
Single-Cell Sequencing: Current Applications in Precision Onco-Genomics and Cancer Therapeutics



Abstract
:Simple Summary
Abstract
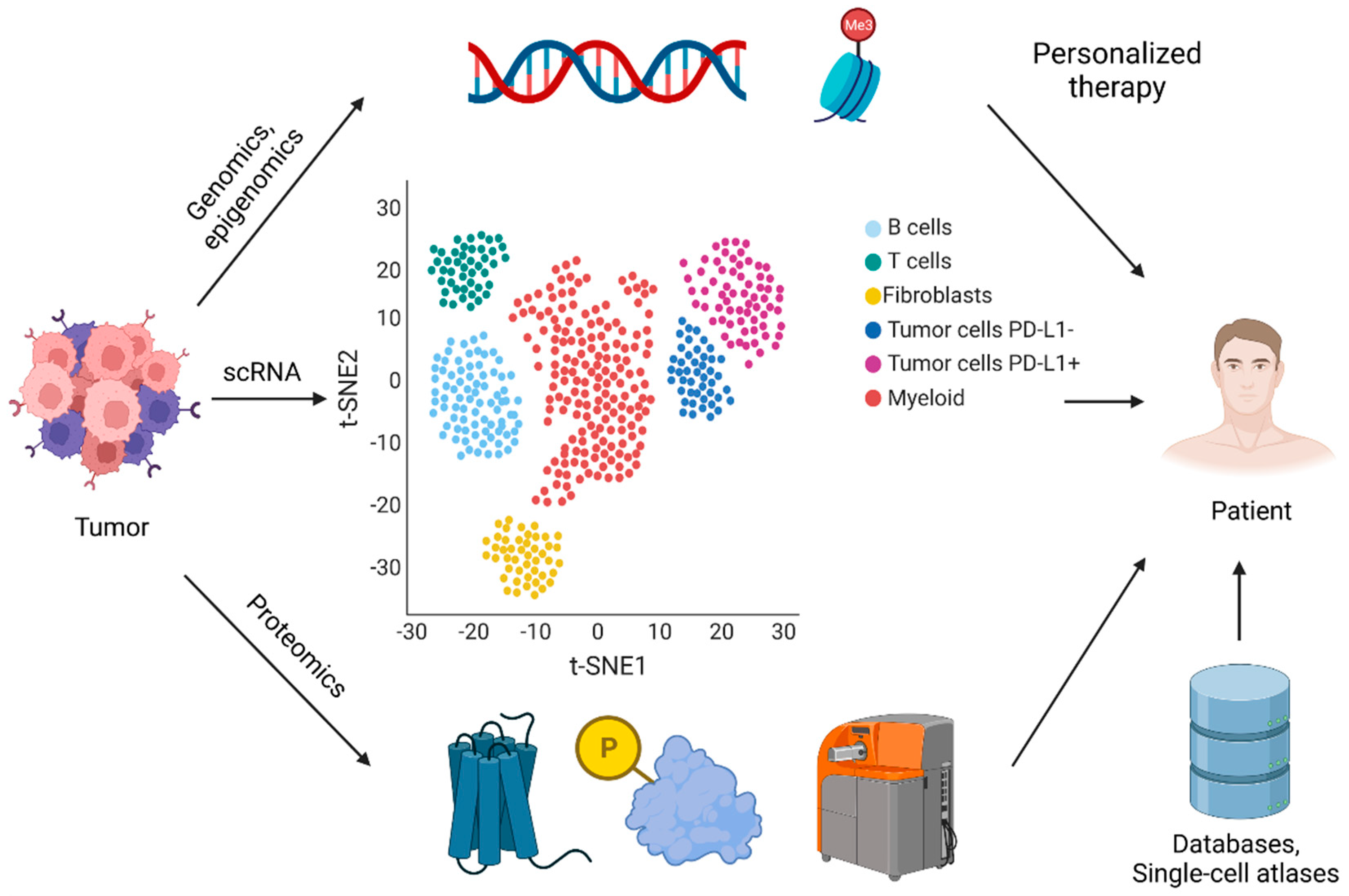
1. Single-Cell Sequencing Technologies and Integrated Databases
2. Translational Relevance of Single-Cell Sequencing Technologies
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, J.; Hyeon, D.Y.; Hwang, D. Single-cell multiomics: Technologies and data analysis methods. Exp. Mol. Med. 2020, 52, 1428–1442. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.L.; MacKay, M.; Afshinnekoo, E.; Melnick, A.M.; Wu, S.; Mason, C.E. The Impact of Heterogeneity on Single-Cell Sequencing. Front. Genet. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashima, Y.; Sakamoto, Y.; Kaneko, K.; Seki, M.; Suzuki, Y.; Suzuki, A. Single-cell sequencing techniques from individual to multiomics analyses. Exp. Mol. Med. 2020, 52, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Engel, J.; Teichmann, S.A.; Lonnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S. Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol. 2017, 14, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 2021, 184, 4734–4752.e4720. [Google Scholar] [CrossRef]
- Davis, R.T.; Blake, K.; Ma, D.; Gabra, M.B.I.; Hernandez, G.A.; Phung, A.T.; Yang, Y.; Maurer, D.; Lefebvre, A.; Alshetaiwi, H.; et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat. Cell Biol. 2020, 22, 310–320. [Google Scholar] [CrossRef]
- Marjanovic, N.D.; Hofree, M.; Chan, J.E.; Canner, D.; Wu, K.; Trakala, M.; Hartmann, G.G.; Smith, O.C.; Kim, J.Y.; Evans, K.V.; et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020, 38, 229–246.e213. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e422. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.I.; Suh, Y.L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Franco, I.; Helgadottir, H.T.; Moggio, A.; Larsson, M.; Vrtacnik, P.; Johansson, A.; Norgren, N.; Lundin, P.; Mas-Ponte, D.; Nordstrom, J.; et al. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biol. 2019, 20, 285. [Google Scholar] [CrossRef] [PubMed]
- Bronner, I.F.; Lorenz, S. Combined Genome and Transcriptome (G&T) Sequencing of Single Cells. Methods Mol. Biol. 2019, 1979, 319–362. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, S.; Hou, Y.; Zhou, X.; Li, X.; Yong, J.; Wang, Y.; Wang, W.; Yan, J.; Hu, B.; Guo, H.; et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 2018, 362, 1060–1063. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Cooper, J.; Ding, Y.; Song, J.; Zhao, K. Genome-wide mapping of DNase I hypersensitive sites in rare cell populations using single-cell DNase sequencing. Nat. Protoc. 2017, 12, 2342–2354. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef]
- Perkel, J.M. Single-cell proteomics takes centre stage. Nature 2021, 597, 580–582. [Google Scholar] [CrossRef]
- Zhou, Z.; Ye, C.; Wang, J.; Zhang, N.R. Surface protein imputation from single cell transcriptomes by deep neural networks. Nat. Commun. 2020, 11, 651. [Google Scholar] [CrossRef] [Green Version]
- Kashima, Y.; Togashi, Y.; Fukuoka, S.; Kamada, T.; Irie, T.; Suzuki, A.; Nakamura, Y.; Shitara, K.; Minamide, T.; Yoshida, T.; et al. Potentiality of multiple modalities for single-cell analyses to evaluate the tumor microenvironment in clinical specimens. Sci. Rep. 2021, 11, 341. [Google Scholar] [CrossRef]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e1018. [Google Scholar] [CrossRef] [PubMed]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jane-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e316. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Kwon, A.T.; Shin, J.W. An era of single-cell genomics consortia. Exp. Mol. Med. 2020, 52, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Guo, L.; Li, S.; Hua, K. Single-cell transcriptomics reveals the landscape of intra-tumoral heterogeneity and transcriptional activities of ECs in CC. Mol. Ther. Nucleic Acids 2021, 24, 682–694. [Google Scholar] [CrossRef] [PubMed]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [Green Version]
- Bhat-Nakshatri, P.; Gao, H.; Sheng, L.; McGuire, P.C.; Xuei, X.; Wan, J.; Liu, Y.; Althouse, S.K.; Colter, A.; Sandusky, G.; et al. A single-cell atlas of the healthy breast tissues reveals clinically relevant clusters of breast epithelial cells. Cell Rep. Med. 2021, 2, 100219. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, D.; Yang, Q.; Lv, X.; Huang, W.; Zhou, Z.; Wang, Y.; Zhang, Z.; Yuan, T.; Ding, X.; et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat. Commun. 2020, 11, 6322. [Google Scholar] [CrossRef]
- Rocque, B.; Barbetta, A.; Singh, P.; Goldbeck, C.; Helou, D.G.; Loh, Y.E.; Ung, N.; Lee, J.; Akbari, O.; Emamaullee, J. Creation of a Single Cell RNASeq Meta-Atlas to Define Human Liver Immune Homeostasis. Front. Immunol. 2021, 12, 679521. [Google Scholar] [CrossRef]
- Brancale, J.; Vilarinho, S. A single cell gene expression atlas of 28 human livers. J. Hepatol. 2021, 75, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, W.; Hua, X.; Chen, X.; Chang, Y.; Hu, Y.; Xu, Z.; Song, J. Single-Cell Transcriptomic Atlas of Different Human Cardiac Arteries Identifies Cell Types Associated With Vascular Physiology. Arter. Thromb. Vasc. Biol. 2021, 41, 1408–1427. [Google Scholar] [CrossRef] [PubMed]
- Conway, B.R.; O‘Sullivan, E.D.; Cairns, C.; O‘Sullivan, J.; Simpson, D.J.; Salzano, A.; Connor, K.; Ding, P.; Humphries, D.; Stewart, K.; et al. Kidney Single-Cell Atlas Reveals Myeloid Heterogeneity in Progression and Regression of Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 2833–2854. [Google Scholar] [CrossRef] [PubMed]
- Lukowski, S.W.; Lo, C.Y.; Sharov, A.A.; Nguyen, Q.; Fang, L.; Hung, S.S.; Zhu, L.; Zhang, T.; Grunert, U.; Nguyen, T.; et al. A single-cell transcriptome atlas of the adult human retina. EMBO J. 2019, 38, e100811. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.; Cambuli, F.; Aparicio, L.; Shibata, M.; Robinson, B.D.; Xuan, S.; Li, W.; Hibshoosh, H.; Loda, M.; Rabadan, R.; et al. A single-cell atlas of the mouse and human prostate reveals heterogeneity and conservation of epithelial progenitors. eLife 2020, 9, e59465. [Google Scholar] [CrossRef]
- He, S.; Wang, L.H.; Liu, Y.; Li, Y.Q.; Chen, H.T.; Xu, J.H.; Peng, W.; Lin, G.W.; Wei, P.P.; Li, B.; et al. Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol. 2020, 21, 294. [Google Scholar] [CrossRef]
- Karlsson, M.; Zhang, C.; Mear, L.; Zhong, W.; Digre, A.; Katona, B.; Sjostedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W.; et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef]
- Lee, H.W.; Chung, W.; Lee, H.O.; Jeong, D.E.; Jo, A.; Lim, J.E.; Hong, J.H.; Nam, D.H.; Jeong, B.C.; Park, S.H.; et al. Single-cell RNA sequencing reveals the tumor microenvironment and facilitates strategic choices to circumvent treatment failure in a chemorefractory bladder cancer patient. Genome Med. 2020, 12, 47. [Google Scholar] [CrossRef]
- Zhang, Y.; Narayanan, S.P.; Mannan, R.; Raskind, G.; Wang, X.; Vats, P.; Su, F.; Hosseini, N.; Cao, X.; Kumar-Sinha, C.; et al. Single-cell analyses of renal cell cancers reveal insights into tumor microenvironment, cell of origin, and therapy response. Proc. Natl. Acad. Sci. USA 2021, 118, e2103240118. [Google Scholar] [CrossRef]
- Shalek, A.K.; Benson, M. Single-cell analyses to tailor treatments. Sci Transl Med. 2017, 9, eaan4730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.H.; Chen, Y.C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.T.; Lee, W.; Hao, Z.; Sahoo, S.; Min Kang, H.; et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat. Commun. 2019, 10, 2163. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.S.; Li, Y.; Mitra, A.K.; Bi, L.; Abyzov, A.; van Wijnen, A.J.; Baughn, L.B.; Van Ness, B.; Rajkumar, V.; Kumar, S.; et al. Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J. 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehgal, K.; Portell, A.; Ivanova, E.V.; Lizotte, P.H.; Mahadevan, N.R.; Greene, J.R.; Vajdi, A.; Gurjao, C.; Teceno, T.; Taus, L.J.; et al. Dynamic single-cell RNA sequencing identifies immunotherapy persister cells following PD-1 blockade. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Yofe, I.; Dahan, R.; Amit, I. Single-cell genomic approaches for developing the next generation of immunotherapies. Nat. Med. 2020, 26, 171–177. [Google Scholar] [CrossRef]
- Wu, B.; Yuan, Y.; Liu, J.; Shang, H.; Dong, J.; Liang, X.; Wang, D.; Chen, Y.; Wang, C.; Zhou, Y.; et al. Single-cell RNA sequencing reveals the mechanism of sonodynamic therapy combined with a RAS inhibitor in the setting of hepatocellular carcinoma. J. Nanobiotechnol. 2021, 19, 177. [Google Scholar] [CrossRef]
- Kim, M.; Min, Y.K.; Jang, J.; Park, H.; Lee, S.; Lee, C.H. Single-cell RNA sequencing reveals distinct cellular factors for response to immunotherapy targeting CD73 and PD-1 in colorectal cancer. J. Immunother. Cancer 2021, 9, e002503. [Google Scholar] [CrossRef]
- Aissa, A.F.; Islam, A.; Ariss, M.M.; Go, C.C.; Rader, A.E.; Conrardy, R.D.; Gajda, A.M.; Rubio-Perez, C.; Valyi-Nagy, K.; Pasquinelli, M.; et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat. Commun. 2021, 12, 1628. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e813. [Google Scholar] [CrossRef] [Green Version]
- Lahnemann, D.; Koster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]


{kind=link}
| Site/Disease | Species | References |
|---|---|---|
| Cervical cancer | Human | [24] |
| Breast cancer | Human | [25,27,28] |
| Lung | Human | [26] |
| Osteosarcoma | Human | [29] |
| Liver | Human | [30,31] |
| Cardiac arteries | Human | [32] |
| Kidney | Human | [33] |
| Retina | Human | [34] |
| Prostate | Human, Mouse | [35] |
| 15 organs | Human | [36] |
| 13 tissues | Human | [37] |
| Clinical Trial ID | Trial Name | Site/Disease |
|---|---|---|
| NCT04927611 | Single-cell Sequencing and Establishment of Models in Neuroendocrine Neoplasm | Brain, neuroendocrine neoplasm |
| NCT04162691 | Single Cell Sequencing Analysis of Thymoma | Thymus, thymoma |
| NCT04568291 | CTC in Lung Cancer Patients With Bone Metastases | Lung cancer, bone metastasis |
| NCT02313623 | MR-US Image Fusion Targeted Biopsy for Single-cell Prostate Cancer Research | Prostate cancer |
| NCT04807127 | A Single-cell Approach to Identify Biomarkers of Pulmonary Toxicity for Immune Checkpoint Blockade | Lung, pneumonitis |
| NCT04807114 | A single-cell Approach to Identify Biomarkers of Efficacy and Toxicity for ICI in NSCLC | Lung, non-small cell lung cancer |
| NCT04434833 | A Single-cell Transcriptome Study in Patients with Non-Hodgkin’s Lymphoma | Lymph nodes, Non-Hodgkin’s Lymphoma |
| NCT04204291 | Project A4sc- An Atlas of Airways at a Single Cell Level (A4sc) | Lung, chronic respiratory diseases |
| NCT04789252 | Heterogeneity of Dendritic Cells in Colon and Non-small Cell Lung Cancer (TUM-DC) | Colon and Non-small cell lung cancers |
| NCT04696692 | Single-cell Map of Immune and Lymphoma Cells in B-cell Non-Hodgkin’s Lymphoma (SIMILY) | B-cell Non-Hodgkin’s Lymphoma |
| NCT04261010 | TNF and IL23 Blocking Agents Gene Expression Ratios in the Psoriatic Arthritis Synovium_(TIGERS) Study (TIGERS) | Psoriatic arthritis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustachio, L.M.; Roszik, J. Single-Cell Sequencing: Current Applications in Precision Onco-Genomics and Cancer Therapeutics. Cancers 2022, 14, 657. https://doi.org/10.3390/cancers14030657
Mustachio LM, Roszik J. Single-Cell Sequencing: Current Applications in Precision Onco-Genomics and Cancer Therapeutics. Cancers. 2022; 14(3):657. https://doi.org/10.3390/cancers14030657
Chicago/Turabian StyleMustachio, Lisa Maria, and Jason Roszik. 2022. "Single-Cell Sequencing: Current Applications in Precision Onco-Genomics and Cancer Therapeutics" Cancers 14, no. 3: 657. https://doi.org/10.3390/cancers14030657
APA StyleMustachio, L. M., & Roszik, J. (2022). Single-Cell Sequencing: Current Applications in Precision Onco-Genomics and Cancer Therapeutics. Cancers, 14(3), 657. https://doi.org/10.3390/cancers14030657