
Rescuing SLAMF3 Expression Restores Sorafenib Response in Hepatocellular Carcinoma Cells through the Induction of Mesenchymal-to-Epithelial Transition

 ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Isolation and Culture of Mesenchymal Stem Cells
2.3. Establishment of Sorafenib-Resistant Cells
2.4. Cell Transfection and Plasmid Construction
2.5. General Experimental Design
2.6. Statistical Analysis
3. Results
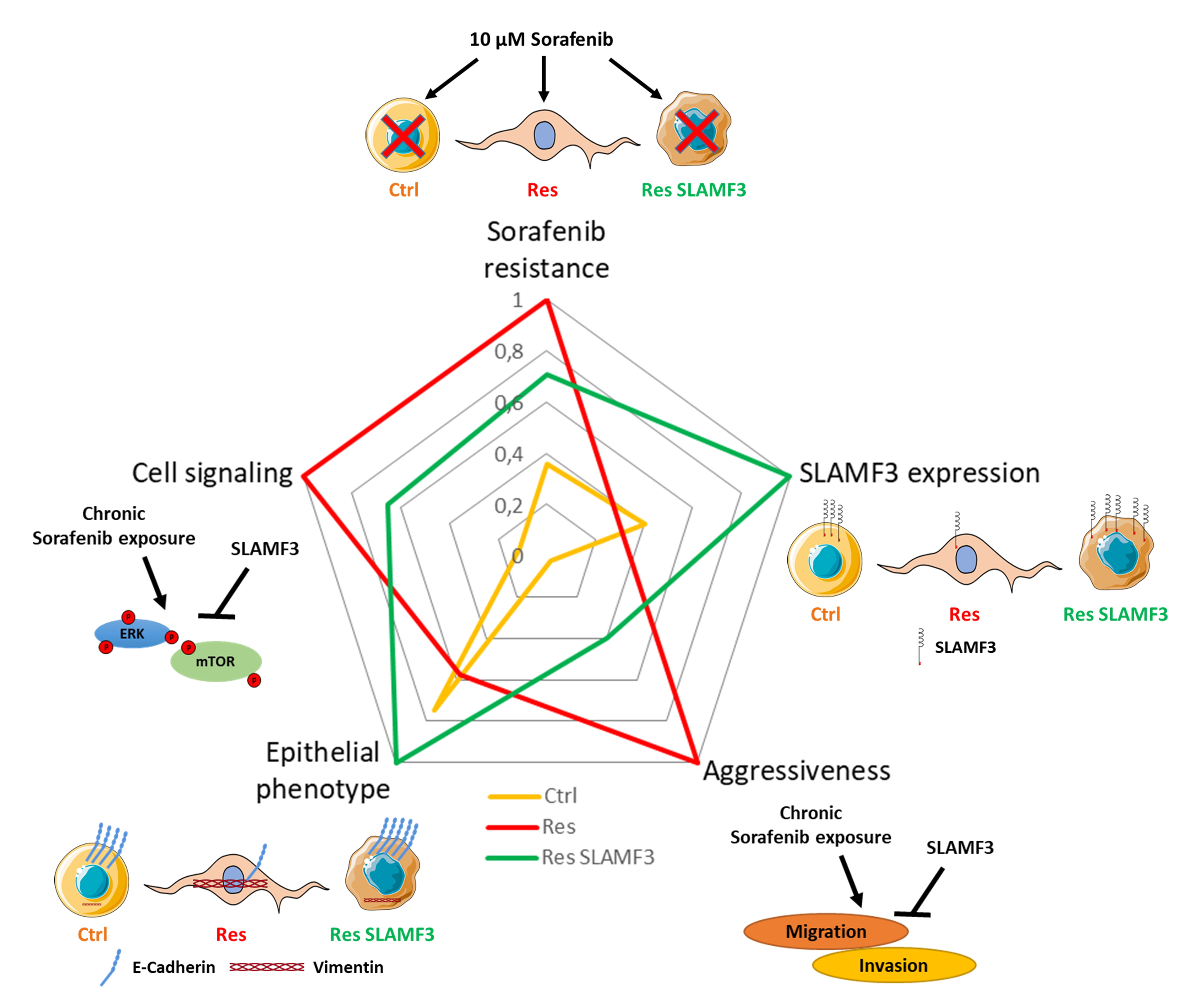
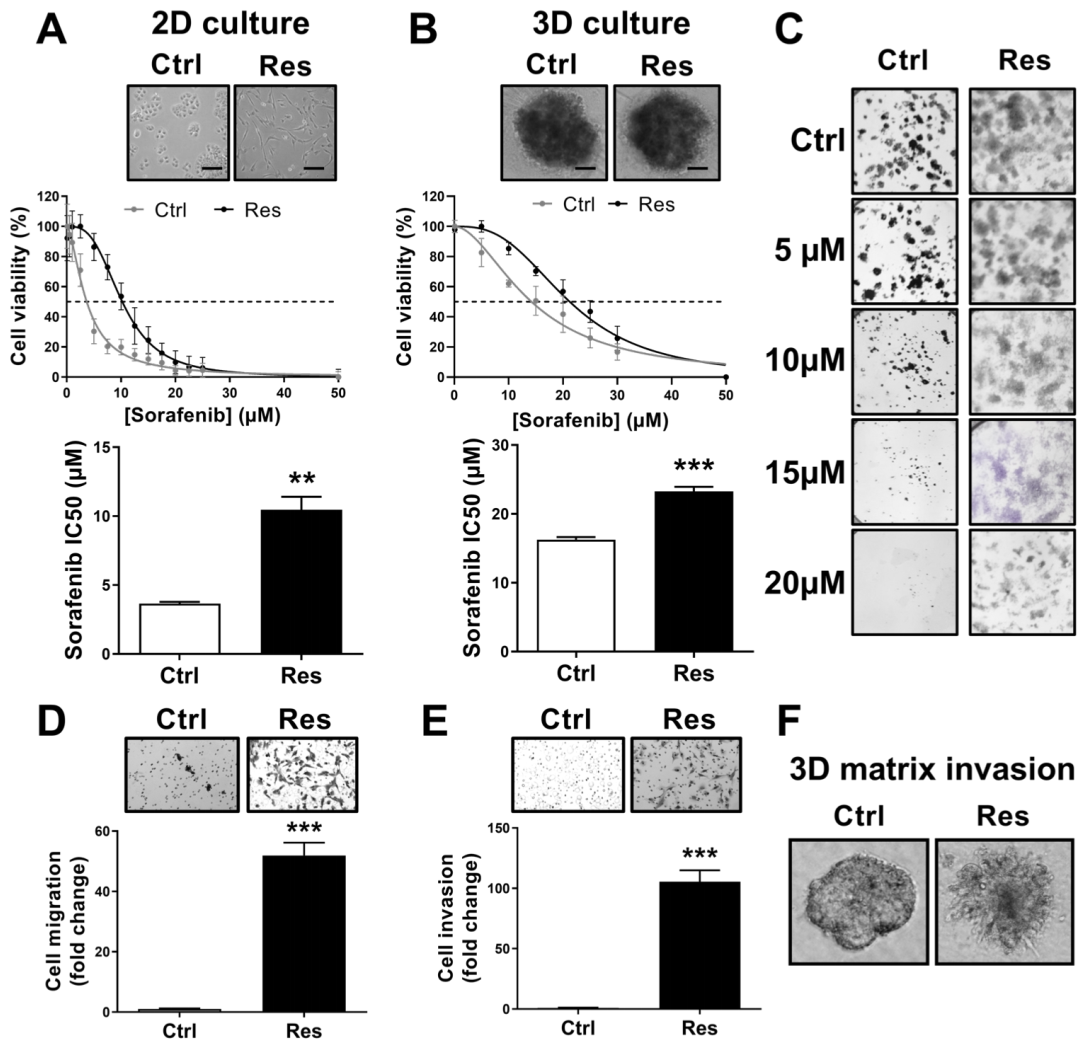
3.1. Long-Term Exposure to Increased Concentrations of Sorafenib Induces Resistance and Aggressive Phenotypes in HCC Cells
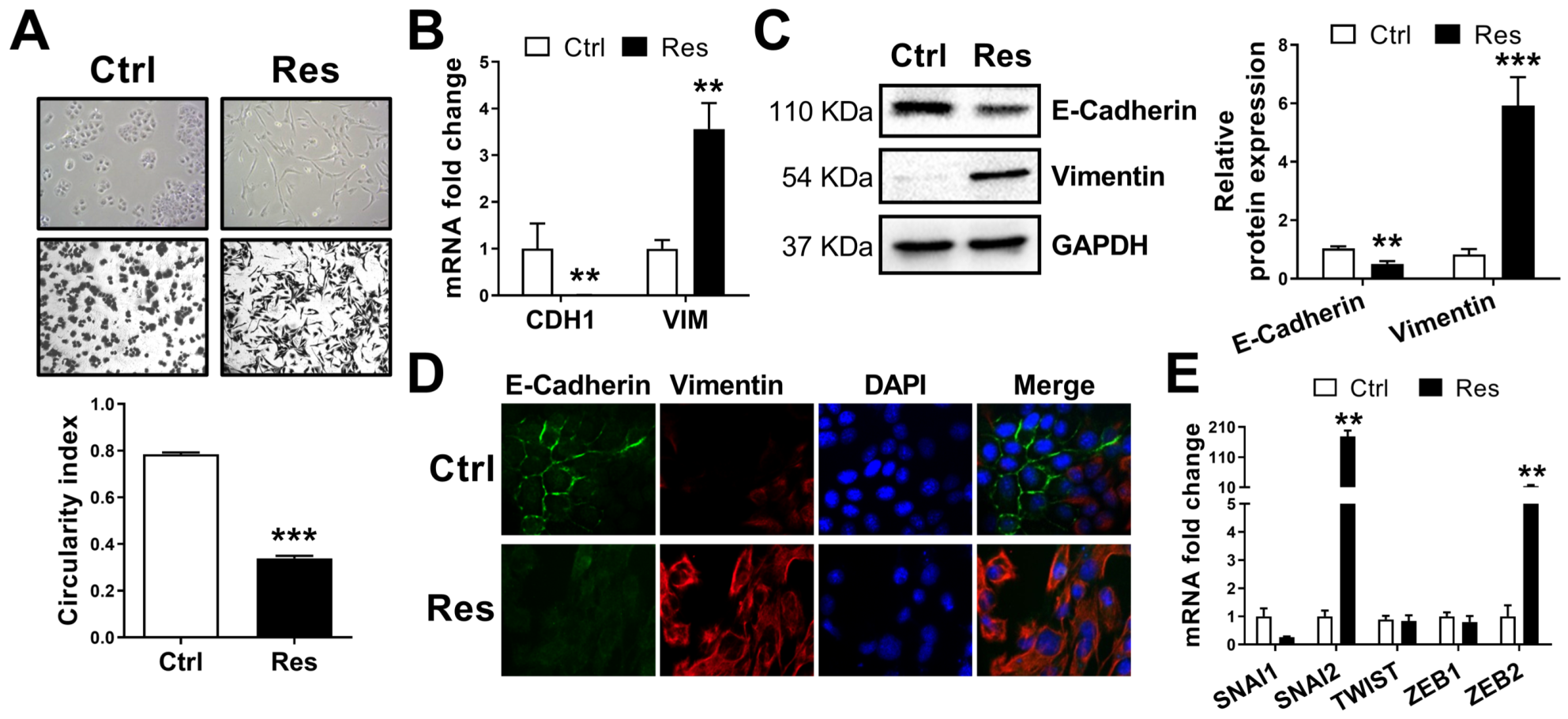
3.2. Resistance to Sorafenib Is Associated with an Epithelial-to-Mesenchymal Transition
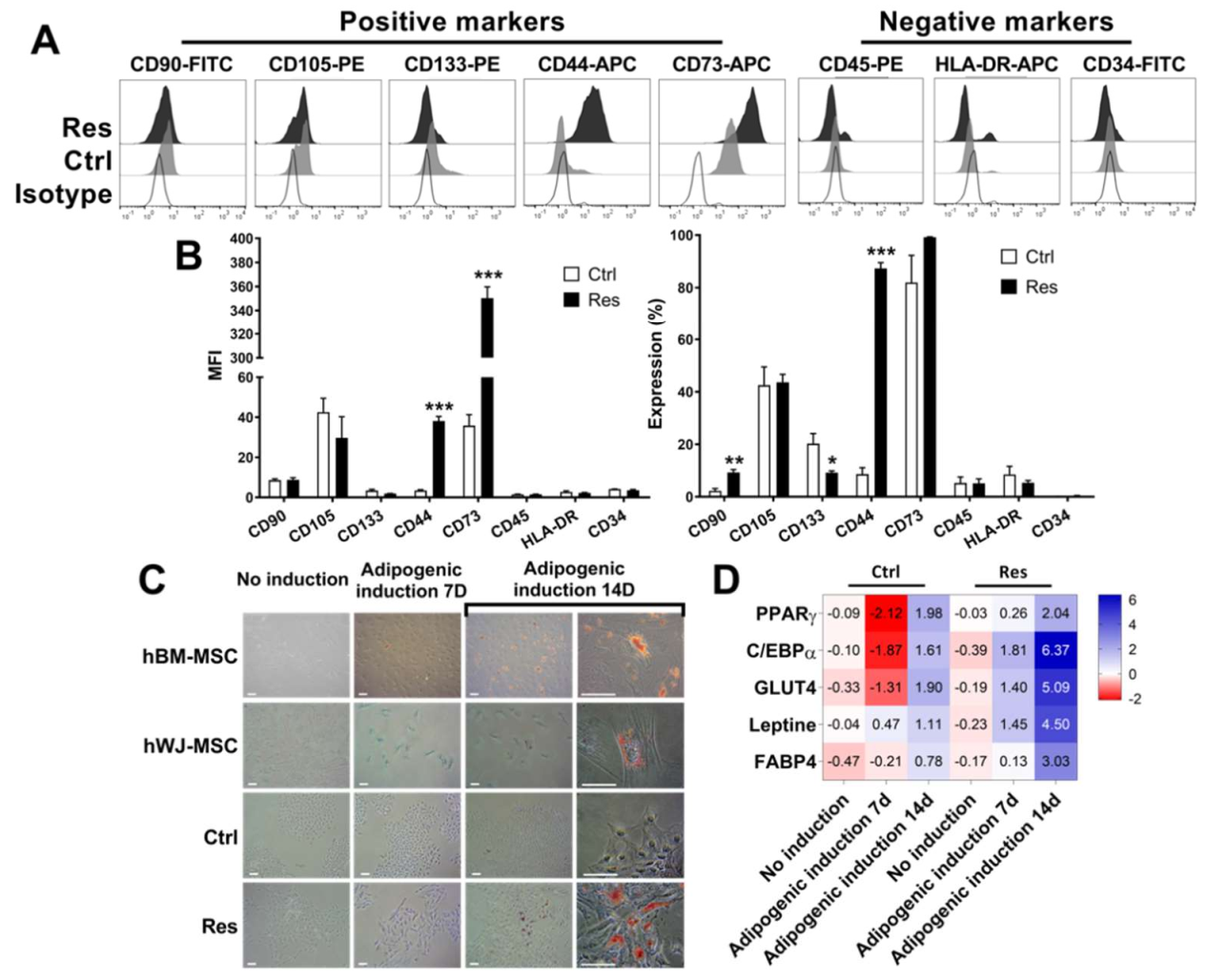
3.3. Sorafenib Resistance Is Associated with Induction of Multipotent MSC Characteristics
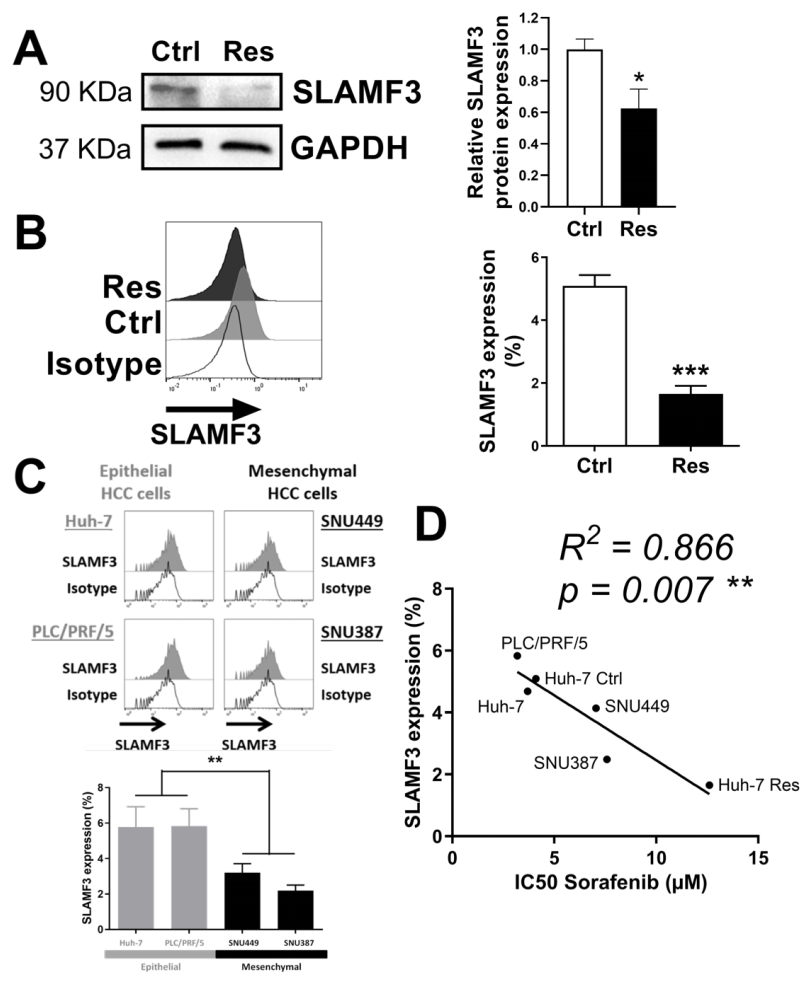
3.4. SLAMF3 Expression Loss Corresponds to Sorafenib Resistance Phenotype
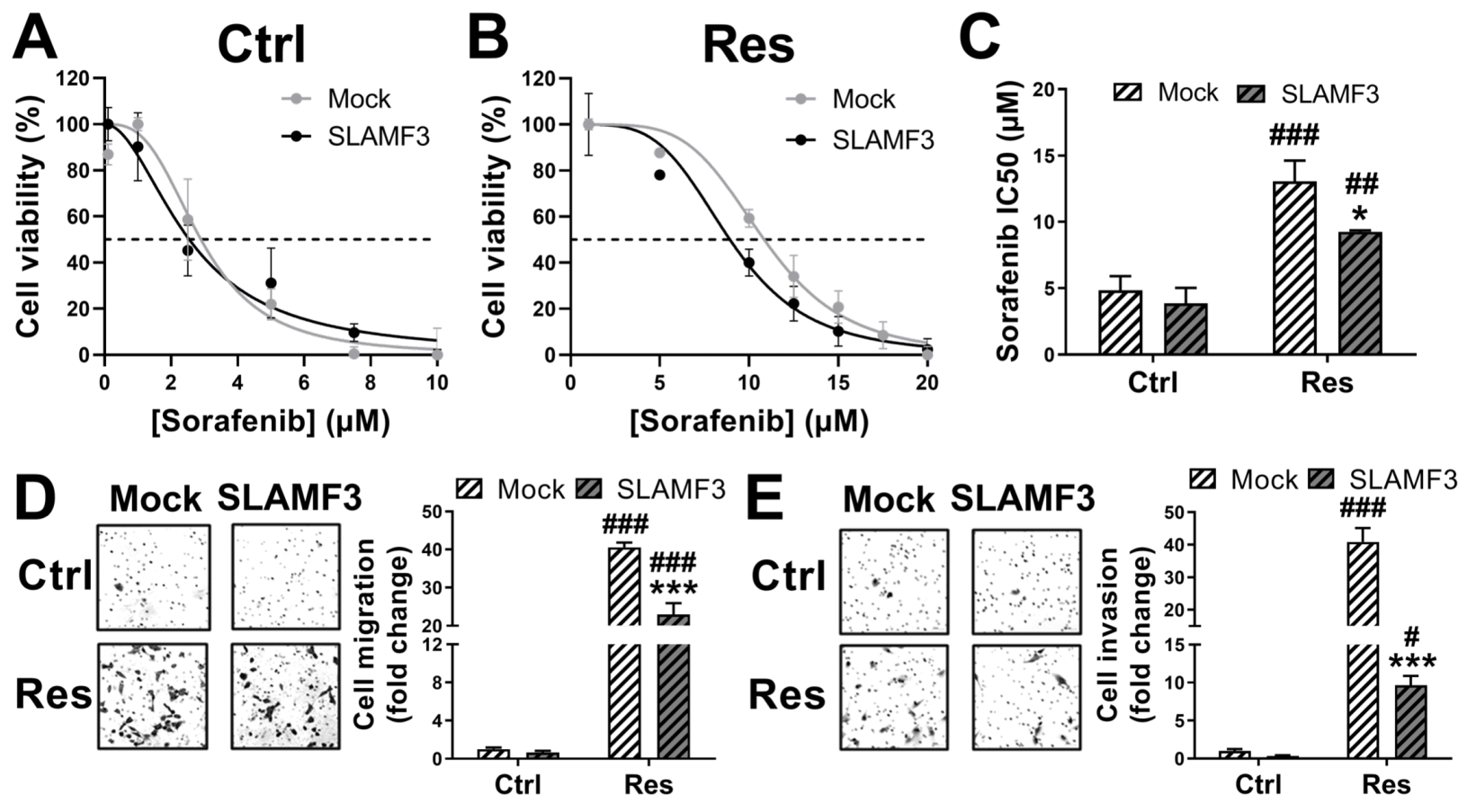
3.5. SLAMF3 Expression Enhances the Sorafenib Efficacy and Inhibits Aggressiveness of HCC Res Cells
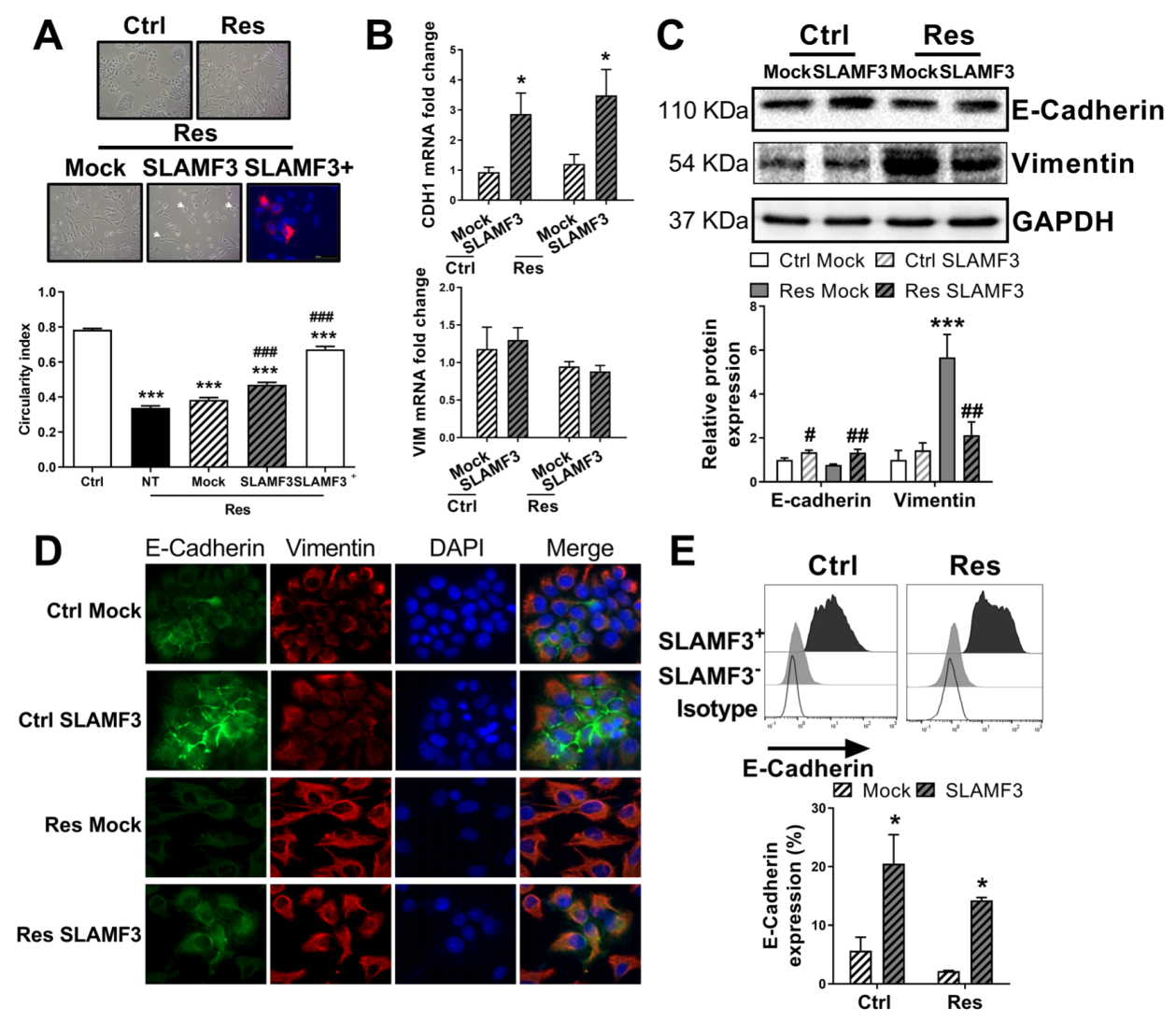
3.6. SLAMF3 Overexpression Reverses the EMT
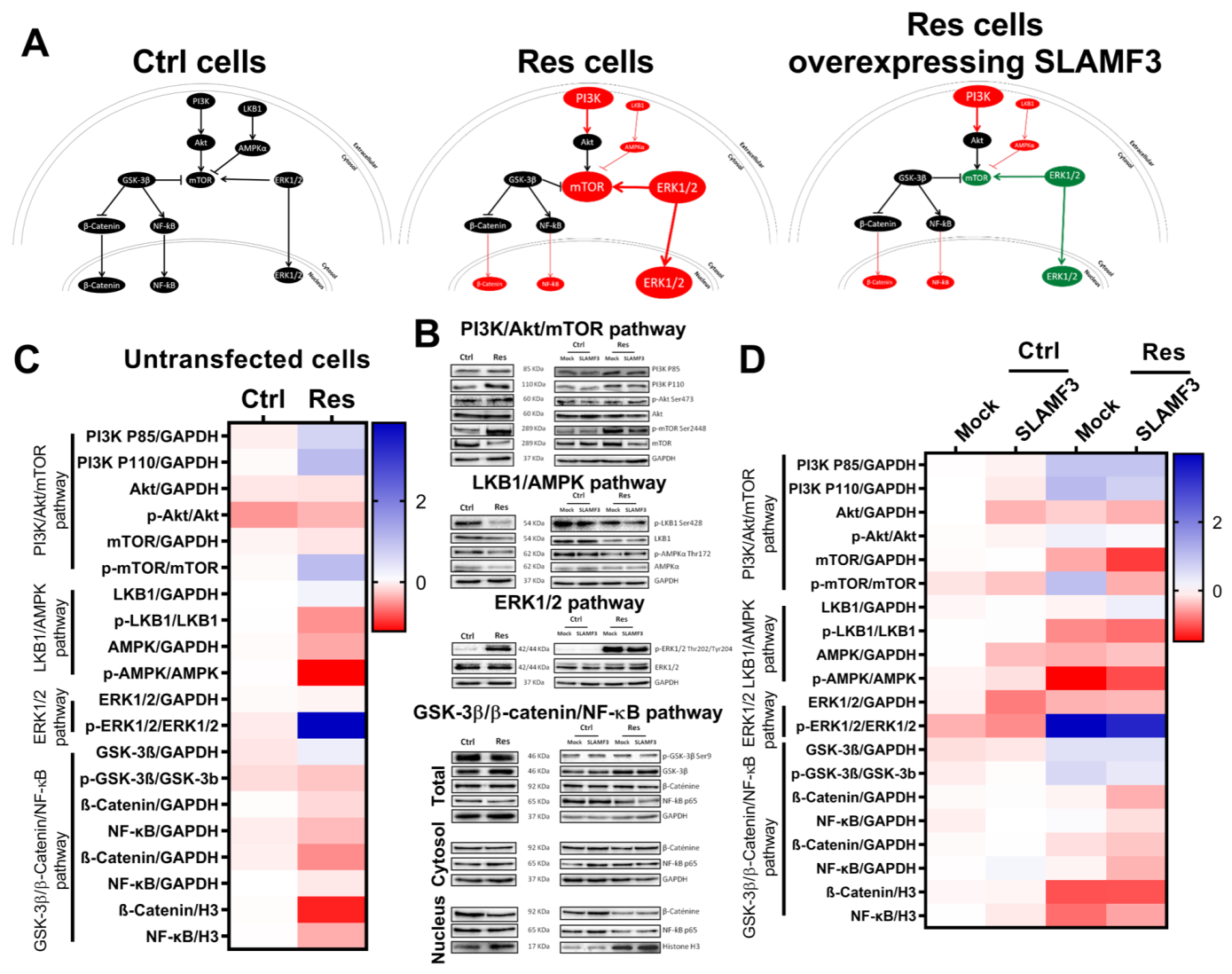
3.7. SLAMF3-Induced Cell Signaling in Sorafenib-Resistant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Chaparro, M.; GonzáLez Moreno, L.; Trapero-MarugáN, M.; Medina, J.; Moreno-Otero, R. Review Article: Pharmacological Therapy for Hepatocellular Carcinoma with Sorafenib and Other Oral Agents. Aliment. Pharmacol. Ther. 2008, 28, 1269–1277. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and Safety of Sorafenib in Patients in the Asia-Pacific Region with Advanced Hepatocellular Carcinoma: A Phase III Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New Insights into Sorafenib Resistance in Hepatocellular Carcinoma: Responsible Mechanisms and Promising Strategies. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Jou, J.; Diehl, A.M. Epithelial-Mesenchymal Transitions and Hepatocarcinogenesis. J. Clin. Investig. 2010, 120, 1031–1034. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Zhai, B.; Sun, W.; Hu, F.; Cheng, H.; Xu, J. Activation of Phosphatidylinositol 3-Kinase/AKT/Snail Signaling Pathway Contributes to Epithelial-Mesenchymal Transition-Induced Multi-Drug Resistance to Sorafenib in Hepatocellular Carcinoma Cells. PLoS One 2017, 12, e0185088. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P. Epithelial-Mesenchymal Transitions in Tumour Progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Sukowati, C.H.; Rosso, N.; Crocè, L.S.; Tiribelli, C. Hepatic Cancer Stem Cells and Drug Resistance: Relevance in Targeted Therapies for Hepatocellular Carcinoma. World J. Hepatol. 2010, 2, 114–126. [Google Scholar] [CrossRef]
- Flores-Téllez, T.N.; Villa-Treviño, S.; Piña-Vázquez, C. Road to Stemness in Hepatocellular Carcinoma. World J. Gastroenterol. 2017, 23, 6750–6776. [Google Scholar] [CrossRef]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, À.; Alvarez-Barrientos, A.; Fernández-Salguero, P.; Fernández-Rodríguez, C.M.; et al. A Mesenchymal-like Phenotype and Expression of CD44 Predict Lack of Apoptotic Response to Sorafenib in Liver Tumor Cells: CD44 and Mesenchymal Traits in Liver Cancer Cell Response to Sorafenib. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef]
- Hagiwara, S.; Kudo, M.; Nagai, T.; Inoue, T.; Ueshima, K.; Nishida, N.; Watanabe, T.; Sakurai, T. Activation of JNK and High Expression Level of CD133 Predict a Poor Response to Sorafenib in Hepatocellular Carcinoma. Br. J. Cancer 2012, 106, 1997–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, A.K.-M.; Ng, L.; Lam, C.S.-C.; Wong, S.K.-M.; Wan, T.M.-H.; Cheng, N.S.-M.; Yau, T.C.-C.; Poon, R.T.-P.; Pang, R.W.-C. The Enhanced Metastatic Potential of Hepatocellular Carcinoma (HCC) Cells with Sorafenib Resistance. PLoS ONE 2013, 8, e78675. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-L.; Shen, M.-N.; Hu, B.; Wang, B.-L.; Yang, W.-J.; Lv, L.-H.; Wang, H.; Zhou, Y.; Jin, A.-L.; Sun, Y.-F.; et al. CD73 Promotes Hepatocellular Carcinoma Progression and Metastasis via Activating PI3K/AKT Signaling by Inducing Rap1-Mediated Membrane Localization of P110β and Predicts Poor Prognosis. J. Hematol. Oncol. 2019, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Dong, K.; Zhang, H. The Roles of CD73 in Cancer. Biomed. Res. Int. 2014, 2014, 460654. [Google Scholar] [CrossRef] [PubMed]
- Marcq, I.; Nyga, R.; Cartier, F.; Amrathlal, R.S.; Ossart, C.; Ouled-Haddou, H.; Ghamlouch, H.; Galmiche, A.; Chatelain, D.; Lamotte, L.; et al. Identification of SLAMF3 (CD229) as an Inhibitor of Hepatocellular Carcinoma Cell Proliferation and Tumour Progression. PLoS ONE 2013, 8, e82918. [Google Scholar] [CrossRef]
- Bouhlal, H.; Ouled-Haddou, H.; Debuysscher, V.; Singh, A.R.; Ossart, C.; Reignier, A.; Hocini, H.; Fouquet, G.; Baghami, M.A.; Eugenio, M.S.; et al. RB/PLK1-Dependent Induced Pathway by SLAMF3 Expression Inhibits Mitosis and Control Hepatocarcinoma Cell Proliferation. Oncotarget 2016, 7, 9832–9843. [Google Scholar] [CrossRef] [Green Version]
- Fouquet, G.; Debuysscher, V.; Ouled-Haddou, H.; Eugenio, M.S.; Demey, B.; Singh, A.R.; Ossart, C.; Al Bagami, M.; Regimbeau, J.-M.; Nguyen-Khac, E.; et al. Hepatocyte SLAMF3 Reduced Specifically the Multidrugs Resistance Protein MRP-1 and Increases HCC Cells Sensitization to Anti-Cancer Drugs. Oncotarget 2016, 7, 32493–32503. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, W.; Zhang, K.; Yin, X.; Lai, J.; Liang, L.; Chen, D. Activation of C-Jun Predicts a Poor Response to Sorafenib in Hepatocellular Carcinoma: Preliminary Clinical Evidence. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Q.; Liu, J.; Cao, H. Inhibition of the PI3K/Akt Signaling Pathway Reverses Sorafenib-Derived Chemo-Resistance in Hepatocellular Carcinoma. Oncol. Lett. 2018, 15, 9377–9384. [Google Scholar] [CrossRef] [PubMed]
- Tomonari, T.; Takeishi, S.; Taniguchi, T.; Tanaka, T.; Tanaka, H.; Fujimoto, S.; Kimura, T.; Okamoto, K.; Miyamoto, H.; Muguruma, N.; et al. MRP3 as a Novel Resistance Factor for Sorafenib in Hepatocellular Carcinoma. Oncotarget 2016, 7, 7207–7215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naudot, M.; Barre, A.; Caula, A.; Sevestre, H.; Dakpé, S.; Mueller, A.A.; Devauchelle, B.; Testelin, S.; Marolleau, J.P.; Le Ricousse, S. Co-Transplantation of Wharton’s Jelly Mesenchymal Stem Cell-Derived Osteoblasts with Differentiated Endothelial Cells Does Not Stimulate Blood Vessel and Osteoid Formation in Nude Mice Models. J. Tissue Eng. Regen. Med. 2020, 14, 257–271. [Google Scholar] [CrossRef]
- Campbell, K. Contribution of Epithelial-Mesenchymal Transitions to Organogenesis and Cancer Metastasis. Curr. Opin. Cell Biol. 2018, 55, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers (Basel) 2017, 9, E127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.-F.; Chen, H.-L.; Tai, W.-T.; Feng, W.-C.; Hsu, C.-H.; Chen, P.-J.; Cheng, A.-L. Activation of Phosphatidylinositol 3-Kinase/Akt Signaling Pathway Mediates Acquired Resistance to Sorafenib in Hepatocellular Carcinoma Cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Chen, C.; Geng, J.; Han, D.; Wang, T.; Xie, T.; Wang, L.; Wang, Y.; Wang, C.; Lei, Z.; et al. Targeting KDM1A Attenuates Wnt/β-Catenin Signaling Pathway to Eliminate Sorafenib-Resistant Stem-like Cells in Hepatocellular Carcinoma. Cancer Lett. 2017, 398, 12–21. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, X.; Shen, H.; Wang, D.; Wang, Y. Phosphorylated ERK Is a Potential Predictor of Sensitivity to Sorafenib When Treating Hepatocellular Carcinoma: Evidence from an in Vitro Study. BMC Med. 2009, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Lo, J.; Lau, E.Y.T.; Ching, R.H.H.; Cheng, B.Y.L.; Ma, M.K.F.; Ng, I.O.L.; Lee, T.K.W. Nuclear Factor Kappa B-Mediated CD47 up-Regulation Promotes Sorafenib Resistance and Its Blockade Synergizes the Effect of Sorafenib in Hepatocellular Carcinoma in Mice. Hepatology 2015, 62, 534–545. [Google Scholar] [CrossRef]
- Bort, A.; Sánchez, B.G.; Mateos-Gómez, P.A.; Vara-Ciruelos, D.; Rodríguez-Henche, N.; Díaz-Laviada, I. Targeting AMP-Activated Kinase Impacts Hepatocellular Cancer Stem Cells Induced by Long-Term Treatment with Sorafenib. Mol. Oncol. 2019, 13, 1311–1331. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, D.; Kurisu, S.; Takenawa, T. Regulation of Cancer Cell Motility through Actin Reorganization. Cancer Sci. 2005, 96, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-N.; Lee, W.-W.; Wang, C.-Y.; Chao, T.-H.; Chen, Y.; Chen, J.H. Regulatory Mechanisms Controlling Human E-Cadherin Gene Expression. Oncogene 2005, 24, 8277–8290. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fouquet, G.; Marié, C.; Collet, L.; Vilpoux, C.; Ouled-Haddou, H.; Nguyen-Khac, E.; Bayry, J.; Naassila, M.; Marcq, I.; Bouhlal, H. Rescuing SLAMF3 Expression Restores Sorafenib Response in Hepatocellular Carcinoma Cells through the Induction of Mesenchymal-to-Epithelial Transition. Cancers 2022, 14, 910. https://doi.org/10.3390/cancers14040910
Fouquet G, Marié C, Collet L, Vilpoux C, Ouled-Haddou H, Nguyen-Khac E, Bayry J, Naassila M, Marcq I, Bouhlal H. Rescuing SLAMF3 Expression Restores Sorafenib Response in Hepatocellular Carcinoma Cells through the Induction of Mesenchymal-to-Epithelial Transition. Cancers. 2022; 14(4):910. https://doi.org/10.3390/cancers14040910
Chicago/Turabian StyleFouquet, Grégory, Constance Marié, Louison Collet, Catherine Vilpoux, Hakim Ouled-Haddou, Eric Nguyen-Khac, Jagadeesh Bayry, Mickaël Naassila, Ingrid Marcq, and Hicham Bouhlal. 2022. "Rescuing SLAMF3 Expression Restores Sorafenib Response in Hepatocellular Carcinoma Cells through the Induction of Mesenchymal-to-Epithelial Transition" Cancers 14, no. 4: 910. https://doi.org/10.3390/cancers14040910
APA StyleFouquet, G., Marié, C., Collet, L., Vilpoux, C., Ouled-Haddou, H., Nguyen-Khac, E., Bayry, J., Naassila, M., Marcq, I., & Bouhlal, H. (2022). Rescuing SLAMF3 Expression Restores Sorafenib Response in Hepatocellular Carcinoma Cells through the Induction of Mesenchymal-to-Epithelial Transition. Cancers, 14(4), 910. https://doi.org/10.3390/cancers14040910