carmona, C.; Nieto, A.; Osinaga, E. O-glycosylation in Echinococcus granulosus: Identification and characterization of the carcinoma-associated Tn antigen. Exp. Parasitol. 2001, 98, 100–109. [Google Scholar] [CrossRef]
- Freire, T.; Casaravilla, C.; Carmona, C.; Osinaga, E. Mucin type O-glycosylation in Fasciola hepatica: Characterization of carcinoma associated Tn and sialyl-Tn antigens and evaluation of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase activity. Int. J. Parasitol. 2003, 33, 47–56. [Google Scholar] [CrossRef]
- Casaravilla, C.; Freire, T.; Malgor, R.; Medeiros, A.; Osinaga, E.; Carmona, C. Mucin-Type O-Glycosylation in helminth parasites from major taxonomic groups: Evidence for widespread distribution of the Tn Antigen (GalNAc-Ser/Thr) and identification of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase activity. J. Parasitol. 2003, 89, 709–714. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, A.; Chiribao, M.L.; Ubillos, L.; Festari, M.F.; Saldaña, J.; Robello, C.; Domínguez, L.; Calvete, J.J.; Osinaga, E. Mucin-type O-glycosylation in Mesocestoides vogae (syn. corti). Int. J. Parasitol. 2008, 38, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Freire, T.; Robello, C.; Soule, S.; Ferreira, F.; Osinaga, E. Sialyl-Tn antigen expression and O-linked GalNAc-Thr synthesis by Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2003, 312, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.M.C.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198. [Google Scholar] [CrossRef]
- Noya, V.; Bay, S.; Festari, M.F.; García, E.P.; Rodriguez, E.; Chiale, C.; Ganneau, C.; Baleux, F.; Astrada, S.; Bollati-Fogolín, M.; et al. Mucin-like peptides from Echinococcus granulosus induce antitumor activity. Int. J. Oncol. 2013, 43, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Berriel, E.; Russo, S.; Monin, L.; Festari, M.F.; Berois, N.; Fernández, G.; Freire, T.; Osinaga, E. Antitumor activity of human hydatid cyst fluid in a murine model of colon cancer. Sci. World J. 2013, 2013, 230176. [Google Scholar] [CrossRef] [Green Version]
- Ubillos, L.; Freire, T.; Berriel, E.; Chiribao, M.L.; Chiale, C.; Festari, M.F.; Medeiros, A.; Mazal, D.; Rondán, M.; Bollati-Fogolín, M.E.; et al. Trypanosoma cruzi extracts elicit protective immune response against chemically induced colon and mammary cancers. Int. J. Cancer 2016, 138, 1719–1731. [Google Scholar] [CrossRef] [Green Version]
- Berriel, E.; Freire, T.; Chiale, C.; Rodríguez, E.; Morón, G.; Fernández-Graña, G.; Crispo, M.; Berois, N.; Osinaga, E. Human hydatid cyst fluid-induced therapeutic anti-cancer immune responses via NK1.1+ cell activation in mice. Cancer Immunol. Immunother. 2021, 70, 3617–3627. [Google Scholar] [CrossRef]
- Blanas, A.; Sahasrabudhe, N.M.; Rodríguez, E.; van Kooyk, Y.; van Vliet, S.J. Fucosylated Antigens in Cancer: An Alliance toward Tumor Progression, Metastasis, and Resistance to Chemotherapy. Front. Oncol. 2018, 8, 39. [Google Scholar] [CrossRef]
- Dotz, V.; Wuhrer, M. Histo-blood group glycans in the context of personalized medicine. Biochim. Biophys. Acta 2016, 1860, 1596–1607. [Google Scholar] [CrossRef] [Green Version]
- Skulimowski, A.; Durczyński, A.; Strzelczyk, J.; Hogendorf, P. Comparison of clinical usefulness of serum Ca125 and CA19-9 in pancreatic adenocarcinoma diagnosis: Meta-analysis and systematic review of literature. Biomarkers 2021, 26, 287–295. [Google Scholar] [CrossRef]
- Fernandes, E.; Sores, J.; Cotton, S.; Peixoto, A.; Ferreira, D.; Freitas, R.; Reis, C.A.; Santos, L.L.; Ferreira, J.A. Esophageal, gastric and colorectal cancers: Looking beyond classical serological biomarkers towards glycoproteomics-assisted precision oncology. Theranostics 2020, 10, 4903–4928. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.G.; Carrascal, M.; Mineiro, A.G.; Bugalho, A.; Borralho, P.; Silva, Z.; Dall'olio, F.; Videira, P.A. Carcinoembryonic antigen is a sialyl Lewis x/a carrier and an E-selectin ligand in non-small cell lung cancer. Int. J. Oncol. 2019, 55, 1033–1048. [Google Scholar] [CrossRef] [PubMed]
- Sawada, R.; Sun, S.M.; Wu, X.; Hong, F.; Ragupathi, G.; Livingston, P.O.; Scholz, W.W. Human monoclonal antibodies to sialyl-Lewis (CA19.9) with potent CDC, ADCC, and antitumor activity. Clin. Cancer Res. 2011, 17, 1024–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O'Reilly, E.M.; Borazanci, E.H.; Yu, K.H.; Varghese, A.M.; Estrella, H.; Kamins, D.; Melink, T.; Dorr, K.; Maffuid, P.; Gutheil, J.; et al. HuMab-5B1 (MVT-5873), a mAb targeting sLea, in combination with first-line gemcitabine plus nab-paclitaxel (gem/nab-P) for patients with pancreatic cancer (PDAC) and other CA19-9 positive malignancies. J. Clin. Oncol. 2018, 36, e16235. [Google Scholar] [CrossRef]
- O'Reilly, E.A.; Lohrmann, C.; O'Donoghue, J.A.; Borazanci, E.; Estrella, H.; Teng, R.; Melink, T.; Dorr, K.; Kearns, C.; Peterson, M.; et al. Phase I dose escalation study of 177Lu-HuMab-5B1 (MVT-1075) in combination with MVT-5873 as radioimmunotherapy (RIT) in subjects with relapsed / refractory pancreatic cancer or other CA19-9+ malignancies. In Proceedings of the American Association for Cancer Research Annual Meeting 2018, Chicago, IL, USA, 14–18 April 2018; AACR: Philadelphia, PA, USA, ; CT140, 2018; Volume 78. [Google Scholar] [CrossRef]
- Tivadar, S.T.; McIntosh, R.S.; Chua, J.X.; Moss, R.; Parsons, T.; Zaitoun, A.M.; Madhusudan, S.; Durrant, L.G.; Vankemmelbeke, M. Monoclonal Antibody Targeting Sialyl-di-Lewisa-Containing Internalizing and Noninternalizing Glycoproteins with Cancer Immunotherapy Development Potential. Mol. Cancer Ther. 2020, 19, 790–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, K.; Stockert, E.; Garin-Chesa, P.; Welt, S.; Lloyd, K.O.; Armour, K.L.; Wallace, T.P.; Harris, W.J.; Carr, F.J.; Old, L.J. Specificity analysis of blood group Lewis-y (Le(y)) antibodies generatedagainst synthetic and natural Le(y) determinants. Proc. Natl. Acad. Sci. USA 1994, 91, 12957–12961. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.M.; Geleick, D.; Rubira, M.; Clarke, K.; Nice, E.C.; Smyth, F.E.; Stockert, E.; Richards, E.C.; Carr, F.J.; Harris, W.J.; et al. Construction, production, and characterization of humanized anti-Lewis Y monoclonal antibody 3S193 for targeted immunotherapy of solid tumors. Cancer Res. 2000, 60, 3254–3261. [Google Scholar]
- Smaletz, O.; Ismael, G.; Del Pilar Estevez-Diz, M.; Nascimento, I.L.O.; de Morais, A.L.G.; Cunha-Junior, G.F.; Azevedo, S.J.; Alves, V.A.; Moro, A.M.; Yeda, F.P.; et al. . Phase II consolidation trial with anti-Lewis-Y monoclonal antibody (hu3S193) in platinum-sensitive ovarian cancer after a second remission. Int. J. Gynecol. Cancer. 2021, 31, 562–568. [Google Scholar] [CrossRef]
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Hönemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Pan, Z.; Han, L.; Zhou, Y.; Zong, H.; Wang, L.; Sun, R.; Jiang, H.; Xie, Y.; Yuan, Y.; et al. A Novel Bispecific Antibody Targeting CD3 and Lewis Y with Potent Therapeutic Efficacy against Gastric Cancer. Biomedicines 2021, 9, 1059. [Google Scholar] [CrossRef]
- Chang, C.Y.; Jeffrey, P.D.; Bajorath, J.; Hellström, I.; Hellström, K.E.; Sheriff, S. Crystallization and preliminary X-ray analysis of the monoclonal anti-tumor antibody BR96 and its complex with the Lewis Y determinant. J. Mol. Biol. 1994, 235, 372–376. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Sugarman, S.; Gelmon, K.A.; Cohen, R.; Saleh, M.; Isaacs, C.; Young, L.; Healey, D.; Onetto, N.; Slichenmyer, W. Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. J. Clin. Oncol. 1999, 17, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Ragupathi, G.; Damani, P.; Srivastava, G.; Srivastava, O.; Sucheck, S.J.; Ichikawa, Y.; Livingston, P.O. Synthesis of sialyl Lewis(a) (sLe (a), CA19-9) and construction of an immunogenic sLe(a) vaccine. Cancer Immunol. Immunother. 2009, 58, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Wang, F. The physiological and pathological roles and applications of sialyl Lewis x, a common carbohydrate ligand of the three selectins. Glycoconj. J. 2020, 37, 277–291. [Google Scholar] [CrossRef]
- Springer, T.A. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 1994, 76, 301–314. [Google Scholar] [CrossRef]
- Bassagañas, S.; Allende, H.; Cobler, L.; Ortiz, M.R.; Llop, E.; de Bolós, C.; Peracaula, R. Inflammatory cytokines regulate the expression of glycosyltransferases involved in the biosynthesis of tumor-associated sialylated glycans in pancreatic cancer cell lines. Cytokine 2015, 75, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, A.; Ohmori, K.; Yoneda, T.; Tsuyuoka, K.; Hasegawa, A.; Kiso, M.; Kannagi, R. Contribution of carbohydrate antigens Sialyl Lewis A and Sialyl Lewis X to adhesion of human cancer cells to vascular endothelium. Cancer Res. 1993, 53, 354–361. [Google Scholar] [PubMed]
- Hiller, K.M.; Mayben, J.P.; Bendt, K.M.; Manousos, G.A.; Senger, K.; Cameron, H.S.; Weston, B.W. Transfection of alpha(1,3)fucosyltransferase antisense sequences impairs the proliferative and tumorigenic ability of human colon carcinoma cells. Mol. Carcinog. 2000, 27, 280–288. [Google Scholar] [CrossRef]
- Fukushi, Y.; Nudelman, E.; Levery, S.B.; Hakomori, S.; Rauvala, H. Novel fucolipids accumulating in human adenocarcinoma. III. A hybridoma antibody (FH6) defining a human cancer-associated difucoganglioside (VI3NeuAcV3III3Fuc2nLc6). J. Biol. Chem. 1984, 259, 10511–10517. [Google Scholar] [CrossRef]
- Fukushima, K.; Hirota, M.; Terasaki, P.I.; Wakisaka, A.; Togashi, H.; Chia, D.; Suyama, N.; Fukushi, Y.; Nudelman, E.; Hakomori, S. Characterization of sialosylated Lewisx as a new tumor-associated antigen. Cancer Res. 1984, 44, 5279–5285. [Google Scholar] [PubMed]
- Hanisch, F.G.; Hanski, C.; Hasegawa, A. Sialyl Lewis(x) antigen as defined by monoclonal antibody AM-3 is a marker of dysplasia in the colonic adenoma-carcinoma sequence. Cancer Res. 1992, 52, 3138–3144. [Google Scholar] [PubMed]
- Dohi T, Nemoto T, Ohta S, Shitara K, Hanai N, Nudelman E, Hakomori S, Oshima M Different binding properties of three monoclonal antibodies to sialyl Le(x) glycolipids in a gastric cancer cell line and normal stomach tissue. Anticancer Res. 1993, 13, 1277–1282.
- Magnani, J.L. The discovery, biology, and drug development of sialyl Lea and sialyl Lex. Arch. Biochem. Biophys. 2004, 426, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.X.; Liang, Y.; Gao, W. Clinicopathological and prognostic significance of sialyl Lewis X overexpression in patients with cancer: A meta-analysis. Onco-Targets Ther. 2016, 9, 3113–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, J.; Kobayashi, I.; Tatematsu, K.; Sezutsu, H.; Noda, K.; Ishihara, H. Sandwich ELISA Using a Mouse/Human Chimeric CSLEX-1 Antibody. Clin. Chem. 2016, 62, 1516–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, D.L.; Foshag, L.J.; Hoon, D.S.; Nizze, J.A.; Famatiga, E.; Wanek, L.A.; Chang, C.; Davtyan, D.G.; Gupta, R.K.; Elashoff, R.; et al. Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma vaccine. Ann. Surg. 1992, 216, 463–482. [Google Scholar] [CrossRef]
- Ravindranath, M.H.; Kelley, M.C.; Jones, R.C.; Amiri, A.A.; Bauer, P.M.; Morton, D.L. Ratio of IgG:IgM antibodies to sialyl Lewis(x) and GM3 correlates with tumor growth after immunization with melanoma-cell vaccine with different adjuvants in mice. Int. J. Cancer 1998, 75, 117–124. [Google Scholar] [CrossRef]
- Kieber-Emmons, T.; Luo, P.; Qiu, J.; Chang, T.Y.; Insug, O.; Blaszczyk-Thurin, M.; Steplewski, Z. Vaccination with carbohydrate peptide mimotopes promotes anti-tumor responses. Nat. Biotechnol. 1999, 17, 660–665. [Google Scholar] [CrossRef]
- Kuznetsova, N.R.; Stepanova, E.V.; Peretolchina, N.M.; Khochenkov, D.A.; Boldyrev, I.A.; Bovin, N.V.; Vodovozova, E.L. Targeting liposomes loaded with melphalan prodrug to tumour vasculature via the Sialyl Lewis X selectin ligand. J. Drug Target. 2014, 22, 242–250. [Google Scholar] [CrossRef]
- Kishimoto, S.; Fujitani, N.; Ohnishi, T.; Aoki, H.; Suzuki, R.; Fukushima, S. Cisplatin-loaded, Sialyl Lewis X-Modified Liposomes: Drug Release, Biodistribution and Antitumor Efficacy. Anticancer Res. 2017, 37, 6055–6061. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Imaeda, Y.; Umemoto, S.; Kobayashi, K.; Suzuki, H.; Okamoto, T. Cimetidine increases survival of colorectal cancer patients with high levels of sialyl Lewis-X and sialyl Lewis-A epitope expression on tumour cells. Br. J. Cancer 2002, 86, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Matsumoto, S.; Morishima, T.; Kawabe, T.; Okamoto, T. Cimetidine inhibits cancer cell adhesion to endothelial cells and prevents metastasis by blocking E-selectin expression. Cancer Res. 2000, 60, 3978–3984. [Google Scholar] [PubMed]
- Liu, F.R.; Jiang, C.G.; Li, Y.S.; Li, J.B.; Li, F. Cimetidine inhibits the adhesion of gastric cancer cells expressing high levels of sialyl Lewis x in human vascular endothelial cells by blocking E-selectin expression. Int. J. Mol. Med. 2011, 27, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Borentain, P.; Carmona, S.; Mathieu, S.; Jouve, E.; El-Battari, A.; Gérolami, R. Inhibition of E-selectin expression on the surface of endothelial cells inhibits hepatocellular carcinoma growth by preventing tumor angiogenesis. Cancer Chemother. Pharmacol. 2016, 77, 847–856. [Google Scholar] [CrossRef]
- Schauer, R.; Kamerling, J.P. Exploration of the Sialic Acid World. Adv. Carbohydr. Chem. Biochem. 2018, 75, 1–213. [Google Scholar] [CrossRef]
- Mindler, K.; Ostertag, E.; Stehle, T. The polyfunctional polysialic acid: A structural view. Carbohydr. Res. 2021, 507, 108376. [Google Scholar] [CrossRef]
- Bhide, G.P.; Zapater, J.L.; Colley, K.J. Autopolysialylation of polysialyltransferases is required for polysialylation and polysialic acid chain elongation on select glycoprotein substrates. J. Biol. Chem. 2018, 293, 701–716. [Google Scholar] [CrossRef]
- Berois, N.; Osinaga, E. Glycobiology of neuroblastoma: Impact on tumor behavior, prognosis, and therapeutic strategies. Front. Oncol. 2014, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, X.; Zeng, Y.N.; He, F.; Yang, X.M.; Guan, F. Enhanced expression of polysialic acid correlates with malignant phenotype in breast cancer cell lines and clinical tissue samples. Int. J. Mol. Med. 2016, 37, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Klobučar, M.; Visentin, S.; Jakovčević, A.; Bilić, M.; Kovač-Bilić, L.; Đanić, D.; Pavelić, K.; Kraljević Pavelić, S. Expression of polysialic acid in primary laryngeal squamous cell carcinoma. Life Sci. 2017, 173, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.C.; Giehl, K.; Kastilan, C.; Hasel, C.; Mühlenhoff, M.; Adler, G.; Wedlich, D.; Menke, A. Polysialylated NCAM represses E-cadherin-mediated cell-cell adhesion in pancreatic tumor cells. Gastroenterology 2008, 134, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Otake, Y.; Nakagawa, T.; Kawano, Y.; Miyahara, R.; Li, M.; Yanagihara, K.; Nakayama, J.; Fujimoto, I.; Ikenaka, K.; et al. Expression of polysialic acid and STX, a human polysialyltransferase, is correlated with tumor progression in non-small cell lung cancer. Cancer Res. 2000, 60, 3072–3080. [Google Scholar] [PubMed]
- Gong, L.; Zhou, X.; Yang, J.; Jiang, Y.; Yang, H. Effects of the regulation of polysialyltransferase ST8SiaII on the invasiveness and metastasis of small cell lung cancer cells. Oncol. Rep. 2017, 37, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Kitajima, K. Polysialylation and disease. Mol. Aspects Med. 2021, 79, 100892. [Google Scholar] [CrossRef]
- Jarahian, M.; Marofi, F.; Maashi, M.S.; Ghaebi, M.; Khezri, A.; Berger, M.R. Re-Expression of Poly/Oligo-Sialylated Adhesion Molecules on the Surface of Tumor Cells Disrupts Their Interaction with Immune-Effector Cells and Contributes to Pathophysiological Immune Escape. Cancers 2021, 13, 5203. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kato, K.; Denda-Nagai, K.; Hanisch, F.G.; Clausen, H.; Irimura, T. The epitope recognized by the unique anti-MUC1 monoclonal antibody MY.1E12 involves sialyl alpha 2-3galactosyl beta 1-3N-acetylgalactosaminide linked to a distinct threonine residue in the MUC1 tandem repeat. J. Immunol. Methods 2002, 270, 199–209. [Google Scholar] [CrossRef]
- Martersteck, C.M.; Kedersha, N.L.; Drapp, D.A.; Tsui, T.G.; Colley, K.J. Unique alpha 2, 8-polysialylated glycoproteins in breast cancer and leukemia cells. Glycobiology 1996, 6, 289–301. [Google Scholar] [CrossRef]
- Klebert, S.; Kratzin, H.D.; Zimmermann, B.; Vaesen, M.; Frosch, M.; Weisgerber, C.; Bitter-Suermann, D.; Hilschmann, N. Primary structure of the murine monoclonal IgG2a antibody mAb735 against alpha (2-8) polysialic acid. 2. Amino acid sequence of the heavy (H-) chain Fd' region. Biol. Chem. Hoppe Seyler 1993, 374, 993–1000. [Google Scholar] [CrossRef]
- Cox, E.C.; Thornlow, D.N.; Jones, M.A.; Fuller, J.L.; Merritt, J.H.; Paszek, M.J.; Alabi, C.A.; DeLisa, M.P. Antibody-Mediated Endocytosis of Polysialic Acid Enables Intracellular Delivery and Cytotoxicity of a Glycan-Directed Antibody-Drug Conjugate. Cancer Res. 2019, 79, 1810–1821. [Google Scholar] [CrossRef] [Green Version]
- Ragupathi, G.; Coltart, D.M.; Williams, L.J.; Koide, F.; Kagan, E.; Allen, J.; Harris, C.; Glunz, P.W.; Livingston, P.O.; Danishefsky, S.J. On the power of chemical synthesis: Immunological evaluation of models for multiantigenic carbohydrate-based cancer vaccines. Proc. Natl. Acad. Sci. USA 2002, 99, 13699–13704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slovin, S.F.; Ragupathi, G.; Fernandez, C.; Diani, M.; Jefferson, M.P.; Wilton, A.; Kelly, W.K.; Morris, M.; Solit, D.; Clausen, H.; et al. A polyvalent vaccine for high-risk prostate patients: "are more antigens better?". Cancer Immunol. Immunother. 2007, 56, 1921–1930. [Google Scholar] [CrossRef]
- Sabbatini, P.J.; Ragupathi, G.; Hood, C.; Aghajanian, C.A.; Juretzka, M.; Iasonos, A.; Hensley, M.L.; Spassova, M.K.; Ouerfelli, O.; Spriggs, D.R.; et al. Pilot study of a heptavalent vaccine-keyhole limpet hemocyanin conjugate plus QS21 in patients with epithelial ovarian, fallopian tube, or peritoneal cancer. Clin. Cancer Res. 2007, 13, 4170–4177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragupathi, G.; Koide, F.; Livingston, P.O.; Cho, Y.S.; Endo, A.; Wan, Q.; Spassova, M.K.; Keding, S.J.; Allen, J.; Ouerfelli, O.; et al. Preparation and evaluation of unimolecular pentavalent and hexavalent antigenic constructs targeting prostate and breast cancer: A synthetic route to anticancer vaccine candidates. J. Am. Chem. Soc. 2006, 128, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; McGuirk, J.P. CAR T cells: Continuation in a revolution of immunotherapy. Lancet Oncol. 2020, 21, e168–e178. [Google Scholar] [CrossRef]
- Alcantara, M.; Du Rusquec, P.; Romano, E. Current Clinical Evidence and Potential Solutions to Increase Benefit of CAR T-Cell Therapy for Patients with Solid Tumors. Oncoimmunology 2020, 9, 1777064. [Google Scholar] [CrossRef] [PubMed]
- Rossig, C.; Bollard, C.M.; Nuchtern, J.G.; Merchant, D.A.; Brenner, M.K. Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int. J. Cancer 2001, 94, 228–236. [Google Scholar] [CrossRef]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Nat Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Huang, L.; Lin, D.; Lai, X.; Wu, L.; Liao, X.; Liu, J.; Zeng, Y.; Liang, L.; Zhang, G.; et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J. Cancer Res. Clin. Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tumino, N.; Weber, G.; Besi, F.; Del Bufalo, F.; Bertaina, V.; Paci, P.; Quatrini, L.; Antonucci, L.; Sinibaldi, M.; Quintarelli, C.; et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J. Hematol. Oncol. 2021, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Yvon, E.; Del Vecchio, M.; Savoldo, B.; Hoyos, V.; Dutour, A.; Anichini, A.; Dotti, G.; Brenner, M.K. Immunotherapy of metastatic melanoma using genetically engineered GD2-specific T cells. Clin. Cancer Res. 2009, 15, 5852–5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Wu, X.; Yan, J.; Yu, H.; Xu, L.; Chi, Z.; Sheng, X.; Si, L.; Cui, C.; Dai, J.; et al. Anti-GD2/4-1BB chimeric antigen receptor T cell therapy for the treatment of Chinese melanoma patients. J. Hematol. Oncol. 2018, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Soltantoyeh, T.; Akbari, B.; Karimi, A.; Mahmoodi, C.G.; Ghahri-Saremi, N.; Hadjati, J.; Hamblin, M.R.; Mirzaei, H.R. Chimeric Antigen Receptor (CAR) T Cell Therapy for Metastatic Melanoma: Challenges and Road Ahead. Cells 2021, 10, 1450. [Google Scholar] [CrossRef]
- Lin, Y.; Chen, S.; Zhong, S.; An, H.; Yin, H.; McGowan, E. Phase I clinical trial of PD-1 knockout anti-MUC1 CAR-T cells in the treatment of patients with non-small cell lung cancer. Ann. Oncol. 2019, 30 (Suppl. 11), xi12–xi15. [Google Scholar] [CrossRef]
- Specht, J.M.; Maloney, D.G.; Yeung, C.; Wu, V.; Bamdad, C. Phase I study of adoptive immunotherapy for advanced MUC1* positive breast cancer with autologous T cells engineered to express a chimeric antigen receptor, huMNC2-CAR44 specific for a cleaved form of MUC1 (MUC1*). Cancer Res. 2020, 80, CT232. [Google Scholar] [CrossRef]
- Wilkie, S.; Picco, G.; Foster, J.; Davies, D.M.; Julien, S.; Cooper, L.; Arif, S.; Mather, S.J.; Taylor-Papadimitriou, J.; Burchell, J.M.; et al. Retargeting of human T cells to tumor-associated MUC1: The evolution of a chimeric antigen receptor. J. Immunol. 2008, 180, 4901–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Lai, Y.; Li, J.; Qin, L.; Xu, Y.; Zhao, R.; Li, B.; Lin, S.; Wang, S.; Wu, Q.; et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology 2017, 6, e1284722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, F.; Jiang, L.; Zhang, B.; Lu, Q.; Zhou, Q.; Liao, X.; Wu, H.; Du, K.; Zhu, Y.; Meng, H.; et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells. Sci. China Life Sci. 2016, 59, 386–397. [Google Scholar] [CrossRef] [Green Version]
- Mei, Z.; Zhang, K.; Lam, A.K.; Huang, J.; Qiu, F.; Qiao, B.; Zhang, Y. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med. 2020, 9, 640–652. [Google Scholar] [CrossRef] [Green Version]
- Nalawade, S.A.; Shafer, P.; Bajgain, P.; McKenna, M.K.; Ali, A.; Kelly, L.; Joubert, J.; Gottschalk, S.; Watanabe, N.; Leen, A.; et al. Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J. Immunother. Cancer 2021, 9, e003237. [Google Scholar] [CrossRef]
- Zhai, X.; You, F.; Xiang, S.; Jiang, L.; Chen, D.; Li, Y.; Fan, S.; Han, Z.; Zhang, T.; An, G.; et al. MUC1-Tn-targeting chimeric antigen receptor-modified Vγ9Vδ2 T cells with enhanced antigen-specific anti-tumor activity. Am. J. Cancer Res. 2021, 11, 79–91. [Google Scholar]
- Gutierrez, R.; Shah, P.D.; Hamid, O.; Garfall, A.L.; Posey, A.; Russell Bishop, M.; Blumenschein, G.R.; Lynne Johnson, M.; Lee, S.; Luke, J.J.; et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. J. Clin. Oncol. 2021, 15, e14513. [Google Scholar] [CrossRef]
- Hombach, A.; Heuser, C.; Sircar, R.; Tillmann, T.; Diehl, V.; Kruis, W.; Pohl, C.; Abken, H. T cell targeting of TAG72+ tumor cells by a chimeric receptor with antibody-like specificity for a carbohydrate epitope. Gastroenterology 1997, 113, 1163–1170. [Google Scholar] [CrossRef]
- McGuinness, R.P.; Ge, Y.; Patel, S.D.; Kashmiri, S.V.; Lee, H.S.; Hand, P.H.; Schlom, J.; Finer, M.H.; McArthur, J.G. Anti-tumor activity of human T cells expressing the CC49-zeta chimeric immune receptor. Hum. Gene Ther. 1999, 10, 165–173. [Google Scholar] [CrossRef]
- Hombach, A.A.; Rappl, G.; Abken, H. Blocking CD30 on T Cells by a Dual Specific CAR for CD30 and Colon Cancer Antigens Improves the CAR T Cell Response against CD30- Tumors. Mol. Ther. 2019, 27, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Murad, J.P.; Kozlowska, A.K.; Lee, H.J.; Ramamurthy, M.; Chang, W.C.; Yazaki, P.; Colcher, D.; Shively, J.; Cristea, M.; Forman, S.J.; et al. Effective Targeting of TAG72 + Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front. Immunol. 2018, 9, 2268. [Google Scholar] [CrossRef] [PubMed]
- Shu, R.; Evtimov, V.J.; Hammett, M.V.; Nguyen, N.N.; Zhuang, J.; Hudson, P.J.; Howard, M.C.; Pupovac, A.; Trounson, A.O.; Boyd, R.L. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol Ther Oncolytics. 2021, 20, 325–341. [Google Scholar] [CrossRef] [PubMed]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Shaikh, A.S.; Wang, F. Recent Advance in Tumor-associated Carbohydrate Antigens (TACAs)-based Antitumor Vaccines. ACS Chem. Biol. 2016, 11, 850–863. [Google Scholar] [CrossRef]
- Pifferi, C.; Ruiz-de-Angulo, A.; Goyard, D.; Tiertant, C.; Sacristán, N.; Barriales, D.; Berthet, N.; Anguita, J.; Renaudet, O.; Fernández-Tejada, A. Chemical synthesis and immunological evaluation of new generation multivalent anticancer vaccines based on a Tn antigen analogue. Chem. Sci. 2020, 11, 4488–4498. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Zheng, X.J.; Guo, H.; Cao, Y.; Zhang, F.; Li, Q.; Ye, X.S.; Zhou, Y. Fluorine-modified sialyl-Tn-CRM197 vaccine elicits a robust immune response. Glycoconj. J. 2019, 36, 399–408. [Google Scholar] [CrossRef]
- Wu, X.; McFall-Boegeman, H.; Rashidijahanabad, Z.; Liu, K.; Pett, C.; Yu, J.; Schorlemer, M.; Ramadan, S.; Behren, S.; Westerlind, U.; et al. Synthesis and immunological evaluation of the unnatural beta-linked mucin-1 Thomsen-Friedenreich conjugate. Org. Biomol. Chem. 2021, 19, 2448–2455. [Google Scholar] [CrossRef]
- Amon, R.; Rosenfeld, R.; Perlmutter, S.; Grant, O.C.; Yehuda, S.; Borenstein-Katz, A.; Alcalay, R.; Marshanski, T.; Yu, H.; Diskin, R. Directed Evolution of Therapeutic Antibodies Targeting Glycosylation in Cancer. Cancers 2020, 12, 2824. [Google Scholar] [CrossRef]
- Gogesch, P.; Dudek, S.; van Zandbergen, G.; Waibler, Z.; Anzaghe, M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. Int. J. Mol. Sci. 2021, 22, 8947. [Google Scholar] [CrossRef]
- Vankemmelbeke, M.; McIntosh, R.S.; Chua, J.X.; Kirk, T.; Daniels, I.; Patsalidou, M.; Moss, R.; Parsons, T.; Scott, D.; Harris, G.; et al. Engineering the Human Fc Region Enables Direct Cell Killing by Cancer Glycan-Targeting Antibodies without the Need for Immune Effector Cells or Complement. Cancer Res. 2020, 80, 3399–3412. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.A.; Stanczak, M.A.; Mantuano, N.R.; Xiao, H.; Pijnenborg, J.F.A.; Malaker, S.A.; Miller, C.L.; Weidenbacher, P.A.; Tanzo, J.T.; Ahn, G.; et al. Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat. Chem. Biol. 2020, 16, 1376–1384. [Google Scholar] [CrossRef] [PubMed]


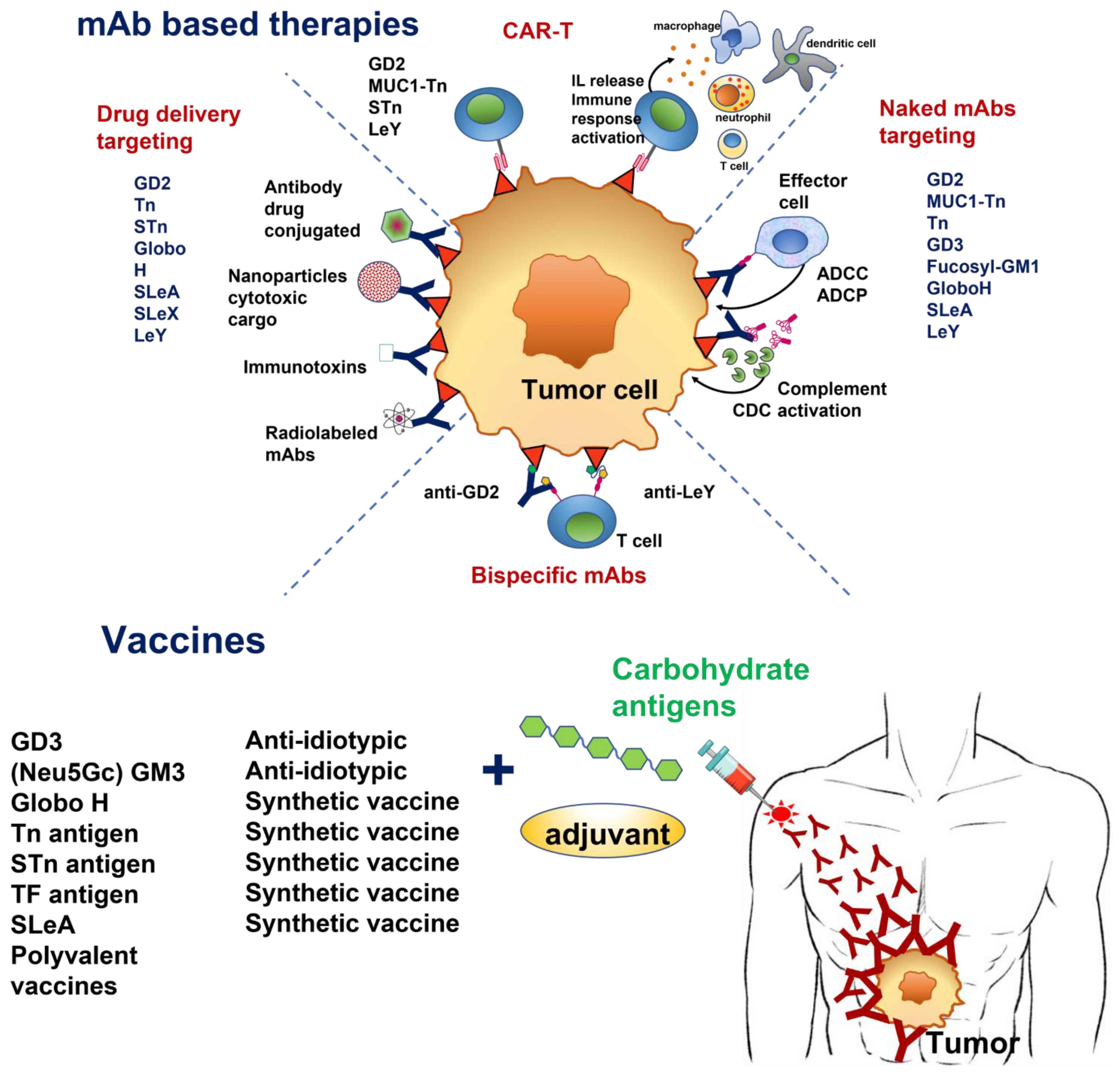{kind=link}
| Tumor Glycan | Glycan Type | Structure * | Type of Cancer | Function in Cancer | Therapeutic Strategy |
|---|---|---|---|---|---|
| GD2 | Ganglioside |  | Glioma Melanoma Neuroblastoma Retinoblastoma | Cell proliferation Motility Apoptosis resistance | mAbs Dinutuximab Naxitamab Bispecific-Abs CAR-T cells |
| GD3 | Ganglioside |  | Glioma Melanoma Breast, Lung | Cell growth Invasion | mAb huR24 Anti-idiotypic mAb BEC2 |
| GM3 (Neu5Gc) | Ganglioside |  | Melanoma Lung Colon Breast | Cell growth Metastasis | mAb 14F7hT Anti-idiotypic mAb Racotumomab Synthetic vaccine |
| Fucosyl-GM1 | Ganglioside |  | SCLC | The role in cancer is unclear | Human mAb BMS-986012 |
| Globo H | Globoside |  | Colon, Ovary, Breast, Prostate, Gastric, Lung, Endometrial, Pancreatic | Angiogenesis Immunosuppression | Vaccine mAb OBI-888 ADC OBI-999 |
| Tn antigen | O-GalNAc mucin-type |  | Carcinomas | Metastasis Immuno- suppression | Antibodies Vaccine CAR-T cells |
| STn antigen | O-GalNAc mucin-type |  | Carcinomas | Metastasis Immuno- suppression | Antibodies Vaccine CAR-T cells |
| TF antigen | O-GalNAc mucin-type |  | Carcinomas | Cell growth Adhesion | Vaccine |
| SLeA | Glycoprotein Gangliosides |  | Breast, Colon, Gastric, Lung, Ovarian, Pancreas | Metastasis | Abs MVT-5873 177Lu- MVT-1075 Vaccine |
| SLeX | Glycoprotein Gangliosides |  | Colon, breast melanoma, lung, liver ovary, pancreas | Invasion Metastasis | SLeX -liposomes anticancer drug delivery |
| LeY | Glycoprotein Gangliosides |  | Lung, Ovarian, Fallopian tube, Breast | Metastasis | mAb hu3S193 m3s193 BsAb DCA SGN-15 CAR-T cells |
| Poly-sialic acid | Glycoprotein Glycolipids |  | Neuroblastoma Breast, Pancreas Lung | Metastasis Cell growth | Abs MY.1E12, OL.28, MAB735 Vaccine |
| Target | Drug Candidate | Phase | Cancer Type | Status Time Period | CT ID Refs |
|---|---|---|---|---|---|
| GD2 | |||||
| hu 14.18 (Dinutuximab) | III | High risk Neuroblastoma | Active, not recruiting October 2010–September 2021 | NCT00026312 [36] | |
| hu3F8 (Naxitamab) + GM-CSF | II | High risk Neuroblastoma | Recruiting 4/2018–11/2027 | NCT03363373 | |
| hu3F8 + GM-CSF | I/II | High risk Neuroblastoma | Active, not recruiting 12/2012–12/2023 | NCT01757626 [43,44] | |
| Naxitamab hu3F8 + GM-CSF + irinotecan and temozolomide | II | Recurrent Neuroblastoma | Recruiting 11/2021–10/2026 | NCT04560166 | |
| Dinutuximab hu 14.18 + GM-CSF | II | Lung cancer Osteosarcoma | Active, not recruiting 11/2015–10/2021 | NCT02484443 | |
| hu3F8 + GM-CSF | II | Osteosarcoma | Recruiting 7/2015–7/2022 | NCT02502786 [45] | |
| hu14.18-IL2 | II | Melanoma | Completed 12/2007–11/2019 | NCT00590824 [46,47] | |
| I 131 mAb 3F8 + bevacizumab | I | Neuroblastoma | Completed 8/2006–9/2015 | NCT00450827 | |
| I 131 mAb 3F8 | I | Central nervous system, Leptomeningeal metastases | Active, not recruiting 1/2006–1/2022 | NCT00445965 | |
| Hu3F8-Bispecific antibody (GD2-CD3) | I/II | Neuroblastoma Osteosarcoma Solid Tumors | Recruiting 2/2019–2/2022 | NCT03860207 [48] | |
| Anti-GD2/CD3 bispecific antibody (Nivatrotamab) | I/II | SCLC | Recruiting 8/2021–12/2024 | NCT04750239 | |
| GD3 | |||||
| PF-06688992 Anti-GD3/drug conjugated | I | Stage III/IV Melanoma | Completed 5/2017–1/2020 | NCT03159117 | |
| Human Chimeric Ab KW2871 (ecromeximab) | I/II | Stage IV Melanoma | Terminated 11/2004–2/2015 | NCT00199342 [49] | |
| Human Chimeric Ab KW2871 (ecromeximab) + high dose IFN-α2b | II | Metastatic Melanoma | Completed 3/2008–2/2018 | NCT00679289 [50] | |
| fucosyl-GM1 | |||||
| BMS-986012 + platinum + etoposide | I/II | SCLC | Active, not recruiting 11/2016–4/2021 | NCT02815592 | |
| BMS-986012 | I/II | Relapsed/refractory SCLC | Active, not recruiting 11/2014–6/2022 | NCT02247349 [51] | |
| BMS-986012 + carboplatin + etoposide + nivolumab | II | Extensive-stage SCLC | Recruiting 3/2021–9/2024 | NCT04702880 | |
| Globo H | |||||
| OBI-888 | I/II | Advanced and metastatic solid tumors | Recruiting 5/2018–12/2022 | NCT03573544 | |
| OBI-999 immunotoxin | I/II | Locally advanced solid tumors | Active, not recruiting 12/2019–12/2023 | NCT04084366 | |
| Tn antigen | |||||
| Anti- TA-MUC1 (PankoMab-GEX™) | I | Solid tumors | Completed 11/2009–5/2021 | NCT01222624 [52] | |
| Anti- TA-MUC1 (PankoMab-GEX™) | II | Recurrent Ovarian cancer, Fallopian cancer, Peritoneal cancer | Completed 9/2013–10/2020 | NCT01899599 [53] | |
| Anti- TA-MUC1 (Gatipotuzumab) + anti-EGFR (Tomuzotuximab) | I | Solid tumors | Completed 11/2017–7/2021 | NCT03360734 [54] | |
| TF antigen | |||||
| yttrium Y 90-m170 cyclosporine paclitaxel | I | Breast cancer | Unknown 3/2001–9/2013 | NCT00009763 [55] | |
| yttrium Y 90-m170 cyclosporine paclitaxel | I | Prostate cancer | Unknown 3/2001–9/2013 | NCT00009750 [55] | |
| LeA | |||||
| hu mAb-5B1 (MVT-5873) + FOLFIRINOX | 1 | Pancreatic cancer or CA19-9 positive malignancies | Recruiting 1/2016–1/2023 | NCT02672917 | |
| hu mAb-5B1 (MVT-5873) | 2 | Operable tumors expressing CA19-9 | Recruiting 11/2019–12/2024 | NCT03801915 [56] | |
| LeY | |||||
| hu3S193 | 1 | SCLC | Completed 2/2004–6/2015 | NCT00084799 [57] | |
| hu3S193 | 2 | Fallopian tube, Ovarian cancer, Primary peritoneal cancer | Completed 5/2008–6/2012 | NCT00617773 [58] | |
| SGN-15 + docetaxel | 2 | NSCLC | Completed 1/2003–10/2011 | NCT00051571 [59] |
| Target | Drug Candidate | Phase | Cancer Type | Status/ Time Period | CT ID Refs |
|---|---|---|---|---|---|
| GD3 | |||||
| BEC2 + BCG | III | Small cell lung cancer | Completed September 1998–April 2010 | NCT00037713 [69] | |
| BEC2 + BCG | III | Lung cancer | Completed September 1999–July 2012 | NCT00006352 [70] | |
| BEC2 + BCG | III | Lung cancer | Completed 3/1998–3/2012 | NCT00003279 [71] | |
| Glycolyl GM3 | |||||
| Racotumomab | I | Pediatric tumors | Completed 2/2011–7/2015 | NCT01598454 [72] | |
| Racotumomab | II | High risk Neuroblastoma | Active, not recruiting 11/2016–9/2022 | NCT02998983 | |
| Racotumomab | II | NSCLC | Completed 9/2009–7/2014 | NCT01240447 | |
| Racotumomab | III | NSCLC | Unknown 9/2010–7/2016 | NCT01460472 | |
| Globo H | |||||
| OPT-822/OPT-821 + cyclophosphamide | II | Metastatic breast cancer | Completed 12/2011–9/2020 | NCT01516307 [73] | |
| OBI 822(adagloxad simolenin)/OBI-821 | III | Triple negative breast cancer | Recruiting 12/2018–12/2027 | NCT03562637 | |
| Tn antigen | |||||
| MAG-Tn3 + AS15 | I | Breast cancer | Active, not recruiting 2/2015–12/2021 | NCT02364492 [74] | |
| STn antigen | |||||
| STn/KLH (THERATOPE®) | III | Metastatic breast cancer | Completed 1/1999–3/2013 | NCT00003638 [75] | |
| STn/KLH (THERATOPE®) | II | Metastatic breast cancer | Completed 8/2002–1/2008 | NCT00046371 [76] | |
| TF antigen | |||||
| TF(c)-KLH + QS21 | 1 | Prostate cancer | Completed 6/1998–3/2013 | NCT00003819 [77] | |
| Lewis | |||||
| sialyl Lewis A-KLH + QS21 | Metastatic breast cancer | Completed 3/2007–1/2020 | NCT00470574 [78] | ||
| Polysialic acid | |||||
| Polysialic acid-KLH + QS21 | II | Small cell lung cancer | Completed 8/1998–6/2013 | NCT00004249 [79] | |
| Polyvalent vaccines | |||||
| Bivalent vaccine (GD2L/GD3L) + OPT-821 | I/II | Neuroblastoma | Active, not recruiting 5/2009–5/2023 | NCT00911560 [67,68] | |
| Trivalent vaccine GM2/GD2L/GD3L-KLH conjugated + OPT-821 | II | Sarcoma | Completed 6/2010–3/2017 | NCT01141491 [68] | |
| Globo-H-GM2-STn-TF-Tn-KLH- QS21 | I | Ovarian cancer | Completed 11/2010–03/2017 | NCT01248273 [80] | |
| Globo-H-GM2-LeY-MUC1-32(aa)-STn(c)-TF(c)-Tn(c)-KLH conjugate vaccine- QS21 | High risk breast cancer | Completed 03/2001–12/2015 | NCT00030823 | ||
| GD2L, GD3L, Globo H, fucosyl GM1, and N-propionylated polysialic acid -KLH + OPT-821 | I | Small cell lung cancer | Completed 05/2011–01/2016 | NCT01349647 |
| Target | Drug Candidate | Phase | Cancer Type | Status/Period | CT ID Refs |
|---|---|---|---|---|---|
| GD2 | |||||
| 1RG-CART + Cyclophosphamide + Fludarabine | I | Relapsed or refractory Neuroblastoma | Completed February 2016–December 2020 | NCT02761915 [313] | |
| 4SCAR-GD2 | I | Neuroblastoma | Suspended January 2016–December 2022 | NCT02765243 [314] | |
| GD2-CART01 | I/II | Neuroblastoma and GD2 positive solid tumors | Recruiting May 2018–December 2027 | NCT03373097 [315] | |
| GD2-CAR NKT cells (GINAKIT) | I | Neuroblastoma | Recruiting 1/2018–8/2034 | NCT03294954 [316,317] | |
| MUC1/MUC1-Tn | |||||
| MUC1-CAR+/PD-1- T cells | I/II | NSCLC | Recruiting 2/2018–1/2022 | NCT03525782 [321] | |
| huMNC2-CAR44 | I | Metastatic breast cancer | Active 1/2020–1/2035 | NCT04020575 [322] | |
| CART-TnMUC1 | I | NSCLC, Ovarian cancer, Fallopian tube cancer, Triple negative breast cancer, Multiple myeloma, Pancreatic ductal adenocarcinoma | Recruiting 10/2019–10/2036 | NCT04025216 | |
| CAR-T cells targeting PSCA, MUC1, TGFβ, HER2, Mesothelin, LeY, GPC3, AXL, EGFR, Claudin18.2, or B7-H3 | I | Lung cancer | Recruiting 7/2017–8/2023 | NCT03198052 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berois, N.; Pittini, A.; Osinaga, E. Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives. Cancers 2022, 14, 645. https://doi.org/10.3390/cancers14030645
Berois N, Pittini A, Osinaga E. Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives. Cancers. 2022; 14(3):645. https://doi.org/10.3390/cancers14030645
Chicago/Turabian StyleBerois, Nora, Alvaro Pittini, and Eduardo Osinaga. 2022. "Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives" Cancers 14, no. 3: 645. https://doi.org/10.3390/cancers14030645
APA StyleBerois, N., Pittini, A., & Osinaga, E. (2022). Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives. Cancers, 14(3), 645. https://doi.org/10.3390/cancers14030645