
Clinical Validity of Circulating Tumor DNA as Prognostic and Predictive Marker for Personalized Colorectal Cancer Patient Management

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
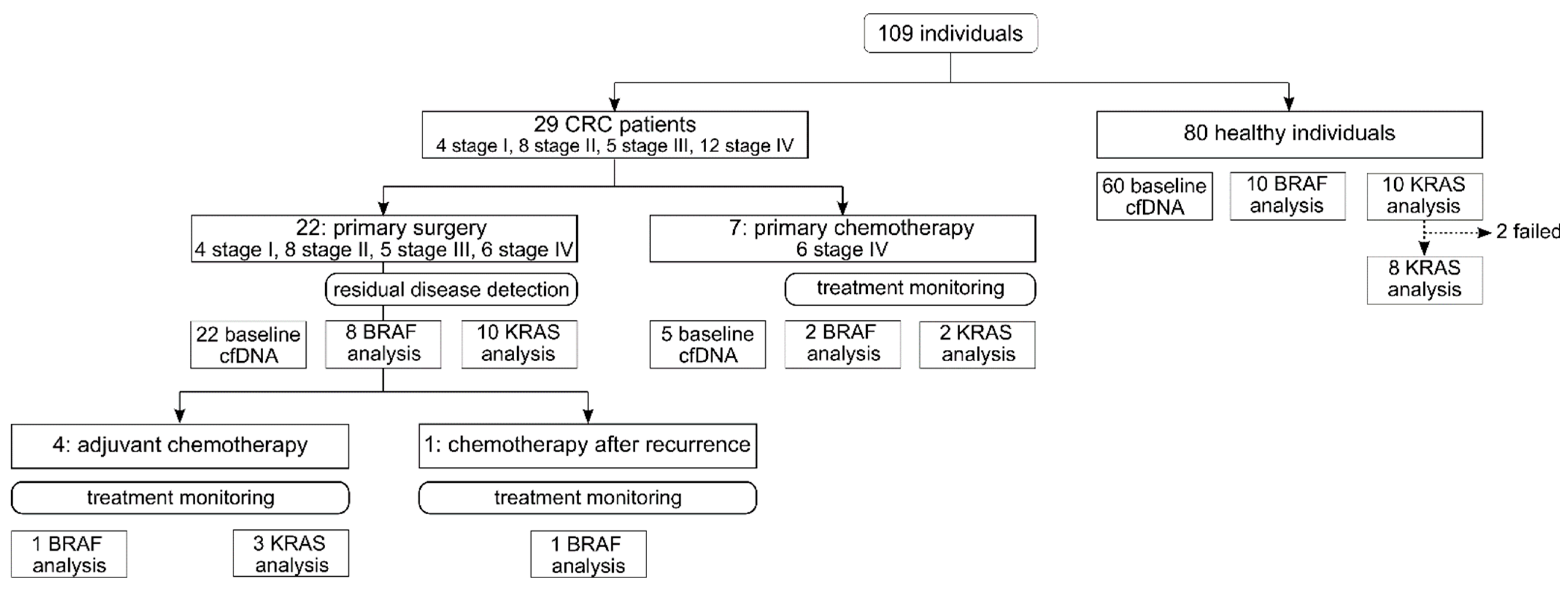
2.1. Study Design and Participants
2.2. Sample Preparation and Droplet Digital PCR
2.3. Droplet Digital™ PCR
2.4. Determination of Cutoffs for ctDNA Positive Status and Quantifiable ctDNA
2.5. Determination of Cutoff for Elevated cfDNA Concentrations
2.6. CEA Analysis
2.7. Statistical Analysis
3. Results
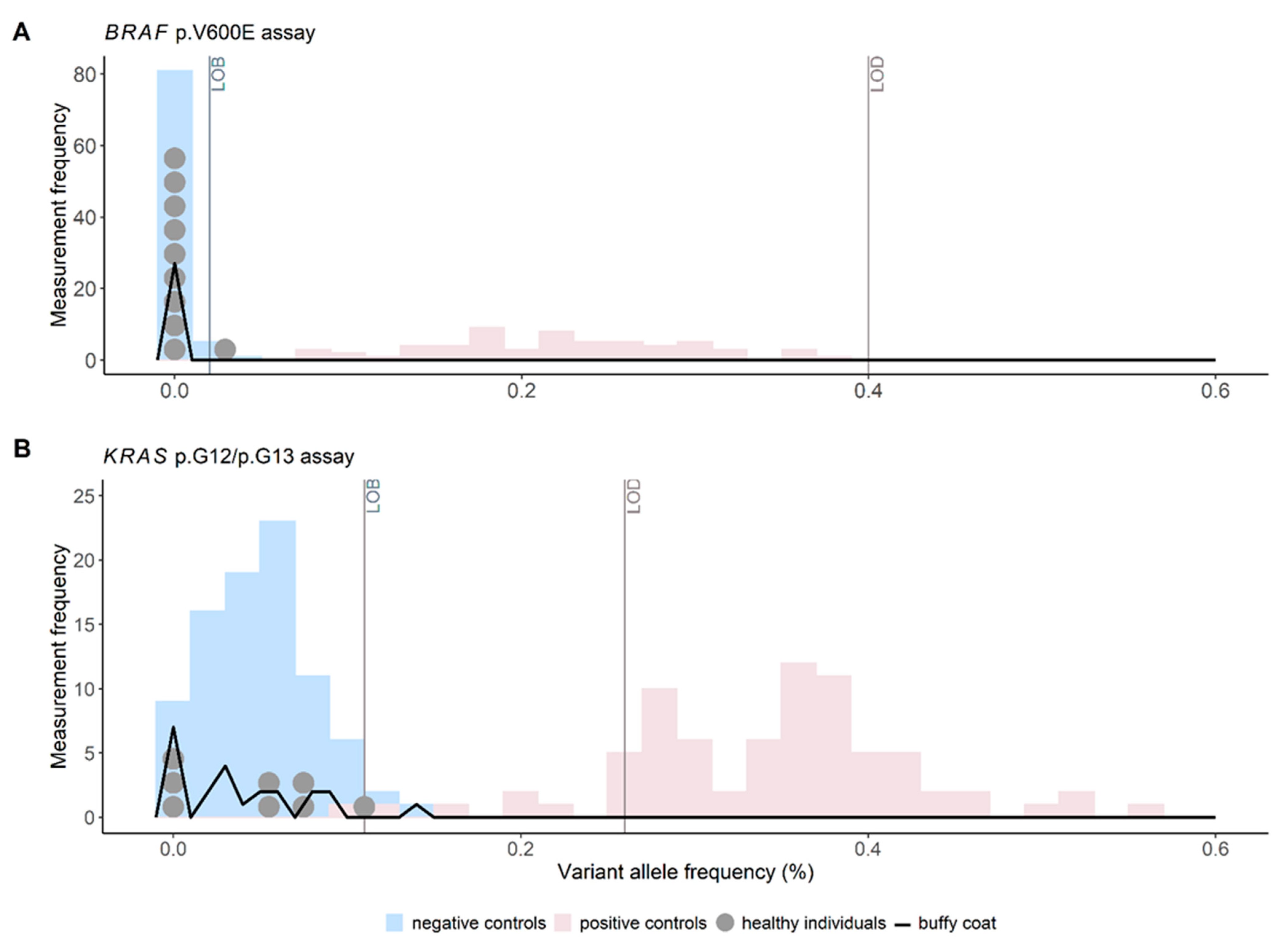
3.1. Cutoff Validation for ctDNA Status and Quantifiable ctDNA VAFs in Reference Materials
3.2. Cutoff Verification for ctDNA Status in Plasma of Healthy Controls
3.3. Positive ctDNA Status Is Tumor Specific
3.4. At Baseline, Elevated cfDNA Concentration Outperform ctDNA Positive Status and CEA Levels
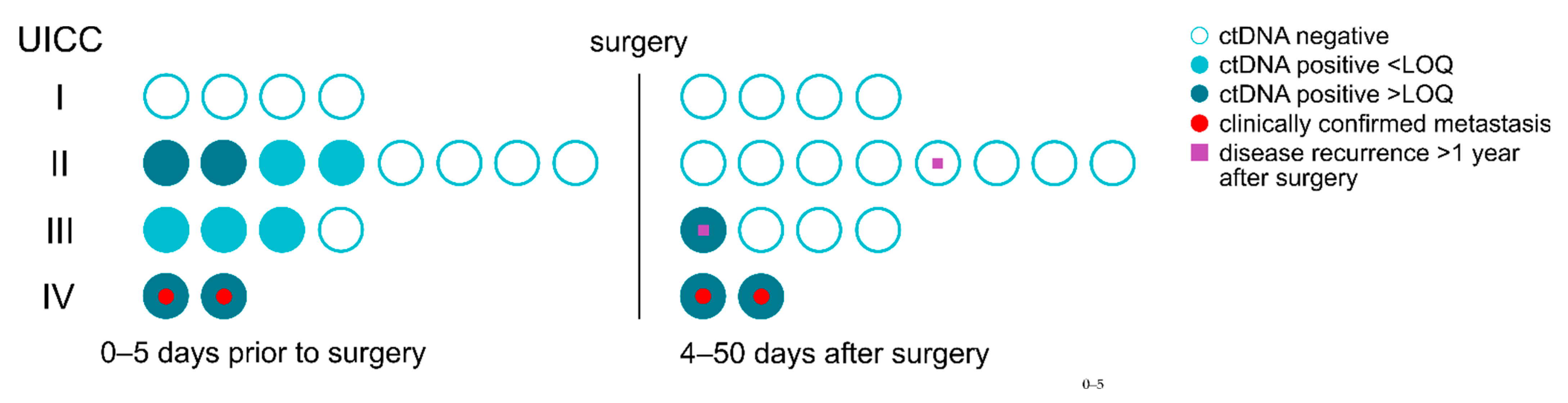
3.5. Residual Disease and Recurrence Are Predicted by ctDNA Positive Status
3.6. Residual Disease and Recurrence Are More Reliably Predicted by Positive ctDNA Status Than by Elevated cfDNA Concentration and CEA Levels
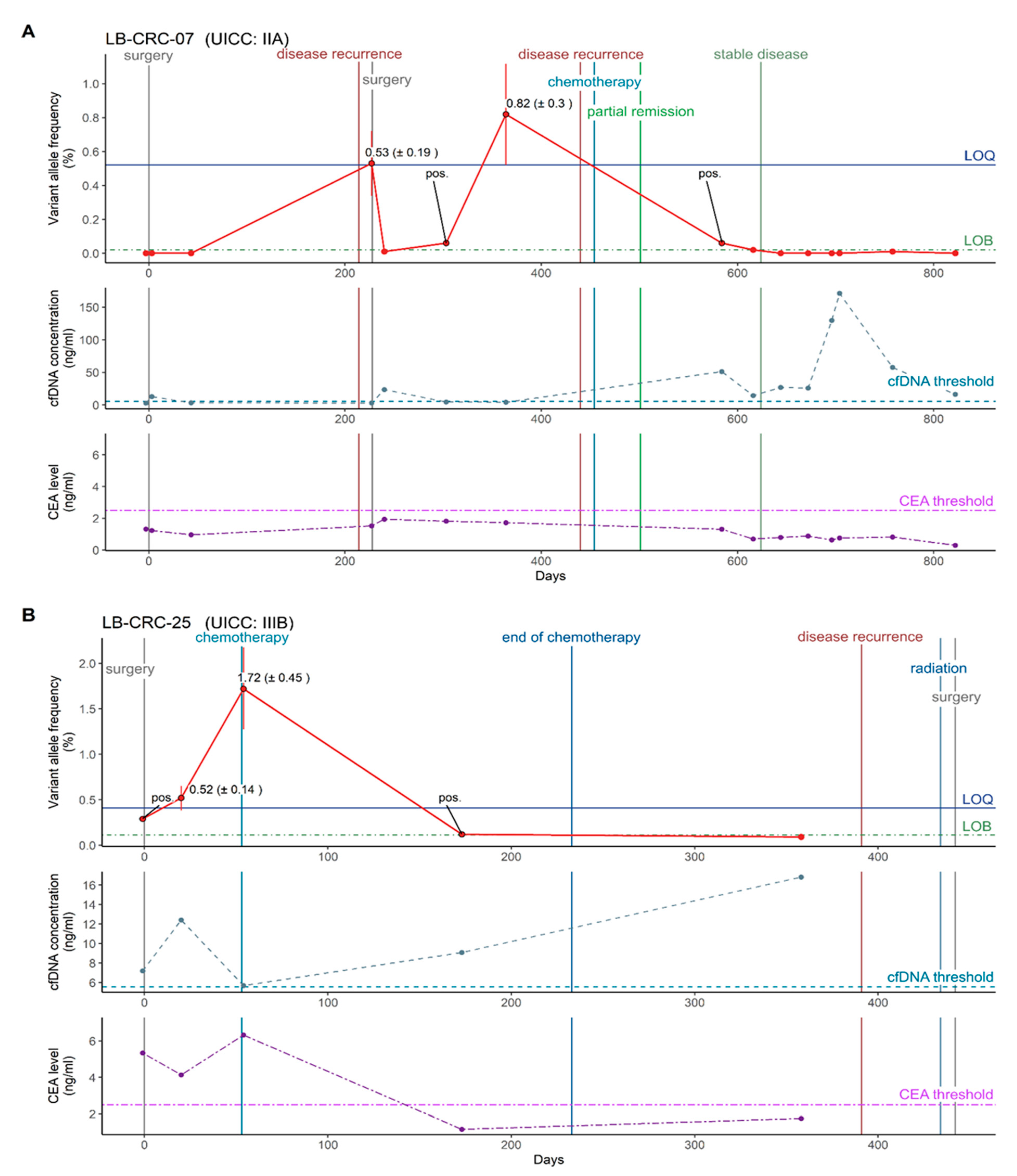
3.7. Chemotherapy Monitoring Possible through Precise ctDNA Quantification
3.8. Chemotherapy Monitoring by Precise ctDNA Quantification Outperforms cfDNA Concentration and CEA Levels
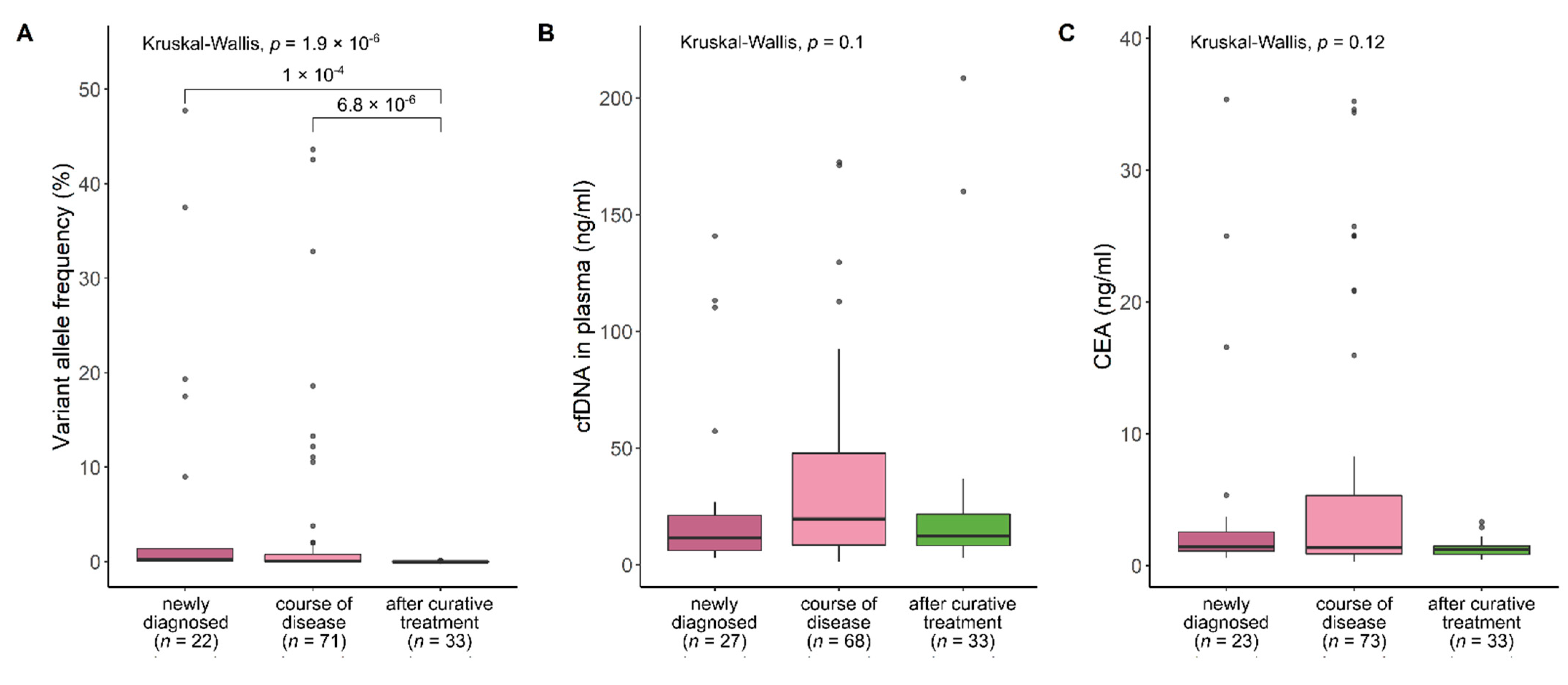
3.9. Significant Differences Depending on Time in Course of Disease in ctDNA VAFs, but Not in cfDNA Concentration and CEA Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lui, Y.Y.N.; Chik, K.-W.; Chiu, R.W.K.; Ho, C.-Y.; Lam, C.W.K.; Lo, Y.D. Predominant Hematopoietic Origin of Cell-free DNA in Plasma and Serum after Sex-mismatched Bone Marrow Transplantation. Clin. Chem. 2002, 48, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Márquez, C.P.; Chu, C.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J.; et al. Cell-free DNA concentration and fragment size as a biomarker for prostate cancer. Sci. Rep. 2021, 11, 5040. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, P.; Gaskell, A.A.; Merghoub, T.; Viale, A.; Chapman, P.B. Correlation of tumor-derived circulating cell free DNA (cfDNA) measured by digital PCR (DigPCR) with tumor burden measured radiographically in patients (pts) with BRAFV600E mutated melanoma (mel) treated with RAF inhibitor (RAFi) and/or ipilimumab (Ipi). J. Clin. Oncol. 2014, 32, 9085. [Google Scholar] [CrossRef]
- Schøler, L.V.; Reinert, T.; Ørntoft, M.-B.W.; Kassentoft, C.G.; Árnadóttir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, L.; Rothwell, D.G.; Mesquita, B.; Smowton, C.; Leong, H.S.; Fernandez-Gutierrez, F.; Li, Y.; Burt, D.J.; Antonello, J.; Morrow, C.J.; et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat. Med. 2017, 23, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Chen, Y.-C.; Lin, E.; Brien, R.; Jung, S.; Chen, Y.-T.; Lee, W.; Hao, Z.; Sahoo, S.; Min Kang, H.; et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat. Commun. 2019, 10, 2163. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Brown, N.A.; Elenitoba-Johnson, K.S.J. Enabling Precision Oncology Through Precision Diagnostics. Annu. Rev. Pathol. 2020, 15, 97–121. [Google Scholar] [CrossRef] [Green Version]
- IJzerman, M.J.; de Boer, J.; Azad, A.; Degeling, K.; Geoghegan, J.; Hewitt, C.; Hollande, F.; Lee, B.; To, Y.H.; Tothill, R.W.; et al. Towards Routine Implementation of Liquid Biopsies in Cancer Management: It Is Always Too Early, until Suddenly It Is Too Late. Diagnostics 2021, 11, 103. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.; Wu, H.-T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients with Urothelial Bladder Carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.R.; Contente-Cuomo, T.; Sammut, S.-J.; Odenheimer-Bergman, A.; Ernst, B.; Perdigones, N.; Chin, S.-F.; Farooq, M.; Mejia, R.; Cronin, P.A.; et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.; van Grieken, N.C.T.; Palsgrove, D.N.; Phallen, J.; Medina, J.E.; Hruban, C.; Broeckaert, M.A.M.; Anagnostou, V.; Adleff, V.; Bruhm, D.C.; et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat. Commun. 2020, 11, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lo, S.N.; Wang, Y.; Li, L.; Christie, M.; Lee, M.; Wong, R.; Kosmider, S.; Skinner, I.; et al. Prognostic significance of postsurgery circulating tumor DNA in nonmetastatic colorectal cancer: Individual patient pooled analysis of three cohort studies. Int. J. Cancer 2020, 148, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Argilés, G.; Tabernero, J.; Labianca, R.; Hochhauser, D.; Salazar, R.; Iveson, T.; Laurent-Puig, P.; Quirke, P.; Yoshino, T.; Taieb, J.; et al. Localised colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1291–1305. [Google Scholar] [CrossRef]
- Sørbye, H.; Dahl, O. Carcinoembryonic Antigen Surge in Metastatic Colorectal Cancer Patients Responding to Oxaliplatin Combination Chemotherapy: Implications for Tumor Marker Monitoring and Guidelines. J. Clin. Oncol. 2003, 21, 4466–4467. [Google Scholar] [CrossRef]
- Goldstein, M.; Mitchell, E.P. Carcinoembryonic Antigen in the Staging and Follow-up of Patients with Colorectal Cancer. Cancer Investig. 2005, 23, 338–351. [Google Scholar] [CrossRef]
- Holdhoff, M.; Schmidt, K.; Donehower, R.; Diaz, L.A. Analysis of circulating tumor DNA to confirm somatic KRAS mutations. J. Natl. Cancer Inst. 2009, 101, 1284–1285. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebs, S.; Keilholz, U.; Kehler, I.; Schweiger, C.; Haybäck, J.; Nonnenmacher, A. Detection of mutations in circulating cell-free DNA in relation to disease stage in colorectal cancer. Cancer Med. 2019, 8, 3761–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godsey, J.H.; Silvestro, A.; Barrett, J.C.; Bramlett, K.; Chudova, D.; Deras, I.; Dickey, J.; Hicks, J.; Johann, D.J.; Leary, R.; et al. Generic Protocols for the Analytical Validation of Next-Generation Sequencing-Based ctDNA Assays: A Joint Consensus Recommendation of the BloodPAC’s Analytical Variables Working Group. Clin. Chem. 2020, 66, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- NCCLS. Protocols for Determination of Limits of Detection and Limits of Quantitation Guideline: Approved Guideline; NCCLS Document EP17-A: Wayne, PA, USA, 2004; ISBN 1-56238-551-8. [Google Scholar]
- Wittekind, C.; Meyer, H.-J. TNM Klassifikation Maligner Tumoren, 7th ed.; Wiley-VCH: Weinheim, Germany, 2010; ISBN 978-3-527-32759-1. [Google Scholar]
- Midic, D.; Rinke, J.; Perner, F.; Müller, V.; Hinze, A.; Pester, F.; Landschulze, J.; Ernst, J.; Gruhn, B.; Matziolis, G.; et al. Prevalence and dynamics of clonal hematopoiesis caused by leukemia-associated mutations in elderly individuals without hematologic disorders. Leukemia 2020, 34, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Hallermayr, A.; Benet-Pagès, A.; Steinke-Lange, V.; Mansmann, U.; Rentsch, M.; Holinski-Feder, E.; Pickl, J.M.A. Liquid Biopsy Hotspot Variant Assays: Analytical Validation for Application in Residual Disease Detection and Treatment Monitoring. Clin. Chem. 2021, 67, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Flamini, E.; Mercatali, L.; Nanni, O.; Calistri, D.; Nunziatini, R.; Zoli, W.; Rosetti, P.; Gardini, N.; Lattuneddu, A.; Verdecchia, G.M.; et al. Free DNA and carcinoembryonic antigen serum levels: An important combination for diagnosis of colorectal cancer. Clin. Cancer Res. 2006, 12, 6985–6988. [Google Scholar] [CrossRef] [Green Version]
- Czeiger, D.; Shaked, G.; Eini, H.; Vered, I.; Belochitski, O.; Avriel, A.; Ariad, S.; Douvdevani, A. Measurement of circulating cell-free DNA levels by a new simple fluorescent test in patients with primary colorectal cancer. Am. J. Clin. Pathol. 2011, 135, 264–270. [Google Scholar] [CrossRef]
- Berger, A.W.; Schwerdel, D.; Welz, H.; Marienfeld, R.; Schmidt, S.A.; Kleger, A.; Ettrich, T.J.; Seufferlein, T. Treatment monitoring in metastatic colorectal cancer patients by quantification and KRAS genotyping of circulating cell-free DNA. PLoS ONE 2017, 12, e0174308. [Google Scholar] [CrossRef]
- Garlan, F.; Laurent-Puig, P.; Sefrioui, D.; Siauve, N.; Didelot, A.; Sarafan-Vasseur, N.; Michel, P.; Perkins, G.; Mulot, C.; Blons, H.; et al. Early Evaluation of Circulating Tumor DNA as Marker of Therapeutic Efficacy in Metastatic Colorectal Cancer Patients (PLACOL Study). Clin. Cancer Res. 2017, 23, 5416–5425. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Ulrich, B.C.; Supplee, J.; Kuang, Y.; Lizotte, P.H.; Feeney, N.B.; Guibert, N.M.; Awad, M.M.; Wong, K.-K.; Jänne, P.A.; et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin. Cancer Res. 2018, 24, 4437–4443. [Google Scholar] [CrossRef] [Green Version]
- Konishi, T.; Shimada, Y.; Hsu, M.; Tufts, L.; Jimenez-Rodriguez, R.; Cercek, A.; Yaeger, R.; Saltz, L.; Smith, J.J.; Nash, G.M.; et al. Association of Preoperative and Postoperative Serum Carcinoembryonic Antigen and Colon Cancer Outcome. JAMA Oncol. 2018, 4, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, C.H. Understanding The CEA Test in Colon Cancer. Available online: https://news.cancerconnect.com/colon-cancer/understanding-the-cea-test-in-colon-cancer-2yuVIMszUkideV0m-kWBwA (accessed on 19 March 2021).
- Larribère, L.; Martens, U.M. Advantages and Challenges of Using ctDNA NGS to Assess the Presence of Minimal Residual Disease (MRD) in Solid Tumors. Cancers 2021, 13, 5698. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, L.; Cohen, J.D.; Kinde, I.; Ptak, J.; Popoli, M.; Schaefer, J.; Silliman, N.; Dobbyn, L.; Tie, J.; et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncol. 2019, 5, 1118–1123. [Google Scholar] [CrossRef]
- Jeffery, M.; Hickey, B.E.; Hider, P.N.; See, A.M. Follow-up strategies for patients treated for non-metastatic colorectal cancer. Cochrane Database Syst. Rev. 2016, 11, CD002200. [Google Scholar] [CrossRef]
- Nicholson, B.D.; Shinkins, B.; Pathiraja, I.; Roberts, N.W.; James, T.J.; Mallett, S.; Perera, R.; Primrose, J.N.; Mant, D. Blood CEA levels for detecting recurrent colorectal cancer. Cochrane Database Syst. Rev. 2015, 12, CD011134. [Google Scholar] [CrossRef] [Green Version]
- Park, G.; Park, J.K.; Son, D.-S.; Shin, S.-H.; Kim, Y.J.; Jeon, H.-J.; Lee, J.; Park, W.-Y.; Lee, K.H.; Park, D. Utility of targeted deep sequencing for detecting circulating tumor DNA in pancreatic cancer patients. Sci. Rep. 2018, 8, 11631. [Google Scholar] [CrossRef]
- Murahashi, S.; Akiyoshi, T.; Sano, T.; Fukunaga, Y.; Noda, T.; Ueno, M.; Zembutsu, H. Serial circulating tumour DNA analysis for locally advanced rectal cancer treated with preoperative therapy: Prediction of pathological response and postoperative recurrence. Br. J. Cancer 2020, 123, 803–810. [Google Scholar] [CrossRef]
- COSMIC Cancer Browser: Large Intestine—Carcinoma—Adenocarcinoma Genes. Available online: https://cancer.sanger.ac.uk/cosmic/browse/tissue?wgs=off&sn=large_intestine&ss=all&hn=carcinoma&sh=adenocarcinoma&in=t&src=tissue&all_data=n (accessed on 17 January 2021).
- van Cutsem, E.; Cervantes, A.; Adam, R.; Sobrero, A.; van Krieken, J.H.; Aderka, D.; Aranda Aguilar, E.; Bardelli, A.; Benson, A.; Bodoky, G.; et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016, 27, 1386–1422. [Google Scholar] [CrossRef]
- Huang, X.; Gao, P.; Song, Y.; Sun, J.; Chen, X.; Zhao, J.; Liu, J.; Xu, H.; Wang, Z. Relationship between circulating tumor cells and tumor response in colorectal cancer patients treated with chemotherapy: A meta-analysis. BMC Cancer 2014, 14, 976. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, S.; Zhang, W.; Wang, J.; Wang, M.; Hu, X.; Liu, F.; Zhang, Y.; Jiang, B.; Yuan, H. Circulating tumor cells as an independent prognostic factor in advanced colorectal cancer: A retrospective study in 121 patients. Int. J. Colorectal Dis. 2019, 34, 589–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Li, W.; Wang, K.; Xu, C.; Hao, M.; Ding, L. Perspectives of the Application of Liquid Biopsy in Colorectal Cancer. Biomed Res. Int. 2020, 2020, 6843180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacante, M.; Ciuni, R.; Basile, F.; Biondi, A. The Liquid Biopsy in the Management of Colorectal Cancer: An Overview. Biomedicines 2020, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Lambros, M.B.; Gil, V.S.; Crespo, M.; Fontes, M.S.; Neves, R.N.; Mahra, N.; Fowler, G.; Ebbs, B.; Flohr, P.; Seed, G.; et al. Abstract 993: Diagnostic leukapheresis (DLA): Molecular characterisation and organoid culture of circulating tumor cells (CTC) from metastatic castration resistant prostate cancer (mCRPC). Cancer Res. 2017, 77 (Suppl. 13). [Google Scholar] [CrossRef]
- Schou, J.V.; Rossi, S.; Jensen, B.V.; Nielsen, D.L.; Pfeiffer, P.; Høgdall, E.; Yilmaz, M.; Tejpar, S.; Delorenzi, M.; Kruhøffer, M.; et al. miR-345 in metastatic colorectal cancer: A non-invasive biomarker for clinical outcome in non-KRAS mutant patients treated with 3rd line cetuximab and irinotecan. PLoS ONE 2014, 9, e99886. [Google Scholar] [CrossRef]
- Tsukamoto, M.; Iinuma, H.; Yagi, T.; Matsuda, K.; Hashiguchi, Y. Circulating Exosomal MicroRNA-21 as a Biomarker in Each Tumor Stage of Colorectal Cancer. Oncology 2017, 92, 360–370. [Google Scholar] [CrossRef]
- Fu, F.; Jiang, W.; Zhou, L.; Chen, Z. Circulating Exosomal miR-17-5p and miR-92a-3p Predict Pathologic Stage and Grade of Colorectal Cancer. Transl. Oncol. 2018, 11, 221–232. [Google Scholar] [CrossRef]
- Jin, G.; Liu, Y.; Zhang, J.; Bian, Z.; Yao, S.; Fei, B.; Zhou, L.; Yin, Y.; Huang, Z. A panel of serum exosomal microRNAs as predictive markers for chemoresistance in advanced colorectal cancer. Cancer Chemother. Pharmacol. 2019, 84, 315–325. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hallermayr, A.; Steinke-Lange, V.; Vogelsang, H.; Rentsch, M.; de Wit, M.; Haberl, C.; Holinski-Feder, E.; Pickl, J.M.A. Clinical Validity of Circulating Tumor DNA as Prognostic and Predictive Marker for Personalized Colorectal Cancer Patient Management. Cancers 2022, 14, 851. https://doi.org/10.3390/cancers14030851
Hallermayr A, Steinke-Lange V, Vogelsang H, Rentsch M, de Wit M, Haberl C, Holinski-Feder E, Pickl JMA. Clinical Validity of Circulating Tumor DNA as Prognostic and Predictive Marker for Personalized Colorectal Cancer Patient Management. Cancers. 2022; 14(3):851. https://doi.org/10.3390/cancers14030851
Chicago/Turabian StyleHallermayr, Ariane, Verena Steinke-Lange, Holger Vogelsang, Markus Rentsch, Maike de Wit, Christopher Haberl, Elke Holinski-Feder, and Julia M. A. Pickl. 2022. "Clinical Validity of Circulating Tumor DNA as Prognostic and Predictive Marker for Personalized Colorectal Cancer Patient Management" Cancers 14, no. 3: 851. https://doi.org/10.3390/cancers14030851
APA StyleHallermayr, A., Steinke-Lange, V., Vogelsang, H., Rentsch, M., de Wit, M., Haberl, C., Holinski-Feder, E., & Pickl, J. M. A. (2022). Clinical Validity of Circulating Tumor DNA as Prognostic and Predictive Marker for Personalized Colorectal Cancer Patient Management. Cancers, 14(3), 851. https://doi.org/10.3390/cancers14030851