
Glycans as Targets for Drug Delivery in Cancer


 ,
,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Glycosylation in Cancer
2.1. Glycans with Specific Expression in Cancer
2.1.1. Truncated O-Glycans
2.1.2. Sialylated Glycans
2.2. Glycan-Binding Proteins
3. Specific Targeting of Cancer Cells
3.1. Monoclonal Antibodies
3.2. Antibody-Drug Conjugates
3.3. Nanoparticles: Application of Nanotherapeutics in Cancer Therapy
Decorating Nanoparticles with Monoclonal Antibodies for Selective and Efficient Cancer Cell Targeting
4. Sweetening Precision Oncology with Glycan-Directed Nanoparticles
4.1. Vaccines Encapsulated in Synthetic Glycan-Targeting Nanoparticles with Glycan Targeting
4.2. Nanoparticle Strategies Using Glycoproteins
4.3. Glycan-Targeting Nanoparticles
4.4. Nanoparticles Strategies Using Glycan-Binding Proteins
5. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-P.; Zheng, C.-C.; Huang, Y.-N.; He, M.-L.; Xu, W.W.; Li, B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm 2021, 2, 315–340. [Google Scholar] [CrossRef]
- Eichler, H.G.; Thomson, A.; Eichler, I.; Schneeweiss, S. Assessing the relative efficacy of new drugs: An emerging opportunity. Nat. Rev. Drug Discov. 2015, 14, 443–444. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, S.; Khurana, S.; Choudhari, R.; Kesari, K.K.; Kamal, M.A.; Garg, N.; Ruokolainen, J.; Das, B.C.; Kumar, D. Specific targeting cancer cells with nanoparticles and drug delivery in cancer therapy. Semin. Cancer Biol. 2021, 69, 166–177. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Smith, B.A.H.; Bertozzi, C.R. The clinical impact of glycobiology: Targeting selectins, Siglecs and mammalian glycans. Nat. Rev. Drug Discov. 2021, 20, 217–243. [Google Scholar] [CrossRef]
- Colley, K.J.; Varki, A.; Kinoshita, T. Cellular Organization of Glycosylation. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 41–49. [Google Scholar]
- Stanley, P.; Taniguchi, N.; Aebi, M. N-Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 99–111. [Google Scholar]
- Brockhausen, I.; Stanley, P. O-GalNAc Glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 113–123. [Google Scholar]
- Schnaar, R.L.; Kinoshita, T. Glycosphingolipids. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 125–135. [Google Scholar]
- Lindahl, U.; Couchman, J.; Kimata, K.; Esko, J.D. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 207–221. [Google Scholar]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: Mechanisms and clinical implications. Nat. Rev. Cancer 2015, 15, 540–555. [Google Scholar] [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 729–749. [Google Scholar] [CrossRef]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol. 2015, 10, 473–510. [Google Scholar] [CrossRef] [Green Version]
- Britain, C.M.; Holdbrooks, A.T.; Anderson, J.C.; Willey, C.D.; Bellis, S.L. Sialylation of EGFR by the ST6Gal-I sialyltransferase promotes EGFR activation and resistance to gefitinib-mediated cell death. J. Ovarian Res. 2018, 11, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mereiter, S.; Magalhães, A.; Adamczyk, B.; Jin, C.; Almeida, A.; Drici, L.; Ibáñez-Vea, M.; Gomes, C.; Ferreira, J.A.; Afonso, L.P.; et al. Glycomic analysis of gastric carcinoma cells discloses glycans as modulators of RON receptor tyrosine kinase activation in cancer. Biochim. Biophys. Acta 2016, 1860, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Duarte, H.O.; Rodrigues, J.G.; Gomes, C.; Hensbergen, P.J.; Ederveen, A.L.H.; de Ru, A.H.; Mereiter, S.; Polonia, A.; Fernandes, E.; Ferreira, J.A.; et al. ST6Gal1 targets the ectodomain of ErbB2 in a site-specific manner and regulates gastric cancer cell sensitivity to trastuzumab. Oncogene 2021, 40, 3719–3733. [Google Scholar] [CrossRef]
- Rodrigues, J.G.; Duarte, H.O.; Gomes, C.; Balmaña, M.; Martins Á, M.; Hensbergen, P.J.; de Ru, A.H.; Lima, J.; Albergaria, A.; van Veelen, P.A.; et al. Terminal α2,6-sialylation of epidermal growth factor receptor modulates antibody therapy response of colorectal cancer cells. Cell Oncol. 2021, 44, 835–850. [Google Scholar] [CrossRef]
- Gomes, C.; Osorio, H.; Pinto, M.T.; Campos, D.; Oliveira, M.J.; Reis, C.A. Expression of ST3GAL4 leads to SLe(x) expression and induces c-Met activation and an invasive phenotype in gastric carcinoma cells. PLoS ONE 2013, 8, e66737. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, T.; Aryal, R.P.; Kudelka, M.R.; Wang, Y.; Cummings, R.D. The Cosmc connection to the Tn antigen in cancer. Cancer Biomark 2014, 14, 63–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewell, R.; Backstrom, M.; Dalziel, M.; Gschmeissner, S.; Karlsson, H.; Noll, T.; Gatgens, J.; Clausen, H.; Hansson, G.C.; Burchell, J.; et al. The ST6GalNAc-I sialyltransferase localizes throughout the Golgi and is responsible for the synthesis of the tumor-associated sialyl-Tn O-glycan in human breast cancer. J. Biol. Chem. 2006, 281, 3586–3594. [Google Scholar] [CrossRef] [Green Version]
- Marcos, N.T.; Bennett, E.P.; Gomes, J.; Magalhaes, A.; Gomes, C.; David, L.; Dar, I.; Jeanneau, C.; DeFrees, S.; Krustrup, D.; et al. ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front. Biosci. 2011, 3, 1443–1455. [Google Scholar] [CrossRef]
- Julien, S.; Videira, P.A.; Delannoy, P. Sialyl-tn in cancer: (how) did we miss the target? Biomolecules 2012, 2, 435–466. [Google Scholar] [CrossRef] [Green Version]
- Munkley, J. The Role of Sialyl-Tn in Cancer. Int. J. Mol. Sci. 2016, 17, 275. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, P.; Dabelsteen, S.; Madsen, F.B.; Francavilla, C.; Kopp, K.L.; Steentoft, C.; Vakhrushev, S.Y.; Olsen, J.V.; Hansen, L.; Bennett, E.P.; et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc. Natl. Acad. Sci. USA 2014, 111, E4066–E4075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.; Sagar, S.; Caffrey, T.; Grandgenett, P.M.; Radhakrishnan, P. Truncated O-glycans promote epithelial-to-mesenchymal transition and stemness properties of pancreatic cancer cells. J. Cell Mol. Med. 2019, 23, 6885–6896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, D.; Campos, D.; Gomes, J.; Pinto, F.; Macedo, J.A.; Matos, R.; Mereiter, S.; Pinto, M.T.; Polonia, A.; Gartner, F.; et al. O-glycans truncation modulates gastric cancer cell signaling and transcription leading to a more aggressive phenotype. EBioMedicine 2019, 40, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Miles, D.W.; Happerfield, L.C.; Smith, P.; Gillibrand, R.; Bobrow, L.G.; Gregory, W.M.; Rubens, R.D. Expression of sialyl-Tn predicts the effect of adjuvant chemotherapy in node-positive breast cancer. Br. J. Cancer 1994, 70, 1272–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mereiter, S.; Martins Á, M.; Gomes, C.; Balmaña, M.; Macedo, J.A.; Polom, K.; Roviello, F.; Magalhães, A.; Reis, C.A. O-glycan truncation enhances cancer-related functions of CD44 in gastric cancer. FEBS Lett. 2019, 593, 1675–1689. [Google Scholar] [CrossRef]
- Pinho, S.; Marcos, N.T.; Ferreira, B.; Carvalho, A.S.; Oliveira, M.J.; Santos-Silva, F.; Harduin-Lepers, A.; Reis, C.A. Biological significance of cancer-associated sialyl-Tn antigen: Modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett. 2007, 249, 157–170. [Google Scholar] [CrossRef]
- D’Addio, M.; Frey, J.; Otto, V.I. The manifold roles of sialic acid for the biological functions of endothelial glycoproteins. Glycobiology 2020, 30, 490–499. [Google Scholar] [CrossRef] [Green Version]
- McEver, R.P. Role of selectins in leukocyte adhesion to platelets and endothelium. Ann. N. Y. Acad. Sci. 1994, 714, 185–189. [Google Scholar] [CrossRef]
- McEver, R.P. Selectins: Initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc. Res. 2015, 107, 331–339. [Google Scholar] [CrossRef] [Green Version]
- Dall’Olio, F.; Pucci, M.; Malagolini, N. The Cancer-Associated Antigens Sialyl Lewis(a/x) and Sd(a): Two Opposite Faces of Terminal Glycosylation. Cancers 2021, 13, 5273. [Google Scholar] [CrossRef]
- Jin, F.; Wang, F. The physiological and pathological roles and applications of sialyl Lewis x, a common carbohydrate ligand of the three selectins. Glycoconj J. 2020, 37, 277–291. [Google Scholar] [CrossRef]
- Borsig, L. Selectins in cancer immunity. Glycobiology 2018, 28, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, C.; Almeida, A.; Barreira, A.; Calheiros, J.; Pinto, F.; Abrantes, R.; Costa, A.; Polonia, A.; Campos, D.; Osorio, H.; et al. Carcinoembryonic antigen carrying SLe(X) as a new biomarker of more aggressive gastric carcinomas. Theranostics 2019, 9, 7431–7446. [Google Scholar] [CrossRef] [PubMed]
- Samraj, A.N.; Läubli, H.; Varki, N.; Varki, A. Involvement of a non-human sialic Acid in human cancer. Front. Oncol. 2014, 4, 33. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Lin, S.L.; Chang, T.; Wu, S.H.; Yao, C.W.; Chu, T.Y.; Troy, F.A., 2nd; Inoue, Y. Identification of free deaminated sialic acid (2-keto-3-deoxy-D-glycero-D-galacto-nononic acid) in human red blood cells and its elevated expression in fetal cord red blood cells and ovarian cancer cells. J. Biol. Chem. 1998, 273, 27199–27204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Xie, B.; Wang, B.; Troy, F.A., II. LC–MS/MS glycomic analyses of free and conjugated forms of the sialic acids, Neu5Ac, Neu5Gc and KDN in human throat cancers. Glycobiology 2015, 25, 1362–1374. [Google Scholar] [CrossRef] [Green Version]
- Macauley, M.S.; Crocker, P.R.; Paulson, J.C. Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol. 2014, 14, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, R.D.; McEver, R.P. C-Type Lectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; pp. 435–452. [Google Scholar]
- Läubli, H.; Varki, A. Sialic acid-binding immunoglobulin-like lectins (Siglecs) detect self-associated molecular patterns to regulate immune responses. Cell Mol. Life Sci. 2020, 77, 593–605. [Google Scholar] [CrossRef]
- Lubbers, J.; Rodriguez, E.; van Kooyk, Y. Modulation of Immune Tolerance via Siglec-Sialic Acid Interactions. Front. Immunol. 2018, 9, 2807. [Google Scholar] [CrossRef] [Green Version]
- RodrÍguez, E.; Schetters, S.T.T.; van Kooyk, Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat. Rev. Immunol. 2018, 18, 204–211. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Schwaber, J.; Cohen, E.P. Human x mouse somatic cell hybrid clone secreting immunoglobulins of both parental types. Nature 1973, 244, 444–447. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Mancini, N.; Carletti, S.; Perotti, M.; Canducci, F.; Mammarella, M.; Sampaolo, M.; Burioni, R. Phage display for the production of human monoclonal antibodies against human pathogens. New MicroBiol. 2004, 27, 315–328. [Google Scholar]
- Thie, H.; Meyer, T.; Schirrmann, T.; Hust, M.; Dübel, S. Phage display derived therapeutic antibodies. Curr. Pharm. Biotechnol. 2008, 9, 439–446. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Catenacci, D.V.T.; Chung, H.C.; Shen, L.; Moehler, M.; Yoon, H.H.; Rosales, M.K.; Kang, Y.K. Safety and efficacy of HER2 blockade by trastuzumab-based chemotherapy-containing combination strategies in HER2+ gastroesophageal adenocarcinoma. ESMO Open 2021, 7, 100360. [Google Scholar] [CrossRef]
- Klinger, M.; Farhan, H.; Just, H.; Drobny, H.; Himmler, G.; Loibner, H.; Mudde, G.C.; Freissmuth, M.; Sexl, V. Antibodies directed against Lewis-Y antigen inhibit signaling of Lewis-Y modified ErbB receptors. Cancer Res. 2004, 64, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Beatson, R.; Tajadura-Ortega, V.; Achkova, D.; Picco, G.; Tsourouktsoglou, T.D.; Klausing, S.; Hillier, M.; Maher, J.; Noll, T.; Crocker, P.R.; et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat. Immunol. 2016, 17, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Sagar, S.; Liu, X.; Lee, H.R.; Grunkemeyer, J.A.; Grandgenett, P.M.; Caffrey, T.; O’Connell, K.A.; Swanson, B.; Marcos-Silva, L.; et al. Isoforms of MUC16 activate oncogenic signaling through EGF receptors to enhance the progression of pancreatic cancer. Mol. Ther. 2021, 29, 1557–1571. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer Res. 2021, 27, 2179–2189. [Google Scholar] [CrossRef] [PubMed]
- Aldeghaither, D.S.; Zahavi, D.J.; Murray, J.C.; Fertig, E.J.; Graham, G.T.; Zhang, Y.W.; O’Connell, A.; Ma, J.; Jablonski, S.A.; Weiner, L.M. A Mechanism of Resistance to Antibody-Targeted Immune Attack. Cancer Immunol. Res. 2019, 7, 230–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrantonio, F.; Fucà, G.; Morano, F.; Gloghini, A.; Corso, S.; Aprile, G.; Perrone, F.; De Vita, F.; Tamborini, E.; Tomasello, G.; et al. Biomarkers of Primary Resistance to Trastuzumab in HER2-Positive Metastatic Gastric Cancer Patients: The AMNESIA Case-Control Study. Clin. Cancer Res. 2018, 24, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishima, Y.; Terui, Y.; Takeuchi, K.; Matsumoto-Mishima, Y.; Matsusaka, S.; Utsubo-Kuniyoshi, R.; Hatake, K. The identification of irreversible rituximab-resistant lymphoma caused by CD20 gene mutations. Blood Cancer J. 2011, 1, e15. [Google Scholar] [CrossRef] [Green Version]
- Sickmier, E.A.; Kurzeja, R.J.; Michelsen, K.; Vazir, M.; Yang, E.; Tasker, A.S. The Panitumumab EGFR Complex Reveals a Binding Mechanism That Overcomes Cetuximab Induced Resistance. PLoS ONE 2016, 11, e0163366. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, X.Y.; Koh, X.H.; Hwang, L.A.; Ferrer, F.J.; Rahmat, S.A.B.; Lama, D.; Lane, D.P. Therapeutic anti-cancer activity of antibodies targeting sulfhydryl bond constrained epitopes on unglycosylated RON receptor tyrosine kinase. Oncogene 2019, 38, 7342–7356. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Lee, H.-H.; Hsu, J.L.; Yu, D.; Hung, M.-C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef]
- Lee, H.H.; Wang, Y.N.; Xia, W.; Chen, C.H.; Rau, K.M.; Ye, L.; Wei, Y.; Chou, C.K.; Wang, S.C.; Yan, M.; et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell 2019, 36, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H. Characterization of glycosylation in monoclonal antibodies and its importance in therapeutic antibody development. Crit. Rev. Biotechnol. 2021, 41, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Meng, Y.G.; Weikert, S.H.; Presta, L.G. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, C.; Stuart, F.; Sondermann, P.; Brünker, P.; Umaña, P. The carbohydrate at FcgammaRIIIa Asn-162. An element required for high affinity binding to non-fucosylated IgG glycoforms. J. Biol. Chem. 2006, 281, 5032–5036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scallon, B.J.; Tam, S.H.; McCarthy, S.G.; Cai, A.N.; Raju, T.S. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol. Immunol. 2007, 44, 1524–1534. [Google Scholar] [CrossRef]
- Hodoniczky, J.; Zheng, Y.Z.; James, D.C. Control of recombinant monoclonal antibody effector functions by Fc N-glycan remodeling in vitro. Biotechnol. Prog. 2005, 21, 1644–1652. [Google Scholar] [CrossRef]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Elshiaty, M.; Schindler, H.; Christopoulos, P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int. J. Mol. Sci. 2021, 22, 5632. [Google Scholar] [CrossRef]
- Lutterbuese, R.; Raum, T.; Kischel, R.; Hoffmann, P.; Mangold, S.; Rattel, B.; Friedrich, M.; Thomas, O.; Lorenczewski, G.; Rau, D.; et al. T cell-engaging BiTE antibodies specific for EGFR potently eliminate KRAS- and BRAF-mutated colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12605–12610. [Google Scholar] [CrossRef] [Green Version]
- Huhalov, A.; Adams, S.; Paragas, V.; Oyama, S.; Overland, R.; Luus, L.; Gibbons, F.; Zhang, B.; Nguyen, S.; Nielsen, U.B.; et al. Abstract 3485: MM-111, an ErbB2/ErbB3 bispecific antibody with potent activity in ErbB2-overexpressing cells, positively combines with trastuzumab to inhibit growth of breast cancer cells driven by the ErbB2/ErbB3 oncogenic unit. Cancer Res. 2010, 70, 3485. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Boni, V.; Sharma, M.R.; Patnaik, A. The Resurgence of Antibody Drug Conjugates in Cancer Therapeutics: Novel Targets and Payloads. Am. Soc. Clin. Oncol. Educ Book 2020, 40, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Goldenson, B.H.; Goodman, A.M.; Ball, E.D. Gemtuzumab ozogamicin for the treatment of acute myeloid leukemia in adults. Expert Opin. Biol. Ther. 2021, 21, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Feldman, T.; Zou, D.; Rebeira, M.; Lee, J.; Fanale, M.; Manley, T.; Rao, S.; Feliciano, J.; Harris, M.; Kansal, A. Cost-effectiveness of brentuximab vedotin with chemotherapy in treatment of CD30-expressing PTCL. Am. J. Manag. Care 2020, 26, e41–e49. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Woods, E.C.; Vukojicic, P.; Bertozzi, C.R. Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2016, 113, 10304–10309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Wang, K.; Oppong-Gyebi, A.; Hu, J. Application of Nanotechnology in Cancer Diagnosis and Therapy—A Mini-Review. Int. J. Med. Sci. 2020, 17, 2964–2973. [Google Scholar] [CrossRef]
- Ali, E.S.; Sharker, S.M.; Islam, M.T.; Khan, I.N.; Shaw, S.; Rahman, M.A.; Uddin, S.J.; Shill, M.C.; Rehman, S.; Das, N.; et al. Targeting cancer cells with nanotherapeutics and nanodiagnostics: Current status and future perspectives. Semin. Cancer Biol. 2021, 69, 52–68. [Google Scholar] [CrossRef]
- Marques, A.C.; Costa, P.J.; Velho, S.; Amaral, M.H. Functionalizing nanoparticles with cancer-targeting antibodies: A comparison of strategies. J. Control. Release 2020, 320, 180–200. [Google Scholar] [CrossRef]
- Parakh, S.; King, D.; Gan, H.K.; Scott, A.M. Current Development of Monoclonal Antibodies in Cancer Therapy. Recent Results Cancer Res. 2020, 214, 1–70. [Google Scholar] [CrossRef]
- Khan, H.; Mirzaei, H.R.; Amiri, A.; Kupeli Akkol, E.; Ashhad Halimi, S.M.; Mirzaei, H. Glyco-nanoparticles: New drug delivery systems in cancer therapy. Semin. Cancer Biol. 2021, 69, 24–42. [Google Scholar] [CrossRef]
- Arslan, F.B.; Ozturk Atar, K.; Calis, S. Antibody-mediated drug delivery. Int. J. Pharm. 2021, 596, 120268. [Google Scholar] [CrossRef]
- Richards, D.A.; Maruani, A.; Chudasama, V. Antibody fragments as nanoparticle targeting ligands: A step in the right direction. Chem. Sci. 2017, 8, 63–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, F.; Rafael, D.; Vilar-Hernández, M.; Montero, S.; Martínez-Trucharte, F.; Seras-Franzoso, J.; Díaz-Riascos, Z.V.; Boullosa, A.; García-Aranda, N.; Cámara-Sánchez, P.; et al. Polymeric micelles targeted against CD44v6 receptor increase niclosamide efficacy against colorectal cancer stem cells and reduce circulating tumor cells in vivo. J. Control. Release 2021, 331, 198–212. [Google Scholar] [CrossRef]
- Baião, A.; Sousa, F.; Oliveira, A.V.; Oliveira, C.; Sarmento, B. Effective intracellular delivery of bevacizumab via PEGylated polymeric nanoparticles targeting the CD44v6 receptor in colon cancer cells. Biomater Sci. 2020, 8, 3720–3729. [Google Scholar] [CrossRef] [PubMed]
- Torres-Pérez, S.A.; Torres-Pérez, C.E.; Pedraza-Escalona, M.; Pérez-Tapia, S.M.; Ramón-Gallegos, E. Glycosylated Nanoparticles for Cancer-Targeted Drug Delivery. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Mateu Ferrando, R.; Lay, L.; Polito, L. Gold nanoparticle-based platforms for vaccine development. Drug Discov. Today Technol. 2020, 38, 57–67. [Google Scholar] [CrossRef]
- Anderluh, M.; Berti, F.; Bzducha-Wróbel, A.; Chiodo, F.; Colombo, C.; Compostella, F.; Durlik, K.; Ferhati, X.; Holmdahl, R.; Jovanovic, D.; et al. Recent advances on smart glycoconjugate vaccines in infections and cancer. FEBS J. 2021. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Yu, F.; Li, M.; Zhu, H.; Wang, K.; Meng, M.; Zhao, W. The Adjuvant of α-Galactosylceramide Presented by Gold Nanoparticles Enhances Antitumor Immune Responses of MUC1 Antigen-Based Tumor Vaccines. Int. J. Nanomed. 2021, 16, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Degliangeli, F.; Palitzsch, B.; Gerlitzki, B.; Kunz, H.; Schmitt, E.; Fiammengo, R.; Westerlind, U. Glycopeptide-functionalized gold nanoparticles for antibody induction against the tumor associated mucin-1 glycoprotein. Bioorg. Med. Chem. 2016, 24, 1132–1135. [Google Scholar] [CrossRef]
- Mocan, T.; Matea, C.; Tabaran, F.; Iancu, C.; Orasan, R.; Mocan, L. In Vitro Administration of Gold Nanoparticles Functionalized with MUC-1 Protein Fragment Generates Anticancer Vaccine Response via Macrophage Activation and Polarization Mechanism. J. Cancer 2015, 6, 583–592. [Google Scholar] [CrossRef]
- Parry, A.L.; Clemson, N.A.; Ellis, J.; Bernhard, S.S.; Davis, B.G.; Cameron, N.R. ‘Multicopy multivalent’ glycopolymer-stabilized gold nanoparticles as potential synthetic cancer vaccines. J. Am. Chem. Soc. 2013, 135, 9362–9365. [Google Scholar] [CrossRef] [Green Version]
- Trabbic, K.R.; Kleski, K.A.; Barchi, J.J., Jr. A Stable Gold Nanoparticle-Based Vaccine for the Targeted Delivery of Tumor-Associated Glycopeptide Antigens. ACS Bio Med. Chem. Au 2021, 1, 31–43. [Google Scholar] [CrossRef]
- Hamdy, S.; Haddadi, A.; Shayeganpour, A.; Samuel, J.; Lavasanifar, A. Activation of antigen-specific T cell-responses by mannan-decorated PLGA nanoparticles. Pharm. Res. 2011, 28, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yang, Y.; Dong, W.; Gao, Y.; Meng, Y.; Wang, H.; Li, L.; Jin, J.; Ji, M.; Xia, X.; et al. Drug-free mannosylated liposomes inhibit tumor growth by promoting the polarization of tumor-associated macrophages. Int. J. Nanomed. 2019, 14, 3203–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Hu, Y.; Duan, J.; Yuan, W.; Wang, C.; Xu, H.; Yang, X.D. Novel aptamer-nanoparticle bioconjugates enhances delivery of anticancer drug to MUC1-positive cancer cells in vitro. PLoS ONE 2011, 6, e24077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayari, E.; Dinarvand, M.; Amini, M.; Azhdarzadeh, M.; Mollarazi, E.; Ghasemi, Z.; Atyabi, F. MUC1 aptamer conjugated to chitosan nanoparticles, an efficient targeted carrier designed for anticancer SN38 delivery. Int. J. Pharm. 2014, 473, 304–315. [Google Scholar] [CrossRef]
- Hanafi-Bojd, M.Y.; Moosavian Kalat, S.A.; Taghdisi, S.M.; Ansari, L.; Abnous, K.; Malaekeh-Nikouei, B. MUC1 aptamer-conjugated mesoporous silica nanoparticles effectively target breast cancer cells. Drug Dev. Ind. Pharm. 2018, 44, 13–18. [Google Scholar] [CrossRef]
- Perepelyuk, M.; Sacko, K.; Thangavel, K.; Shoyele, S.A. Evaluation of MUC1-Aptamer Functionalized Hybrid Nanoparticles for Targeted Delivery of miRNA-29b to Nonsmall Cell Lung Cancer. Mol. Pharm. 2018, 15, 985–993. [Google Scholar] [CrossRef]
- Bashir, A.; Yang, Q.; Wang, J.; Hoyer, S.; Chou, W.; McLean, C.; Davis, G.; Gong, Q.; Armstrong, Z.; Jang, J.; et al. Machine learning guided aptamer refinement and discovery. Nat. Commun. 2021, 12, 2366. [Google Scholar] [CrossRef]
- Jafari, R.; Majidi Zolbanin, N.; Majidi, J.; Atyabi, F.; Yousefi, M.; Jadidi-Niaragh, F.; Aghebati-Maleki, L.; Shanehbandi, D.; Soltani Zangbar, M.S.; Rafatpanah, H. Anti-Mucin1 Aptamer-Conjugated Chitosan Nanoparticles for Targeted Co-Delivery of Docetaxel and IGF-1R siRNA to SKBR3 Metastatic Breast Cancer Cells. Iran. Biomed. J. 2019, 23, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Skandalis, S.S.; Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Advances and advantages of nanomedicine in the pharmacological targeting of hyaluronan-CD44 interactions and signaling in cancer. Adv. Cancer Res. 2014, 123, 277–317. [Google Scholar] [CrossRef]
- Ahir, M.; Upadhyay, P.; Ghosh, A.; Sarker, S.; Bhattacharya, S.; Gupta, P.; Ghosh, S.; Chattopadhyay, S.; Adhikary, A. Delivery of dual miRNA through CD44-targeted mesoporous silica nanoparticles for enhanced and effective triple-negative breast cancer therapy. Biomater. Sci. 2020, 8, 2939–2954. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Wang, Y.; Wang, L.; Liang, Z.; Li, D.; Xu, X.; Chen, Y.; Yang, X.; Zhang, H.; Niu, H. Self-crosslinkable chitosan-hyaluronic acid dialdehyde nanoparticles for CD44-targeted siRNA delivery to treat bladder cancer. Bioact. Mater. 2021, 6, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.J.; Sousa, F.; Ferreira, D.; Pereira, C.; Nestor, M.; Oliveira, C.; Granja, P.L.; Sarmento, B. Fab-conjugated PLGA nanoparticles effectively target cancer cells expressing human CD44v6. Acta Biomater. 2018, 81, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Correa, T.D.S.; Bocca, A.L.; Figueiredo, F.; Lima, E.C.O.; Almeida Santos, M.F.M.; Lacava, Z.G.M.; Campos-da-Paz, M. Anti-CEA tagged iron nanoparticles for targeting triple-negative breast cancer. Biomed. Mater. 2021. [Google Scholar] [CrossRef]
- Pereira, I.; Sousa, F.; Kennedy, P.; Sarmento, B. Carcinoembryonic antigen-targeted nanoparticles potentiate the delivery of anticancer drugs to colorectal cancer cells. Int. J. Pharm. 2018, 549, 397–403. [Google Scholar] [CrossRef]
- Fernandes, E.; Ferreira, D.; Peixoto, A.; Freitas, R.; Relvas-Santos, M.; Palmeira, C.; Martins, G.; Barros, A.; Santos, L.L.; Sarmento, B.; et al. Glycoengineered nanoparticles enhance the delivery of 5-fluoroucil and paclitaxel to gastric cancer cells of high metastatic potential. Int. J. Pharm. 2019, 570, 118646. [Google Scholar] [CrossRef]
- Palma-Chavez, J.A.; Fuentes, K.; Applegate, B.E.; Jo, J.A.; Charoenphol, P. Development and Characterization of PLGA-Based Multistage Delivery System for Enhanced Payload Delivery to Targeted Vascular Endothelium. Macromol. Biosci. 2021, 21, e2000377. [Google Scholar] [CrossRef]
- Alhallak, K.; Sun, J.; Muz, B.; Jeske, A.; Yavner, J.; Bash, H.; Park, C.; Lubben, B.; Adebayo, O.; Achilefu, S.; et al. Nanoparticle T cell engagers for the treatment of acute myeloid leukemia. Oncotarget 2021, 12, 1878–1885. [Google Scholar] [CrossRef]
- Niu, F.; Yan, J.; Ma, B.; Li, S.; Shao, Y.; He, P.; Zhang, W.; He, W.; Ma, P.X.; Lu, W. Lanthanide-doped nanoparticles conjugated with an anti-CD33 antibody and a p53-activating peptide for acute myeloid leukemia therapy. Biomaterials 2018, 167, 132–142. [Google Scholar] [CrossRef]
- Hansel, T.T.; Kropshofer, H.; Singer, T.; Mitchell, J.A.; George, A.J. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 2010, 9, 325–338. [Google Scholar] [CrossRef]
- Li, H.; Xu, S.; Quan, J.; Yung, B.C.; Pang, J.; Zhou, C.; Cho, Y.A.; Zhang, M.; Liu, S.; Muthusamy, N.; et al. CD33-Targeted Lipid Nanoparticles (aCD33LNs) for Therapeutic Delivery of GTI-2040 to Acute Myelogenous Leukemia. Mol. Pharm. 2015, 12, 2010–2018. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.C.; Kawasaki, N.; Nycholat, C.M.; Han, S.; Pilotte, J.; Crocker, P.R.; Paulson, J.C. Antigen delivery to macrophages using liposomal nanoparticles targeting sialoadhesin/CD169. PLoS ONE 2012, 7, e39039. [Google Scholar] [CrossRef]
- Martínez-Carmona, M.; Lozano, D.; Colilla, M.; Vallet-Regí, M. Lectin-conjugated pH-responsive mesoporous silica nanoparticles for targeted bone cancer treatment. Acta Biomater. 2018, 65, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Huang, J.; Chen, J.; Yang, M.; Wang, H.; Qiao, H.; Chen, Z.; Hu, L.; Di, L.; Li, J. Enhanced anti-colon cancer efficacy of 5-fluorouracil by epigallocatechin-3- gallate co-loaded in wheat germ agglutinin-conjugated nanoparticles. Nanomedicine 2019, 21, 102068. [Google Scholar] [CrossRef]
- Bhat, R.; García, I.; Aznar, E.; Arnaiz, B.; Martínez-Bisbal, M.C.; Liz-Marzán, L.M.; Penadés, S.; Martínez-Máñez, R. Lectin-gated and glycan functionalized mesoporous silica nanocontainers for targeting cancer cells overexpressing Lewis X antigen. Nanoscale 2017, 10, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Giovampaola, C.; Capone, A.; Ermini, L.; Lupetti, P.; Vannuccini, E.; Finetti, F.; Donnini, S.; Ziche, M.; Magnani, A.; Leone, G.; et al. Formulation of liposomes functionalized with Lotus lectin and effective in targeting highly proliferative cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 860–870. [Google Scholar] [CrossRef]
- Dutta Chowdhury, A.; Ganganboina, A.B.; Tsai, Y.-c.; Chiu, H.-c.; Doong, R.-a. Multifunctional GQDs-Concanavalin A@Fe3O4 nanocomposites for cancer cells detection and targeted drug delivery. Anal. Chim. Acta 2018, 1027, 109–120. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
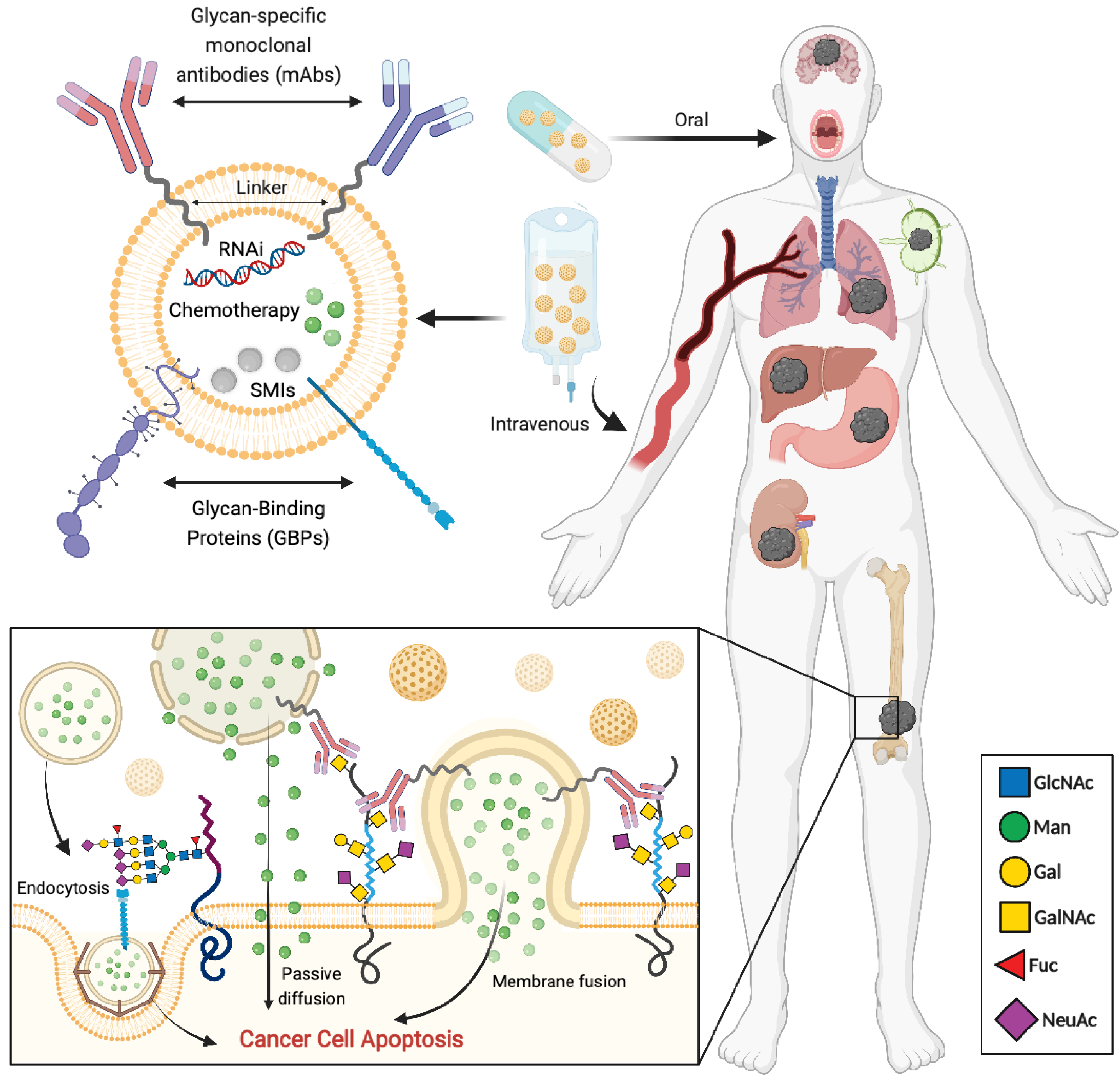{kind=link}
| ADC Formulation | Target Antigen | Antibody | Approved Clinical Application 1 | Year of Approval | Approving Regulatory Entity |
|---|---|---|---|---|---|
| Brentuximab-vedotin (SGN-35, Adcetris) | CD30 | Chimeric IgG1 | Hodgkin’s lymphoma, ALCL, PTCL, MF | 2011/2012 | FDA and EMA |
| Trastuzumab-emtansine (T-DM1, Kadcyla) | ErbB2 | Humanized IgG1 | ErbB2-positive metastatic breast cancer | 2013 | FDA and EMA |
| Inotuzumab-ozogamicin (Besponsa) | CD22 | Recombinant humanized IgG4 | B cell precursor ALL | 2017 | FDA and EMA |
| Gemtuzumab-ozogamicin (Mylotarg) 2 | CD33 | Humanized IgG4 | CD33-positive AML | 2017/2018 | FDA and EMA |
| Polatuzumab-vedotin (Polivy) | CD79 | Humanized IgG1 | DLBCL | 2019/2020 | FDA and EMA |
| Enfortumab-vedotin (ASG-22ME, Padcev) | Nectin-4 | Human IgG1 | Advanced urothelial cancer | 2019 | FDA |
| Trastuzumab-deruxtecan (DS-8201a, Enhertu) | ErbB2 | Humanized IgG1 | Metastatic ErbB2-positive breast cancer | 2019 | FDA |
| Sacituzumab-govitecan (IMMU-132, Trodelvy) | TROP2 | Humanized IgG1 | Triple-negative breast cancer | 2020 | FDA |
| Belantamab mafodotin (GSK2857916, Blenrep) | BCMA | Humanized IgG1 | Relapsed or refractory multiple myeloma | 2020 (orphan drug since 2017 by the EMA) | FDA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diniz, F.; Coelho, P.; Duarte, H.O.; Sarmento, B.; Reis, C.A.; Gomes, J. Glycans as Targets for Drug Delivery in Cancer. Cancers 2022, 14, 911. https://doi.org/10.3390/cancers14040911
Diniz F, Coelho P, Duarte HO, Sarmento B, Reis CA, Gomes J. Glycans as Targets for Drug Delivery in Cancer. Cancers. 2022; 14(4):911. https://doi.org/10.3390/cancers14040911
Chicago/Turabian StyleDiniz, Francisca, Pedro Coelho, Henrique O. Duarte, Bruno Sarmento, Celso A. Reis, and Joana Gomes. 2022. "Glycans as Targets for Drug Delivery in Cancer" Cancers 14, no. 4: 911. https://doi.org/10.3390/cancers14040911
APA StyleDiniz, F., Coelho, P., Duarte, H. O., Sarmento, B., Reis, C. A., & Gomes, J. (2022). Glycans as Targets for Drug Delivery in Cancer. Cancers, 14(4), 911. https://doi.org/10.3390/cancers14040911