
Glioblastoma Microenvironment and Cellular Interactions

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Interactions between Glioblastoma Cells and Tumor Microenvironment
2.1. Cell-to-Cell Communication
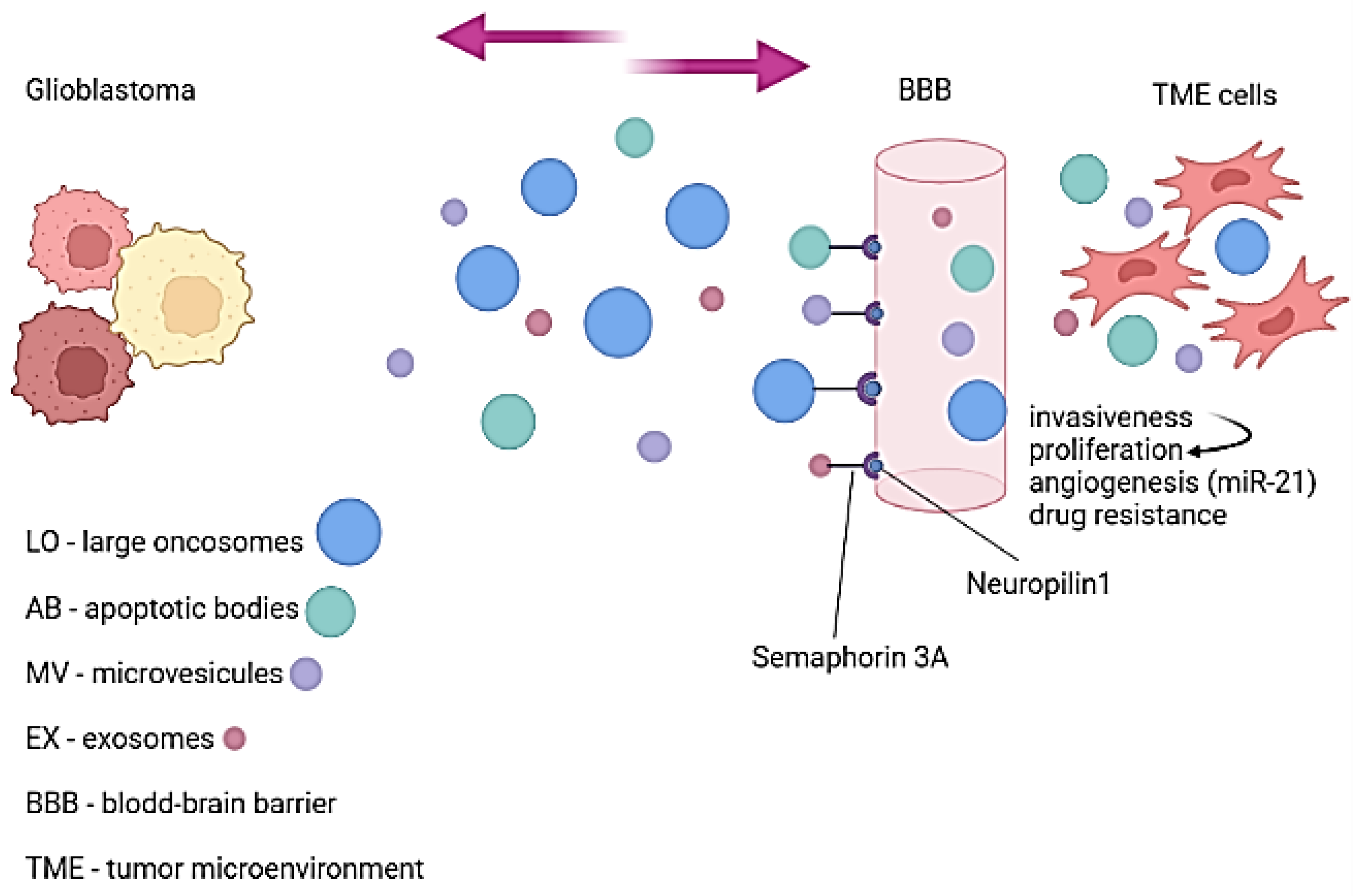
2.1.1. Extracellular Vesicles—Carriers Passing through the Blood–Brain Barrier
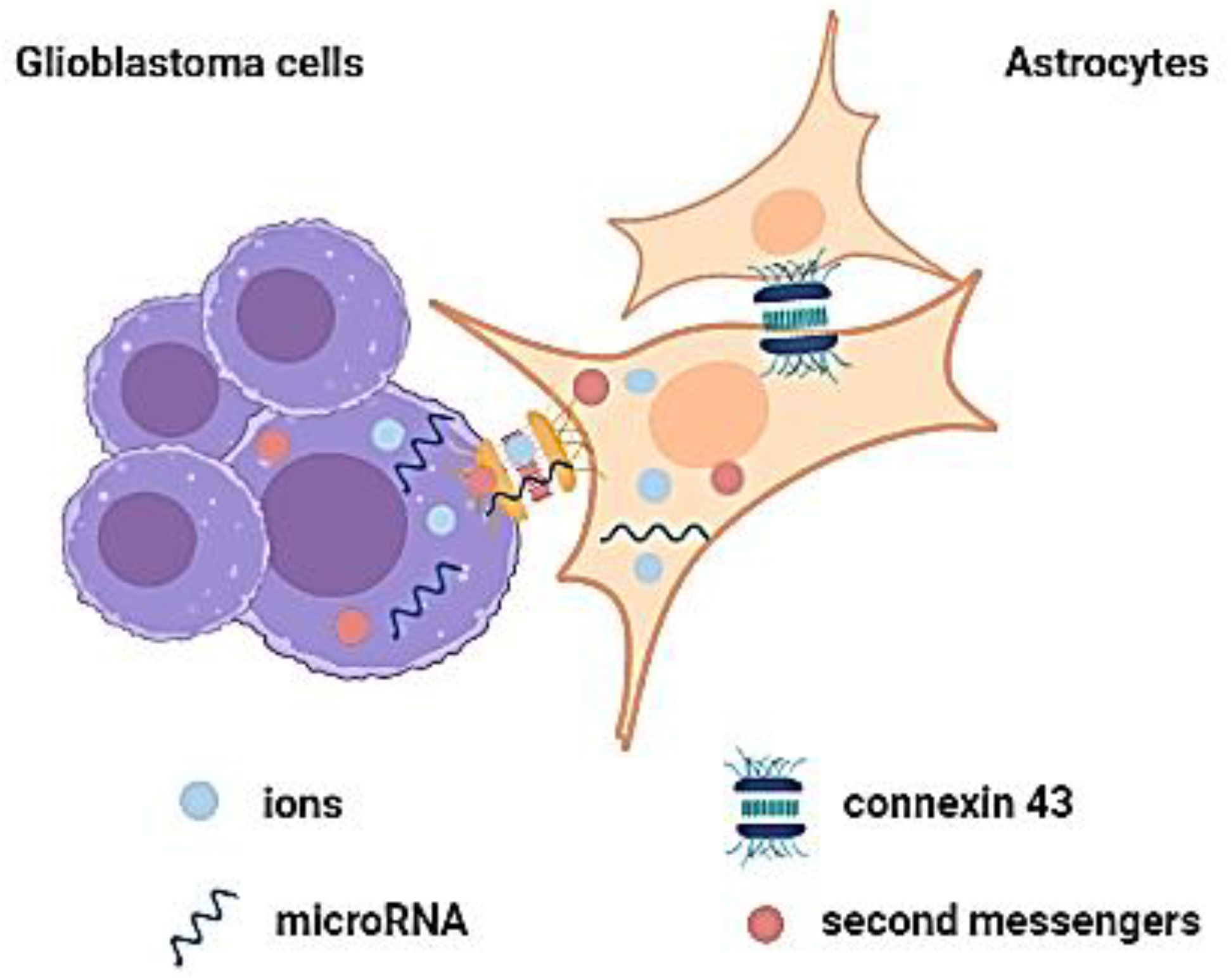
2.1.2. Gap Junctions and Their Role in Cell Communication
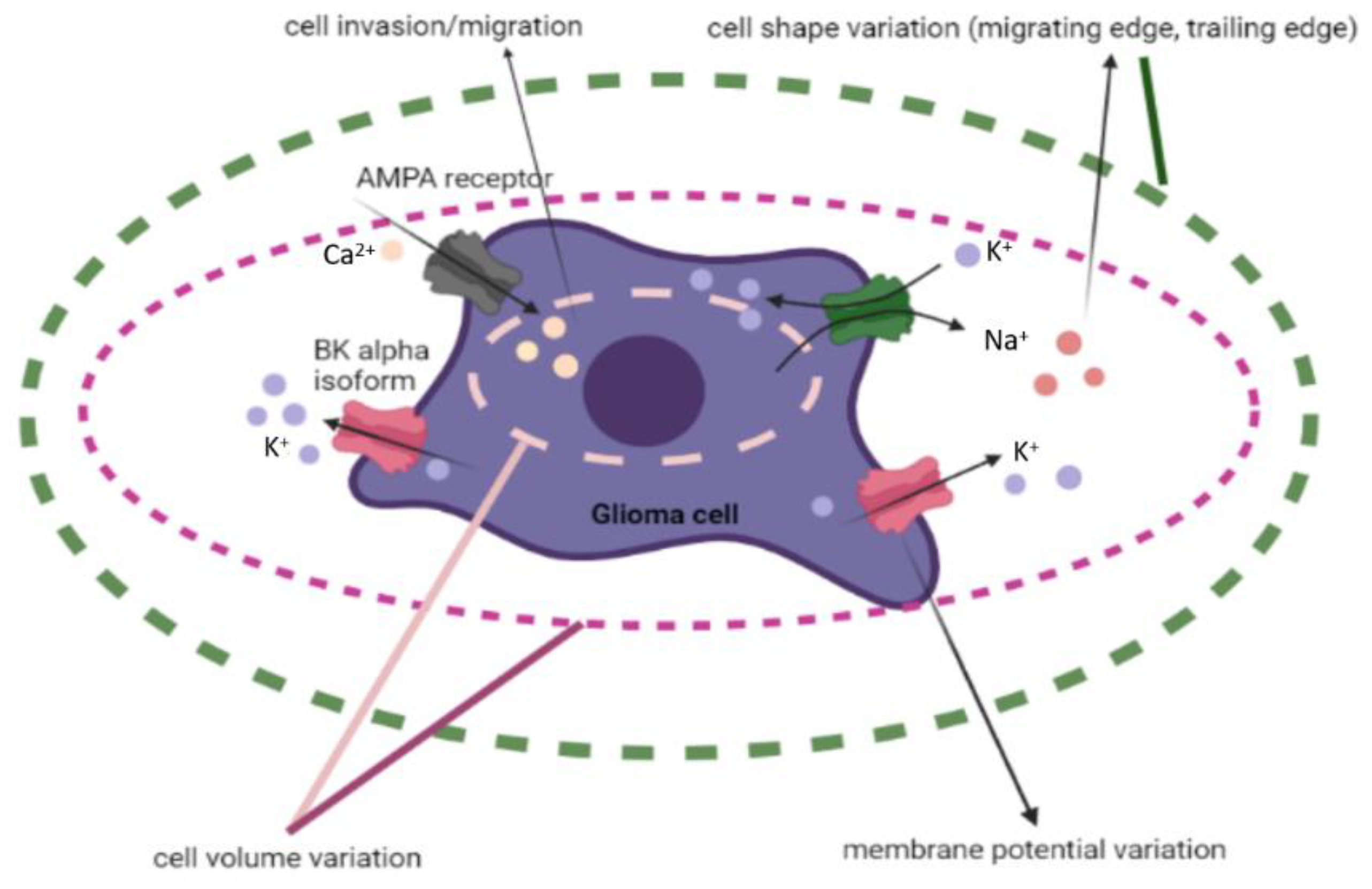
2.1.3. Ion Channels and Transporters—Influencing GBM Cell Communication, Polarization, Shape, and Size
2.2. Dynamic Shape-Shifting Cellular Process Influencing GBM Characteristics
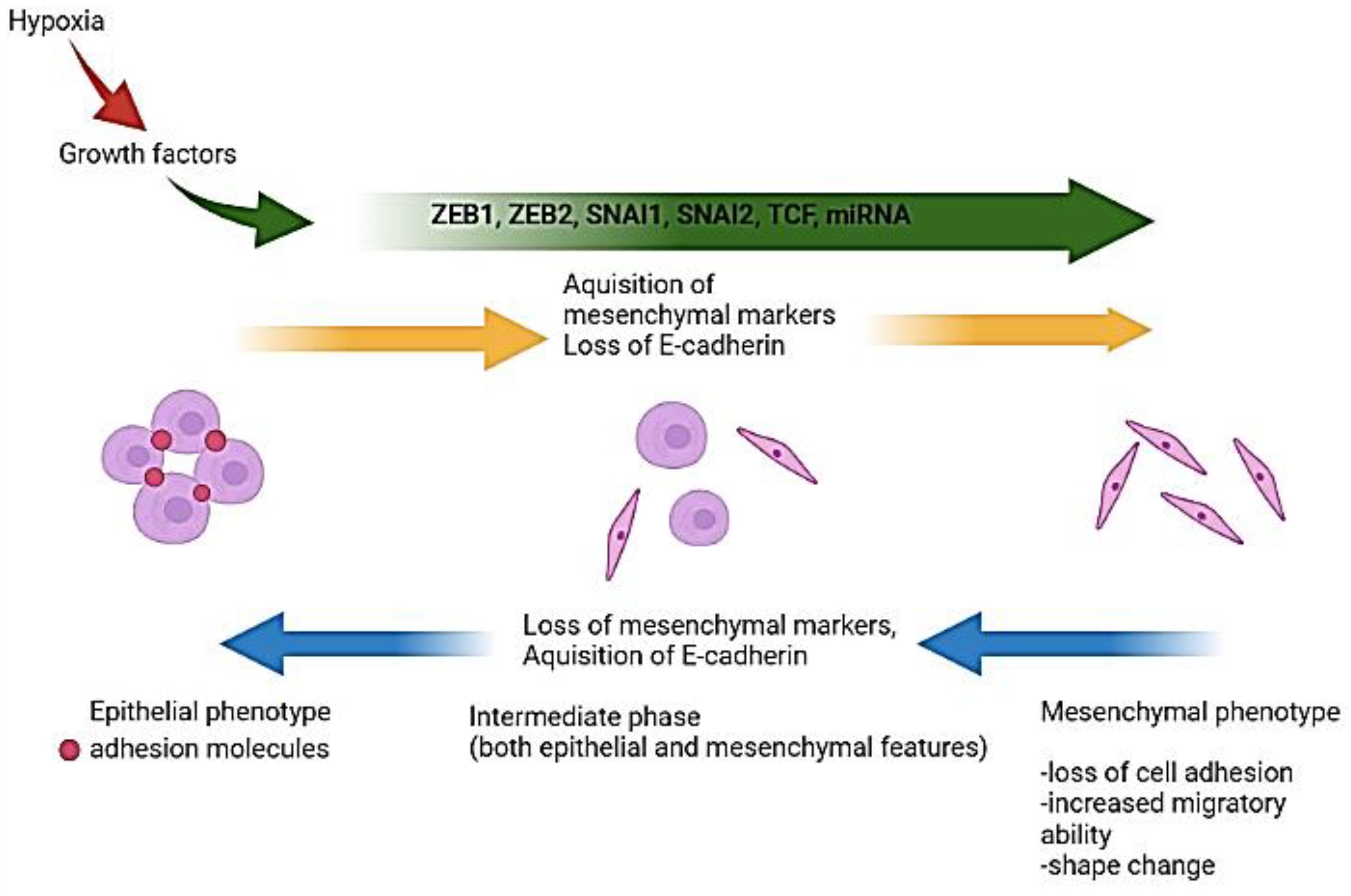
Epithelial-to-Mesenchymal Transition
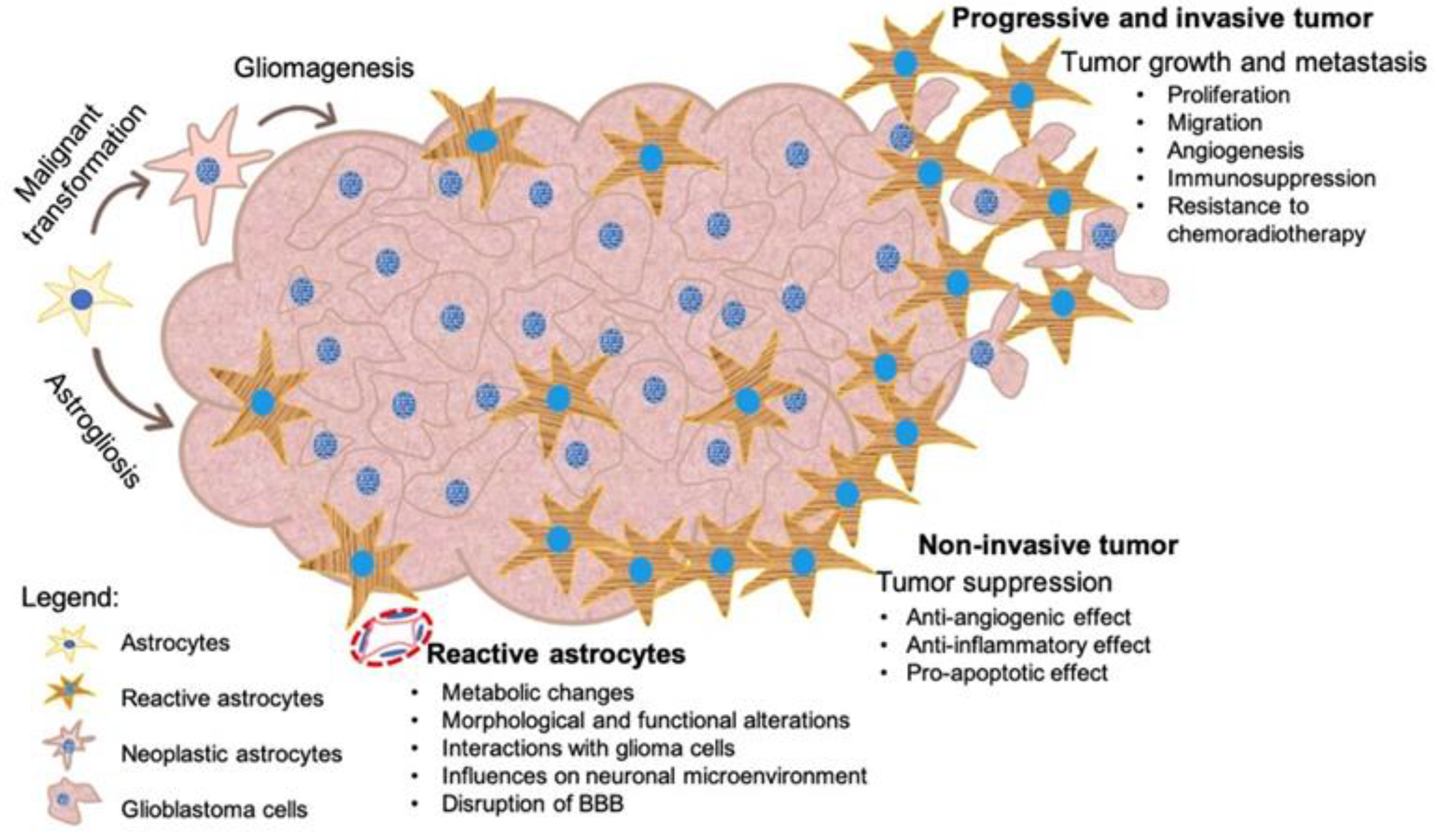
3. Tumoral and Reactive Astrocytes
4. Glioma Stem Cells
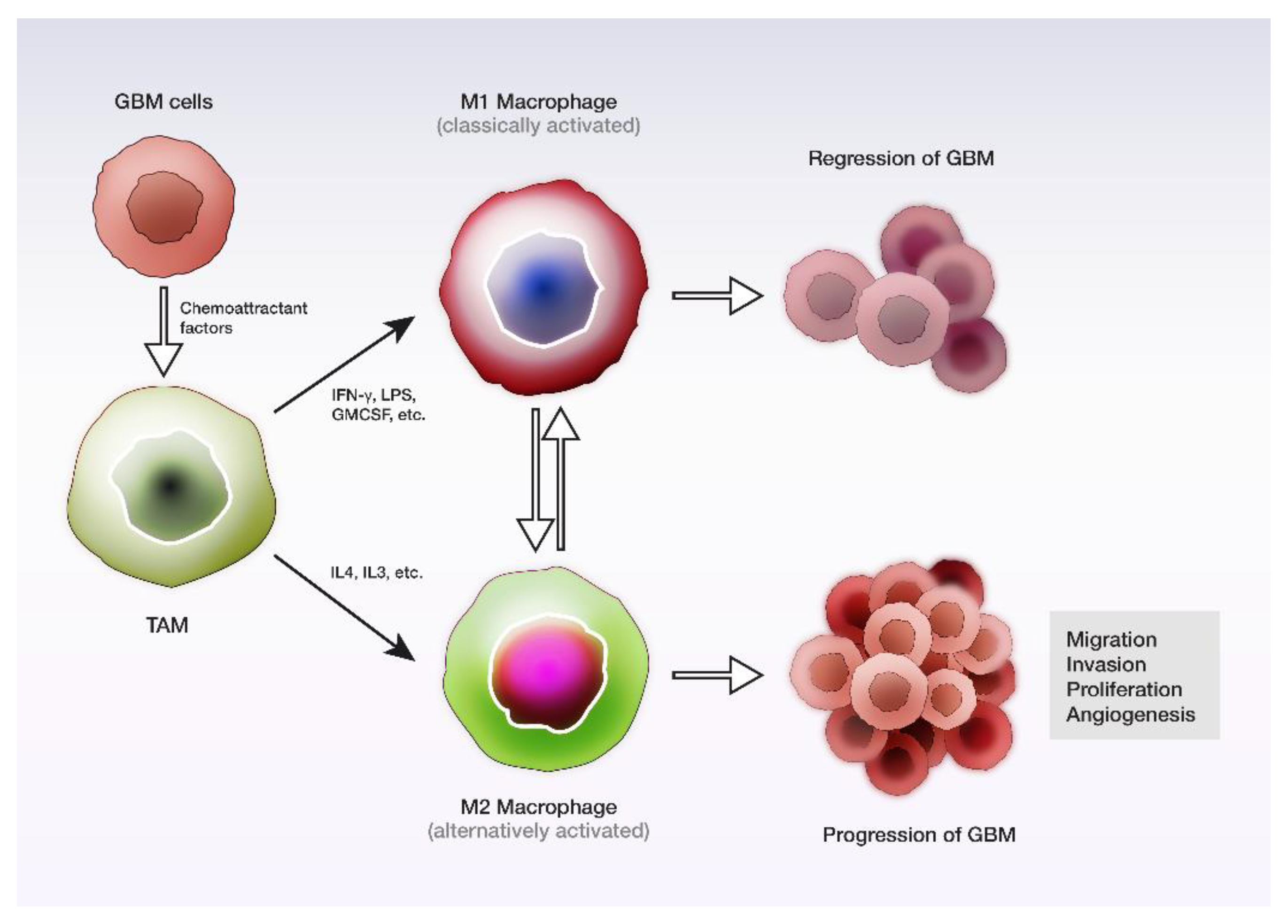
5. Tumor Microenvironment and Tumor-Associated Macrophages
6. Other Immune Cells in Tumor Microenvironment and New Techniques for Cellular Mapping
6.1. Aspects of Neutrophils’ Involvement in GBM
6.2. Aspects of Dendritic Cells’ Involvement in GBM
6.3. Aspects of Lymphoid Cells’ Involvement in GBM—T Lymphocytes and NK Cells
7. Cell-Targeting Therapies
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Ho, M.S.; Verkhratsky, A.; Duan, S.; Parpura, V. Editorial: Glia in Health and Disease. Front. Mol. Neurosci. 2019, 12, 63. [Google Scholar] [CrossRef] [Green Version]
- Zülch, K.J. (Ed.) Histologic Typing of Tumours of the Central Nervous System; World Health Organization: Geneva, Switzerland, 1979.
- Kleihues, P.; Burger, P.C.; Scheithauer, B.W. (Eds.) Histological typing of tumours of the central nervous system. In World Health Organization International Histological Classification of Tumours, 2nd ed.; Springer: Berlin/Heidelberg, Germany, 1993. [Google Scholar]
- Kleihues, P.; Cavenee, W.K. (Eds.) World Health Organisation Classification of Tumours: Pathology and Genetics of Tumours of the Nervous System; IARC Press: Lyon, France, 2000. [Google Scholar]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System, 4th ed.; IARC Press: Lyon, France, 2007. [Google Scholar]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System, Revise, 4th ed.; IARC Press: Lyon, France, 2016. [Google Scholar]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Virtuoso, A.; Giovannoni, R.; De Luca, C.; Gargano, F.; Cerasuolo, M.; Maggio, N.; Lavitrano, M.; Papa, M. The Glioblastoma Microenvironment: Morphology, Metabolism, and Molecular Signature of Glial Dynamics to Discover Metabolic Rewiring Sequence. Int. J. Mol. Sci. 2021, 22, 3301. [Google Scholar] [CrossRef]
- Qiu, R.; Zhong, Y.; Li, Q.; Li, Y.; Fan, H. Metabolic Remodeling in Glioma Immune Microenvironment: Intercellular Interactions Distinct from Peripheral Tumors. Front. Cell Dev. Biol. 2021, 9, 693215. [Google Scholar] [CrossRef]
- Mostofa, A.G.; Punganuru, S.R.; Madala, H.R.; Al-Obaide, M.; Srivenugopal, K.S. The Process and Regulatory Components of Inflammation in Brain Oncogenesis. Biomolecules 2017, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Haksoyler, V.; Besen, A.A.; Koseci, T.; Olgun, P.; Bayram, E.; Topkan, E. Neutrophil-to-lymphocyte ratio is prognostic in recurrent glioblastoma multiforme treated with bevacizumab plus irinotecan. Biomark. Med. 2021, 15, 851–859. [Google Scholar] [CrossRef]
- Ali, S.; Borin, T.F.; Piranlioglu, R.; Ara, R.; Lebedyeva, I.; Angara, K.; Achyut, B.R.; Arbab, A.S.; Rashid, M.H. Changes in the tumor microenvironment and outcome for TME-targeting therapy in glioblastoma: A pilot study. PLoS ONE 2021, 16, e0246646. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Y.; Cui, B.; Liu, Z.; Shen, H. Novel insights into astrocyte-mediated signaling of proliferation, invasion and tumor immune microenvironment in glioblastoma. Biomed. Pharmacother. 2020, 126, 110086. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebraihimi-Kalan, A.; JAymad, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.; Rao, A.; Braganhol, E.; Whiteside, T.L. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neurooncol. Adv. 2020, 2, vdaa056. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Maresh, G.; Zhang, X.; Salomon, C.; Hooper, J.; Margolin, D.; Li, L. The Emerging Roles of Extracellular Vesicles As Communication Vehicles within the Tumor Microenvironment and Beyond. Front. Endocrinol. 2017, 8, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Zivkovic, A.M.; Zocco, D.; Zuba-Surma, E.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Lam, E.W.; Sun, Y. Extracellular vesicles in the tumor microenvironment: Old stories, but new tales. Mol. Cancer 2019, 18, 59. [Google Scholar] [CrossRef] [Green Version]
- André-Grégoire, G.; Gavard, J. Spitting out the demons: Extracellular vesicles in glioblastoma. Cell Adh. Migr. 2017, 11, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumor growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef] [Green Version]
- Treps, L.; Edmond, S.; Harford-Wright, E.; Galan-Moya, E.M.; Schmitt, A.; Azzi, S.; Citerne, A.; Bidère, N.; Ricard, D.; Gavard, J. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 2016, 35, 2615–2623. [Google Scholar] [CrossRef]
- Simon, T.; Pinioti, S.; Schellenberger, P.; Rajeeve, V.; Wendler, F.; Cutillas, P.R.; King, A.; Stebbing, J.; Giamas, G. Shedding of bevacizumab in tumour cells-derived extracellular vesicles as a new therapeutic escape mechanism in glioblastoma. Mol. Cancer 2018, 17, 132. [Google Scholar] [CrossRef]
- Mondal, A.; Kumari Singh, D.; Panda, S.; Shiras, A. Extracellular Vesicles as Modulators of Tumor Microenvironment and Disease Progression in Glioma. Front. Oncol. 2017, 7, 144. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nareshkumar, R.N.; Sulochana, K.N.; Coral, K. Inhibition of angiogenesis in endothelial cells by Human Lysyl oxidase propeptide. Sci. Rep. 2018, 8, 10426. [Google Scholar] [CrossRef] [PubMed]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 6809105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Ma, X.; Wang, J.; Zhao, Y.; Wang, Y.; Bihl, J.C.; Chen, Y.; Jiang, C. Glioma stem cells-derived exosomes promote the angiogenic ability of endothelial cells through miR-21/VEGF signal. Oncotarget 2017, 8, 36137–36148. [Google Scholar] [CrossRef] [Green Version]
- Bronisz, A.; Wang, Y.; Nowicki, M.O.; Peruzzi, P.; Ansari, K.; Ogawa, D.; Balaj, L.; De Rienzo, G.; Mineo, M.; Nakano, I.; et al. Extracellular vesicles modulate the glioblastoma microenvironment via a tumor suppression signaling network directed by miR-1. Cancer Res. 2014, 74, 738–750. [Google Scholar] [CrossRef] [Green Version]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringnér, M.; Mörgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Svensson, K.J.; Kucharzewska, P.; Christianson, H.C.; Sköld, S.; Löfstedt, T.; Johansson, M.C.; Mörgelin, M.; Bengzon, J.; Ruf, W.; Belting, M. Hypoxia triggers a proangiogenic pathway involving cancer cell microvesicles and PAR-2-mediated heparin-binding EGF signaling in endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13147–13152. [Google Scholar] [CrossRef] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4, eaar2766. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.-c.; Wang, L.-l.; Zhang, X.-d.; Xu, J.-i.; Li, P.-f.; Liang, H.; Zhang, X.-b.; Xie, L.; Zhou, Z.-h.; Yang, J.; et al. The relationship between expression of PD-L1 and HIF-1α in glioma cells under hypoxia. J. Hematol. Oncol. 2021, 14, 92. [Google Scholar] [CrossRef] [PubMed]
- Noronha, C.; Ribeiro, A.S.; Taipa, R.; Castro, D.S.; Reis, J.; Faria, C.; Paredes, J. Cadherin Expression and EMT: A Focus on Gliomas. Biomedicines 2021, 9, 1328. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive astrocytes in glioblastoma multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Vanhaecke, T.; Papeleu, P.; Snykers, S.; Henkens, T.; Rogiers, V. Connexins and their channels in cell growth and cell death. Cell. Signal. 2006, 18, 592–600. [Google Scholar] [CrossRef]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Kar, R.; Batra, N.; Riquelme, M.A.; Jiang, J.X. Biological role of connexin intercellular channels and hemichannels. Arch. Biochem. Biophys. 2012, 524, 2–15. [Google Scholar] [CrossRef] [Green Version]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front. Oncol. 2018, 8, 646. [Google Scholar] [CrossRef] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstman, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro-Oncology 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia 2001, 33, 107–117. [Google Scholar] [CrossRef]
- Pu, P.; Xia, Z.; Yu, S.; Huang, Q. Altered expression of Cx43 in astrocytic tumors. Clin. Neurol. Neurosurg. 2004, 107, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Lund-Johansen, M.; Bjervig, R.; Humphrey, P.A.; Bigner, S.H.; Bigner, D.D.; Laerum, O.D. Effect of epidermal growth factor on glioma cell growth, migration and invasion in vitro. Cancer Res. 1990, 50, 6039–6044. [Google Scholar] [PubMed]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 15, 9188–9196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potthoff, A.L.; Heiland, D.H.; Evert, B.O.; Almeida, F.R.; Behringer, S.P.; Dolf, A.; Güresir, Á.; Güresir, E.; Joseph, K.; Pietsch, T.; et al. Inhibition of Gap Junctions Sensitizes Primary Glioblastoma Cells for Temozolomide. Cancers 2019, 11, 858. [Google Scholar] [CrossRef] [Green Version]
- Dermietzel, R.; Spray, D.C. Gap junctions in the brain: Where, what type, how many and why? Trends Neurosci. 1993, 16, 186–192. [Google Scholar] [CrossRef]
- Zhang, W.; Couldwell, W.T.; Simard, M.F.; Song, H.; Lin, J.H.; Nedergaard, M. Direct gap junction communication between malignant glioma cells and astrocytes. Cancer Res. 1999, 59, 1994–2003. [Google Scholar]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Steinhäuser, C. Ion channels in glial cells. Brain Res. Rev. 2000, 32, 380–412. [Google Scholar] [CrossRef]
- Molenaar, R.J. Ion channels in glioblastoma. ISRN Neurol. 2011, 2011, 590249. [Google Scholar] [CrossRef] [Green Version]
- Ransom, C.B.; Sontheimer, H. BK channels in human glioma cells. J. Neurophysiol. 2001, 85, 790–803, Erratum in J. Neurophysiol. 2001, 85, 4. [Google Scholar] [CrossRef] [Green Version]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef]
- Masi, A.; Becchetti, A.; Restano-Cassulini, R.; Polvani, S.; Hofmann, G.; Buccoliero, A.M.; Paglierani, M.; Pollo, B.; Taddei, G.L.; Gallina, P.; et al. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br. J. Cancer 2005, 93, 781–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godard, S.; Getz, G.; Delorenzi, M.; Farmer, P.; Kobayashi, H.; Desbaillets, I.; Nozaki, M.; Diserens, A.C.; Hamou, M.F.; Dietrich, P.Y.; et al. Classification of human astrocytic gliomas on the basis of gene expression: A correlated group of genes with angiogenic activity emerges as a strong predictor of subtypes. Cancer Res. 2003, 63, 6613–6625. [Google Scholar] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.D.; Parsons, D.W.; Velculescu, V.E.; Riggins, G.J. Sodium ion channel mutations in glioblastoma patients correlate with shorter survival. Mol. Cancer 2011, 10, 17. [Google Scholar] [CrossRef] [Green Version]
- Iser, I.C.; Lenz, G.; Wink, M.R. EMT-like process in glioblastomas and reactive astrocytes. Neurochem. Int. 2019, 122, 139–143. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Kinker, G.S.; Rodman, C.; et al. Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 2021, 39, 779–792.e11. [Google Scholar] [CrossRef]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: New insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Pez, F.; Dayan, F.; Durivault, J.; Kaniewski, B.; Aimond, G.; Le Provost, G.S.; Deux, B.; Clézardin, P.; Sommer, P.; Pouysségur, J.; et al. The HIF-1-inducible lysyl oxidase activates HIF-1 via the Akt pathway in a positive regulation loop and synergizes with HIF-1 in promoting tumor cell growth. Cancer Res. 2011, 71, 1647–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signaling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierie, B.; Moses, H.L. Tumor microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Ye, X.Z.; Xu, S.L.; Xin, Y.H.; Yu, S.C.; Ping, Y.F.; Chen, L.; Xiao, H.L.; Wang, B.; Yi, L.; Wang, Q.L.; et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Piao, Y.; Liang, J.; Holmes, L.; Zurita, A.J.; Henry, V.; Heymach, J.V.; de Groot, J.F. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro-Oncology 2012, 14, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, L.A.; Gutman, D.A.; Chisolm, C.; Appin, C.; Kong, J.; Rong, Y.; Kurc, T.; Van Meir, E.G.; Saltz, J.H.; Moreno, C.S.; et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. Am. J. Pathol. 2012, 180, 2108–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.; Hartmann, R.; Schröter, F.; Suwala, A.K.; Maciaczyk, D.; Krüger, A.C.; Willbold, D.; Kahlert, U.D.; Maciaczyk, J. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 2016, 7, 73414–73431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Canc. 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Hulpiau, P.; van Roy, F. Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol. 2009, 41, 349–369. [Google Scholar] [CrossRef]
- Majc, B.; Sever, T.; Zarić, M.; Breznik, B.; Turk, B.; Lah, T.T. Epithelial-to-mesenchymal transition as the driver of changing carcinoma and glioblastoma microenvironment. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118782. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Iser, I.C.; Pereira, M.B.; Lenz, G.; Wink, M.R. The Epithelial-to-Mesenchymal Transition-Like Process in Glioblastoma: An Updated Systematic Review and In Silico Investigation. Med. Res. Rev. 2017, 37, 271–313. [Google Scholar] [CrossRef]
- Iser, I.C.; Ceschini, S.M.; Onzi, G.R.; Bertoni, A.P.; Lenz, G.; Wink, M.R. Conditioned Medium from Adipose-Derived Stem Cells (ADSCs) Promotes Epithelial-to-Mesenchymal-Like Transition (EMT-Like) in Glioma Cells In vitro. Mol. Neurobiol. 2016, 53, 7184–7199. [Google Scholar] [CrossRef]
- Shimoyama, Y.; Tsujimoto, G.; Kitajima, M.; Natori, M. Identification of three human type-II classic cadherins and frequent heterophilic interactions between different subclasses of type-II classic cadherins. Biochem. J. 2000, 349, 159–167. [Google Scholar] [CrossRef]
- Noh, M.G.; Oh, S.J.; Ahn, E.J.; Kim, Y.J.; Jung, T.Y.; Jung, S.; Kim, K.K.; Lee, J.H.; Lee, K.H.; Moon, K.S. Prognostic significance of E-cadherin and N-cadherin expression in Gliomas. BMC Cancer 2017, 17, 583. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Tian, Y.; Wan, H.; Ma, J.; Song, Y.; Wang, Y.; Zhang, L. Expression of beta-catenin and E- and N-cadherin in human brainstem gliomas and clinicopathological correlations. Int. J. Neurosci. 2013, 123, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef] [PubMed]
- Camand, E.; Peglion, F.; Osmani, N.; Sanson, M.; Etienne-Manneville, S. N-cadherin expression level modulates integrin-mediated polarity and strongly impacts on the speed and directionality of glial cell migration. J. Cell Sci. 2012, 125, 844–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, K.; Duntsch, C.D.; Zhou, Q.; Weimar, J.D.; Bordelon, D.; Robertson, J.H.; Pourmotabbed, T. Correlation of N-cadherin expression in high grade gliomas with tissue invasion. J. Neurooncol. 2004, 70, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Utsuki, S.; Oka, H.; Miyajima, Y.; Kijima, C.; Yasui, Y.; Fujii, K. Adult cerebellar glioblastoma cases have different characteristics from supratentorial glioblastoma. Brain Tumor Pathol. 2012, 29, 87–95. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-associated reactive astrocytes aid the evolution of immunosuppressive environment in glioblastoma. Nat. Commun. 2019, 10, 2541. [Google Scholar] [CrossRef] [Green Version]
- Zong, H.; Verhaak, R.G.; Canoll, P. The cellular origin for malignant glioma and prospects for clinical advancements. Expert Rev. Mol. Diagn. 2012, 12, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katz, A.M.; Amankulor, N.M.; Pitter, K.; Helmy, K.; Squatrito, M.; Holland, E.C. Astrocyte-specific expression patterns associated with the PDGF-induced glioma microenvironment. PLoS ONE 2012, 7, e32453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial Molecular Mechanisms and Microenvironmental Factors. Front. Cell Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irvin, D.M.; McNeill, R.S.; Bash, R.E.; Miller, C.R. Intrinsic Astrocyte Heterogeneity Influences Tumor Growth in Glioma Mouse Models. Brain Pathol. 2017, 27, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmigiani, E.; Scalera, M.; Mori, E.; Tantillo, E.; Vannini, E. Old Stars and New Players in the Brain Tumor Microenvironment. Front. Cell Neurosci. 2021, 15, 709917. [Google Scholar] [CrossRef]
- Gril, B.; Palmieri, D.; Qian, Y.; Anwar, T.; Liewehr, D.J.; Steinberg, S.M.; Andreu, Z.; Masana, D.; Fernandez, P.; Steeg, P.S.; et al. Pazopanib inhibits the activation of PDGFRβ-expressing astrocytes in the brain metastatic microenvironment of breast cancer cells. Am. J. Pathol. 2013, 182, 2368–2379. [Google Scholar] [CrossRef] [Green Version]
- Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Na’ara, S.; Gil, Z. Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial- derived neurotrophic factor. Int. J. Cancer 2015, 137, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Rempel, S.A.; Golembieski, W.A.; Ge, S.; Lemke, N.; Elisevich, K.; Mikkelsen, T.; Gutiérrez, J.A. SPARC: A signal of astrocytic neoplastic transformation and reactive response in human primary and xenograft gliomas. J. Neuropathol. Exp. Neurol. 1998, 57, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Laird, M.D.; Vender, J.R.; Dhandapani, K.M. Opposing roles for reactive astrocytes following traumatic brain injury. Neurosignals 2008, 16, 154–164. [Google Scholar] [CrossRef]
- Buffo, A.; Rite, I.; Tripathi, P.; Lepier, A.; Colak, D.; Horn, A.P.; Mori, T.; Götz, M. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc. Natl. Acad. Sci. USA 2008, 105, 3581–3586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robel, S.; Berninger, B.; Götz, M. The stem cell potential of glia: Lessons from reactive gliosis. Nat. Rev. Neurosci. 2011, 12, 88–104. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.D.; Doan, N.B.; Imura, T.; Bush, T.G.; Sofroniew, M.V. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat. Neurosci. 2004, 7, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, B.; Liu, H.L.; Liu, R.; Feng, G.D.; Jiao, X.Y.; Ju, G. Astrocytes in injured adult rat spinal cord may acquire the potential of neural stem cells. Neuroscience 2004, 128, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Wisniewski, P.; Kijewska, M.; Gajdanowicz, P.; Pszczolkowska, D.; Przanowski, P.; Dabrowski, M.; Maleszewska, M.; Kaminska, B. Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene 2016, 35, 6366–6377. [Google Scholar] [CrossRef]
- Turkowski, K.; Brandenburg, S.; Mueller, A.; Kremenetskaia, I.; Bungert, A.D.; Blank, A.; Felsenstein, M.; Vajkoczy, P. VEGF as a modulator of the innate immune response in glioblastoma. Glia 2018, 66, 161–174. [Google Scholar] [CrossRef]
- Laudati, E.; Currò, D.; Navarra, P.; Lisi, L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 2017, 108, 100–108. [Google Scholar] [CrossRef]
- Rickert, U.; Grampp, S.; Wilms, H.; Spreu, J.; Knerlich-Lukoschus, F.; Held-Feindt, J.; Lucius, R. Glial cell line-derived neurotrophic factor family members reduce microglial activation via inhibiting p38MAPKs-mediated inflammatory responses. J. Neurodegener. Dis. 2014, 369468. [Google Scholar] [CrossRef]
- Kim, Y.; Jeon, H.; Othmer, H. The role of the tumor microenvironment in glioblastoma: A mathematical model. IEEE Trans. Biomed. Eng. 2017, 64, 519–527. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef] [PubMed]
- Bouzier-Sore, A.K.; Pellerin, L. Unraveling the complex metabolic nature of astrocytes. Front. Cell. Neurosci. 2013, 7, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kros, J.M.; Mustafa, D.M.; Dekker, L.J.M.; Sillevis Smitt, P.A.E.; Luider, T.M.; Zheng, P.P. Circulating glioma biomarkers. Neuro-Oncology 2014, 17, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Okolie, O.; Bago, J.R.; Schmid, R.S.; Irvin, D.M.; Bash, R.E.; Miller, C.R.; Hingtgen, S.D. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro-Oncology 2016, 18, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silver, D.J.; Siebzehnrubl, F.A.; Schildts, M.J.; Yachnis, A.T.; Smith, G.M.; Smith, A.A.; Scheffler, B.; Reynolds, B.A.; Silver, J.; Steindler, D.A. Chondroitin sulfate proteoglycans potently inhibit invasion and serve as a central organizer of the brain tumor microenvironment. J. Neurosci. 2013, 33, 15603–15617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eder, K.; Kalman, B. The Dynamics of Interactions among Immune and Glioblastoma Cells. Neuromol. Med. 2015, 17, 335–352. [Google Scholar] [CrossRef]
- Kim, S.J.; Lee, H.J.; Kim, M.S.; Choi, H.J.; He, J.; Wu, Q.; Aldape, K.; Weinberg, J.S.; Yung, W.K.; Conrad, C.A.; et al. Macitentan, a Dual Endothelin Receptor Antagonist, in Combination with Temozolomide Leads to Glioblastoma Regression and Long-term Survival in Mice. Clin. Cancer Res. 2015, 21, 4630–4641. [Google Scholar] [CrossRef] [Green Version]
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Investig. 2017, 127, 3136–3151. [Google Scholar] [CrossRef]
- Imai, S.; Ikegami, D.; Yamashita, A.; Shimizu, T.; Narita, M.; Niikura, K.; Furuya, M.; Kobayashi, Y.; Miyashita, K.; Okutsu, D.; et al. Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long-lasting neuropathic pain. Brain 2013, 136, 828–843. [Google Scholar] [CrossRef] [Green Version]
- Jackson, J.G.; Robinson, M.B. Regulation of mitochondrial dynamics in astrocytes: Mechanisms, consequences, and unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef] [PubMed]
- DiNuzzo, M.; Maraviglia, B.; Giove, F. Why does the brain (not) have glycogen? Bioessays 2011, 33, 319–326. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.C.; Jackson, J.G.; Robinson, M.B. Transient Oxygen/Glucose Deprivation Causes a Delayed Loss of Mitochondria and Increases Spontaneous Calcium Signaling in Astrocytic Processes. J. Neurosci. 2016, 36, 7109–7127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blázquez, C.; Woods, A.; de Ceballos, M.L.; Carling, D.; Guzmán, M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J. Neurochem. 1999, 73, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. [Google Scholar] [CrossRef] [Green Version]
- Yip, J.; Geng, X.; Shen, J.; Ding, Y. Cerebral Gluconeogenesis and Diseases. Front. Pharmacol. 2017, 7, 521. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, T.N.; Flores, R.; Poff, A.M.; D’Agostino, D.P.; Mukherjee, P. Metabolic therapy: A new paradigm for managing malignant brain cancer. Cancer Lett. 2015, 356, 289–300. [Google Scholar] [CrossRef]
- Goldberg, G.S.; Moreno, A.P.; Lampe, P.D. Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and ATP. J. Biol. Chem. 2002, 277, 36725–36730. [Google Scholar] [CrossRef] [Green Version]
- Gagliano, N.; Costa, F.; Cossetti, C.; Pettinari, L.; Bassi, R.; Chiriva-Internati, M.; Cobos, E.; Gioia, M.; Pluchino, S. Glioma-astrocyte interaction modifies the astrocyte phenotype in a co-culture experimental model. Oncol. Rep. 2009, 22, 1349–1356. [Google Scholar] [CrossRef] [Green Version]
- Barbero, S.; Bajetto, A.; Bonavia, R.; Porcile, C.; Piccioli, P.; Pirani, P.; Ravetti, J.L.; Zona, G.; Spaziante, R.; Florio, T.; et al. Expression of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1 in human brain tumors and their involvement in glial proliferation in vitro. Ann. N. Y. Acad. Sci. 2002, 973, 60–69. [Google Scholar] [CrossRef]
- Kim, J.K.; Jin, X.; Sohn, Y.W.; Jin, X.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; De Tribolet, N.; Radovanovic, I.; i Altaba, A.R. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Biasoli, D.; Sobrinho, M.F.; da Fonseca, A.C.; de Matos, D.G.; Romão, L.; de Moraes Maciel, R.; Rehen, S.K.; Moura-Neto, V.; Borges, H.L.; Lima, F.R. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis 2014, 3, e123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.K.; Lee, K.; Radisky, D.C.; Nelson, C.M. Extracellular matrix proteins regulate epithelial-mesenchymal transition in mammary epithelial cells. Differentiation 2013, 86, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Piperi, C.; Papavassiliou, K.A.; Papavassiliou, A.G. Pivotal Role of STAT3 in Shaping Glioblastoma Immune Microenvironment. Cells 2019, 8, 1398. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Shirai, K.; Oka, K.; Mobaraki, A.; Yoshida, Y.; Noda, S.E.; Okamoto, M.; Suzuki, Y.; Itoh, J.; Itoh, H.; et al. Higher pAkt expression predicts a significant worse prognosis in glioblastomas. J. Radiat. Res. 2010, 51, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Seike, T.; Fujita, K.; Yamakawa, Y.; Kido, M.A.; Takiguchi, S.; Teramoto, N.; Iguchi, H.; Noda, M. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin. Exp. Metastasis 2011, 28, 13–25. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell. Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef]
- Le, D.M.; Besson, A.; Fogg, D.K.; Choi, K.S.; Waisman, D.M.; Goodyer, C.G.; Rewcastle, B.; Yong, V.W. Exploitation of astrocytes by glioma cells to facilitate invasiveness: A mechanism involving matrix metalloproteinase-2 and the urokinase-type plasminogen activator-plasmin cascade. J. Neurosci. 2003, 23, 4034–4043. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, G.; Deng, L.; Liu, Q.; Dai, J.; Shen, J.; Zhang, J. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol. Rep. 2010, 23, 1553–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Y.; Cheng, Y.J.; Lin, Y.P.; Lin, H.C.; Su, C.C.; Juliano, R.; Yang, B.C. Extracellular matrix of glioblastoma inhibits polarization and transmigration of T cells: The role of tenascin-C in immune suppression. J. Immunol. 2010, 185, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Yu, H.; Zhang, Y.; Zhao, D.; Lv, P.; Zhong, Y.; Xu, Y. IL-10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget 2016, 7, 71673–71685. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Alizadeh, D.; Van Handel, M.; Kortylewski, M.; Yu, H.; Badie, B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia 2009, 57, 1458–1467. [Google Scholar] [CrossRef]
- Gabrilovich, D.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Placone, A.L.; Quiñones-Hinojosa, A.; Searson, P.C. The role of astrocytes in the progression of brain cancer: Complicating the picture of the tumor microenvironment. Tumor Biol. 2016, 37, 61–69. [Google Scholar] [CrossRef]
- Mega, A.; Hartmark Nilsen, M.; Leiss, L.W.; Tobin, N.P.; Miletic, H.; Sleire, L.; Strell, C.; Nelander, S.; Krona, C.; Hägerstrand, D.; et al. Astrocytes enhance glioblastoma growth. Glia 2020, 68, 316–327. [Google Scholar] [CrossRef]
- Lin, Q.; Liu, Z.; Ling, F.; Xu, G. Astrocytes protect glioma cells from chemotherapy and upregulate survival genes via gap junctional communication. Mol. Med. Rep. 2016, 13, 1329–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ding, K.; Wang, J.; Li, X.; Zhao, P. Chemoresistance caused by the microenvironment of glioblastoma and the corresponding solutions. Biomed. Pharmacother. 2019, 109, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, B.H.; Wahba, A.; Camphausen, K.; Tofilon, P.J. Coculture with astrocytes reduces the radiosensitivity of glioblastoma stem-like cells and identifies additional targets for radiosensitization. Cancer Med. 2015, 4, 1705–1716. [Google Scholar] [CrossRef]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [Green Version]
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S.J.; Hsieh, J.K.; Manjila, S.; Lathia, J.D.; Sloan, A. The role of cancer stem cells in glioblastoma. Neurosurg. Focus 2014, 37, E6. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Thomson, J.A. Pluripotent stem cell lines. Genes Dev. 2008, 22, 1987–1997. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019, 234, 116781. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, N.; Rangarajan, A. Cancer Stem Cells: Formidable Allies of Cancer. Indian J. Surg. Oncol. 2015, 6, 400–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piper, K.; DePledge, L.; Karsy, M.; Cobbs, C. Glioma Stem Cells as Immunotherapeutic Targets: Advancements and Challenges. Front. Oncol. 2021, 11, 615704. [Google Scholar] [CrossRef]
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef]
- Wang, X. Stem cells in tissues, organoids, and cancers. Cell Mol. Life Sci. 2019, 76, 4043–4070. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Gómez-López, S.; Lerner, R.G.; Petritsch, C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell. Mol. Life Sci. 2014, 71, 575–597. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Bhattacharya, K.; Mandal, C. Nutritional stress reprograms dedifferention in glioblastoma multiforme driven by PTEN/Wnt/Hedgehog axis: A stochastic model of cancer stem cells. Cell Death Discov. 2018, 4, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Rosińska, S.; Gavard, J. Tumor Vessels Fuel the Fire in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6514. [Google Scholar] [CrossRef]
- Garnier, D.; Renoult, O.; Alves-Guerra, M.C.; Paris, F.; Pecqueur, C. Glioblastoma Stem-Like Cells, Metabolic Strategy to Kill a Challenging Target. Front. Oncol. 2019, 9, 118. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef]
- Alves, A.L.V.; Gomes, I.N.F.; Carloni, A.C.; Rosa, M.N.; da Silva, L.S.; Evangelista, A.F.; Reis, R.M.; Silva, V.A.O. Role of glioblastoma stem cells in cancer therapeutic resistance: A perspective on antineoplastic agents from natural sources and chemical derivatives. Stem Cell Res. Ther. 2021, 12, 206. [Google Scholar] [CrossRef]
- Brescia, P.; Richichi, C.; Pelicci, G. Current strategies for identification of glioma stem cells: Adequate or unsatisfactory? J. Oncol. 2012, 376894. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.S.; Qin, X.L.; Zong, H.L.; He, X.G.; Cao, L. Cancer stem cell markers in glioblastoma—An update. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3207–3211. [Google Scholar] [PubMed]
- Lombard, A.; Digregorio, M.; Delcamp, C.; Rogister, B.; Piette, C.; Coppieters, N. The Subventricular Zone; A Hideout for Adult and Pediatric High-Grade Glioma Stem Cells. Front. Oncol. 2021, 10, 614930. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Xu, S.; Liu, Z.; Cheng, Q. The adaptive transition of glioblastoma stem cells and its implications on treatments. Sig. Transduct. Target. Ther. 2021, 6, 124. [Google Scholar] [CrossRef] [PubMed]
- Pinto, G.; Saenz-de-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Ho, I.A.W.; Shim, W.S.N. Contribution of the Microenvironmental Niche to Glioblastoma Heterogeneity. Biomed. Res. Int. 2017, 2017, 9634172. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Medarova, Z.; Moore, A. Role of microRNAs in glioblastoma. Oncotarget 2021, 12, 1707–1723. [Google Scholar] [CrossRef]
- Wu, P.; Gao, W.; Su, M.; Nice, E.C.; Zhang, W.; Lin, J.; Xie, N. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 641469. [Google Scholar] [CrossRef]
- Auffinger, B.; Spencer, D.; Pytel, P.; Ahmed, A.U.; Lesniak, M.S. The role of glioma stem cells in chemotherapy resistance and glioblastoma multiforme recurrence. Expert Rev. Neurother. 2015, 15, 741–752. [Google Scholar] [CrossRef] [Green Version]
- Iwadate, Y. Plasticity in Glioma Stem Cell Phenotype and Its Therapeutic Implication. Neurol. Med. Chir. 2018, 58, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baisiwala, S.; Auffinger, B.; Caragher, S.P.; Shireman, J.M.; Ahsan, R.; Lee, G.; Hasan, T.; Park, C.; Saathoff, M.R.; Christensen, A.C.; et al. Chemotherapeutic Stress Induces Transdifferentiation of Glioblastoma Cells to Endothelial Cells and Promotes Vascular Mimicry. Stem Cells Int. 2019, 6107456. [Google Scholar] [CrossRef] [PubMed]
- Șovrea, A.S.; Boșca, B.; Melincovici, C.S.; Constantin, A.-M.; Crintea, A.; Mărginean, M.; Dronca, E.; Jianu, M.E.; Suflețel, R.; Gonciar, D.; et al. Multiple Faces of the Glioblastoma Microenvironment. Int. J. Mol. Sci. 2022, 23, 595. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Van Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Markovic, D.S.; Vinnakota, K.; van Rooijen, N.; Kiwit, J.; Synowitz, M.; Glass, R.; Kettenmann, H. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain. Behav. Immun. 2011, 25, 624–628. [Google Scholar] [CrossRef]
- Zhai, H.; Heppner, F.L.; Tsirka, S.E. Microglia/macrophages promote glioma progression. Glia 2011, 59, 472–485. [Google Scholar] [CrossRef] [Green Version]
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. [Google Scholar] [CrossRef] [Green Version]
- Poon, C.C.; Gordon, P.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J. Differential microglia and macrophage profiles in human IDH-mutant and -wild type glioblastoma. Oncotarget 2019, 10, 3129–3143. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.-Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. Clin. Investig. 2019, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.V.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Saavedra-López, E.; Roig-Martínez, M.; Cribaro, G.P.; Casanova, P.V.; Gallego, J.M.; Pérez-Vallés, A.; Barcia, C. Phagocytic glioblastoma-associated microglia and macrophages populate invading pseudopalisades. Brain Commun. 2020, 2, fcz043. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Zhan, D.; Fu, Y.; Wei, S.; Lal, B.; Wang, J.; Li, Y.; Lopez-Bertoni, H.; Yalcin, F.; Dzaye, O.; et al. Mutant IDH1 promotes phagocytic function of microglia/macrophages in gliomas by downregulating ICAM1. Cancer Lett. 2021, 517, 35–45. [Google Scholar] [CrossRef]
- Brandenburg, S.; Blank, A.; Bungert, A.D.; Vajkoczy, P. Distinction of Microglia and Macrophages in Glioblastoma: Close Relatives, Different Tasks? Int. J. Mol. Sci. 2020, 22, 194. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [Green Version]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Landry, A.P.; Balas, M.; Alli, S.; Spears, J.; Zador, Z. Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci. Rep. 2020, 10, 19542. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Hu, Y.; Wu, F.; Guo, Q.; Qian, Z.; Hu, W.; Chen, J.; Wang, K.; Fan, X.; Wu, X.; et al. Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. Natl. Sci. Rev. 2020, 7, 1306–1318. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Qiu, S.; Li, X.; Huang, B.; Xu, Y.; Peng, Y. EZH2 suppression in glioblastoma shifts microglia toward M1 phenotype in tumor microenvironment. J. Neuroinflam. 2017, 14, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Ciotti, G.M.; Braun, D.; Kalinin, S.; Currò, D.; Dello Russo, C.; Coli, A.; Mangiola, A.; Anile, C.; Feinstein, D.L.; et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci. Lett. 2017, 645, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Leblond, M.M.; Gérault, A.N.; Corroyer-Dulmont, A.; MacKenzie, E.T.; Petit, E.; Bernaudin, M.; Valable, S. Hypoxia induces macrophage polarization and re-education toward an M2 phenotype in U87 and U251 glioblastoma models. Oncoimmunology 2015, 5, e1056442. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Grégoire, H.; Roncali, L.; Rousseau, A.; Chérel, M.; Delneste, Y.; Jeannin, P.; Hindré, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef] [Green Version]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Wallmann, T.; Zhang, X.M.; Wallerius, M.; Bolin, S.; Joly, A.L.; Sobocki, C.; Leiss, L.; Jiang, Y.; Bergh, J.; Holland, E.C.; et al. Microglia Induce PDGFRB Expression in Glioma Cells to Enhance Their Migratory Capacity. iScience 2018, 9, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-Oncology 2017, 19, 1435–1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Morales, R.T.; Qian, W.; Wang, H.; Gagner, J.P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef]
- Nusblat, L.M.; Carroll, M.J.; Roth, C.M. Crosstalk between M2 macrophages and glioma stem cells. Cell Oncol. 2017, 40, 471–482. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, L.; Dang, W.Q.; Cao, M.F.; Xiao, J.F.; Lv, S.Q.; Jiang, W.J.; Yao, X.H.; Lu, H.M.; Miao, J.Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 2020, 100, 619–629. [Google Scholar] [CrossRef]
- Caruso, F.P.; Garofano, L.; D’Angelo, F.; Yu, K.; Tang, F.; Yuan, J.; Zhang, J.; Cerulo, L.; Pagnotta, S.M.; Bedognetti, D.; et al. A map of tumor-host interactions in glioma at single-cell resolution. Gigascience 2020, 9, giaa109. [Google Scholar] [CrossRef]
- Wu, S.; Yang, W.; Zhang, H.; Ren, Y.; Fang, Z.; Yuan, C.; Yao, Z. The Prognostic Landscape of Tumor-Infiltrating Immune Cells and Immune Checkpoints in Glioblastoma. Technol. Cancer Res. Treat. 2019, 18, 1533033819869949. [Google Scholar] [CrossRef] [Green Version]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef]
- Locarno, C.V.; Simonelli, M.; Carenza, C.; Capucetti, A.; Stanzani, E.; Lorenzi, E.; Persico, P.; Della Bella, S.; Passoni, L.; Mavilio, D.; et al. Role of myeloid cells in the immunosuppressive microenvironment in gliomas. Immunobiology 2020, 225, 151853. [Google Scholar] [CrossRef] [PubMed]
- Ocana, A.; Nieto-Jiménez, C.; Pandiella, A.; Templeton, A.J. Neutrophils in cancer: Prognostic role and therapeutic strategies. Mol. Cancer 2017, 16, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopo, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozel, I.; Duerig, I.; Domnich, M.; Lang, S.; Pylaeva, E.; Jablonska, J. The Good, the Bad, and the Ugly: Neutrophils, Angiogenesis, and Cancer. Cancers 2022, 14, 536. [Google Scholar] [CrossRef] [PubMed]
- Magod, P.; Mastandrea, I.; Rousso-Noori, L.; Agemy, L.; Shapira, G.; Shomron, N.; Friedmann-Morvinski, D. Exploring the longitudinal glioma microenvironment landscape uncovers reprogrammed pro-tumorigenic neutrophils in the bone marrow. Cell Rep. 2021, 36, 109480. [Google Scholar] [CrossRef]
- Feng, X.; Wang, S.; Sun, Z.; Dong, H.; Yu, H.; Huang, M.; Gao, X. Ferroptosis Enhanced Diabetic Renal Tubular Injury via HIF-1α/HO-1 Pathway in db/db Mice. Front. Endocrinol. 2021, 12, 626390. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Khan, S.; Mittal, S.; McGee, K.; Alfaro-Munoz, K.D.; Majd, N.; Balasubramaniyan, V.; de Groot, J.F. Role of Neutrophils and Myeloid-Derived Suppressor Cells in Glioma Progression and Treatment Resistance. Int. J. Mol. Sci. 2020, 21, 1954. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.J.; Wei, K.C.; Chen, P.Y.; Lim, M.; Hwang, T.L. Roles of Neutrophils in Glioma and Brain Metastases. Front. Immunol. 2021, 12, 701383. [Google Scholar] [CrossRef]
- Yalon, M.; Toren, A.; Jabarin, D.; Fadida, E.; Constantini, S.; Mehrian-Shai, R. Elevated NLR May Be a Feature of Pediatric Brain Cancer Patients. Front. Oncol. 2019, 9, 327. [Google Scholar] [CrossRef]
- Liu, K. Dendritic Cells. Encycl. Cell Biol. 2016, 3, 741–749. [Google Scholar] [CrossRef]
- Srivastava, S.; Jackson, C.; Kim, T.; Choi, J.; Lim, M. A Characterization of Dendritic Cells and Their Role in Immunotherapy in Glioblastoma: From Preclinical Studies to Clinical Trials. Cancers 2019, 11, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datsi, A.; Sorg, R.V. Dendritic Cell Vaccination of Glioblastoma: Road to Success or Dead End. Front. Immunol. 2021, 12, 770390. [Google Scholar] [CrossRef] [PubMed]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira da Costa, M.; Reis, E.; Sousa, C. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Giles, D.A.; Duncker, P.C.; Wilkinson, N.M.; Washnock-Schmid, J.M.; Segal, B.M. CNS-resident classical DCs play a critical role in CNS autoimmune disease. J. Clin. Investig. 2018, 128, 5322–5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löhr, M.; Freitag, B.; Technau, A.; Krauss, J.; Monoranu, C.M.; Rachor, J.; Lutz, M.B.; Hagemann, C.; Kessler, A.F.; Linsenmann, T.; et al. High-grade glioma associated immunosuppression does not prevent immune responses induced by therapeutic vaccines in combination with Treg depletion. Cancer Immunol. Immunother. 2018, 67, 1545–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer 2018, 4, 784–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Vijayan, D.; Putz, E.M.; Aguilera, A.R.; Markey, K.A.; Straube, J.; Kazakoff, S.; Nutt, S.L.; Takeda, K.; Hill, G.R.; et al. Interleukin-12 from CD103 + Batf3-Dependent Dendritic Cells Required for NK-Cell Suppression of Metastasis. Cancer Immunol. Res. 2017, 5, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Grassini, D.; Ortenzi, V.; Pasqualetti, F.; Montemurro, N.; Perrini, P.; Naccarato, A.G.; Scatena, C. Decipher the Glioblastoma Microenvironment: The First Milestone for New Groundbreaking Therapeutic Strategies. Genes 2021, 12, 445. [Google Scholar] [CrossRef]
- Yu, J.; Sun, H.; Cao, W.; Song, Y.; Jiang, Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp. Hematol. Oncol. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468, Erratum in Nat. Med. 2019, 25, 529. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorrey, S.J.; Chongsathidkiet, P.; Wilkinson, D.S.; Champion, C.; Fecci, P. Intracranial tumors lead to sequestration of T cells in the bone marrow. J. Immunol. 2020, 204, 165.26. [Google Scholar]
- Woroniecka, K.; Fecci, P.E. T-cell exhaustion in glioblastoma. Oncotarget 2018, 9, 35287–35288. [Google Scholar] [CrossRef]
- Persico, P.; Lorenzi, E.; Dipasquale, A.; Pessina, F.; Navarria, P.; Politi, L.S.; Santoro, A.; Simonelli, M. Checkpoint Inhibitors as High-Grade Gliomas Treatment: State of the Art and Future Perspectives. J. Clin. Med. 2021, 10, 1367. [Google Scholar] [CrossRef]
- Wu, Y.; Tian, Z.; Wei, H. Developmental and Functional Control of Natural Killer Cells by Cytokines. Front. Immunol. 2017, 8, 930. [Google Scholar] [CrossRef]
- Bellora, F.; Castriconi, R.; Dondero, A.; Reggiardo, G.; Moretta, L.; Mantovani, A.; Moretta, A.; Bottino, C. The interaction of human natural killer cells with either unpolarized or polarized macrophages results in different functional outcomes. Proc. Natl. Acad. Sci. USA 2010, 107, 21659–21664. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, J.; Zimmer, J.; Chekenya, M. Natural killer cells in intracranial neoplasms: Presence and therapeutic efficacy against brain tumours. J. Neurooncol. 2014, 116, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lobinger, D.; Gempt, J.; Sievert, W.; Barz, M.; Schmitt, S.; Nguyen, H.T.; Stangl, S.; Werner, C.; Wang, F.; Wu, Z.; et al. Potential Role of Hsp70 and Activated NK Cells for Prediction of Prognosis in Glioblastoma Patients. Front. Mol. Biosci. 2021, 8, 669366. [Google Scholar] [CrossRef]
- Na, H.Y.; Park, Y.; Nam, S.K.; Koh, J.; Kwak, Y.; Ahn, S.H.; Park, D.J.; Kim, H.H.; Lee, K.S.; Lee, H.S. Prognostic significance of natural killer cell-associated markers in gastric cancer: Quantitative analysis using multiplex immunohistochemistry. J. Transl. Med. 2021, 19, 529. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Toregrosa-Allen, S.; Elzey, B.D.; Utturkar, S.; Lanman, N.A.; Bernal-Crespo, V.; Behymer, M.M.; Knipp, G.T.; Yun, Y.; Veronesi, M.C.; et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2107507118. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with γδ T cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Lamb, L.S.; Pereboeva, L.; Youngblood, S.; Gillespie, G.Y.; Nabors, L.B.; Markert, J.M.; Dasgupta, A.; Langford, C.; Spencer, H.T. A combined treatment regimen of MGMT-modified γδ T cells and temozolomide chemotherapy is effective against primary high-grade gliomas. Sci. Rep. 2021, 11, 21133. [Google Scholar] [CrossRef]
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, C. The Role of Human γδ T Cells in Anti-Tumor Immunity and Their Potential for Cancer Immunotherapy. Cells 2020, 9, 1206. [Google Scholar] [CrossRef]
- Yue, C.; Yang, K.; Dong, W.; Hu, F.; Zhao, S.; Liu, S. γδ T Cells in Peripheral Blood of Glioma Patients. Med. Sci. Monit. 2018, 24, 1784–1792. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef]
- Palit, S.; Heuser, C.; de Almeida, G.P.; Theis, F.J.; Zielinski, C.E. Meeting the Challenges of High-Dimensional Single-Cell Data Analysis in Immunology. Front. Immunol. 2019, 10, 1515. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Li, Y.; Cheng, S.; Li, J.; He, C.; Li, J. A Prognostic Microenvironment-Related Immune Signature via ESTIMATE (PROMISE Model) Predicts Overall Survival of Patients with Glioma. Front. Oncol. 2020, 10, 580263. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomized phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Ohkuri, T.; Ghosh, A.; Kosaka, A.; Zhu, J.; Ikeura, M.; David, M.; Watkins, S.C.; Sarkar, S.N.; Okada, H. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 1199–1208. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Wei, J.; Wang, F.; Kong, L.-Y.; Ling, X.-Y.; Nduom, E.; Gabrusiewicz, K.; Doucette, T.; Yang, Y.; Yaghi, N.K.; et al. Effect of miR-142–3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J. Natl. Cancer Inst. 2014, 106, 8. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglioli, A.; Efe, I.E.; Guneykaya, D.; Ivanov, A.; Huang, Y.; Orlowski, E.; Krüger, C.; Deisz, R.A.; Markovic, D.; Flüh, C.; et al. let-7 MicroRNAs regulate microglial function and suppress glioma growth through toll-like receptor 7. Cell Rep. 2019, 29, 3460–3471.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domino, J.S.; Alvarado, A.; Arnold, L.; Gunewardena, S.; Akhavan, D.; Thomas, S.M. Targeting tumor-associated astrocyte dependence in glioblastoma treatment [abstract]. In Proceedings of the American Association for Cancer Research Annual Meeting 2021, 10–15 April and 17–21 May 2021; AACR: Philadelphia, PA, USA. Cancer Res. 2021, 81, 3170. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crivii, C.-B.; Boșca, A.B.; Melincovici, C.S.; Constantin, A.-M.; Mărginean, M.; Dronca, E.; Suflețel, R.; Gonciar, D.; Bungărdean, M.; Șovrea, A. Glioblastoma Microenvironment and Cellular Interactions. Cancers 2022, 14, 1092. https://doi.org/10.3390/cancers14041092
Crivii C-B, Boșca AB, Melincovici CS, Constantin A-M, Mărginean M, Dronca E, Suflețel R, Gonciar D, Bungărdean M, Șovrea A. Glioblastoma Microenvironment and Cellular Interactions. Cancers. 2022; 14(4):1092. https://doi.org/10.3390/cancers14041092
Chicago/Turabian StyleCrivii, Carmen-Bianca, Adina Bianca Boșca, Carmen Stanca Melincovici, Anne-Marie Constantin, Mariana Mărginean, Eleonora Dronca, Rada Suflețel, Diana Gonciar, Maria Bungărdean, and Alina Șovrea. 2022. "Glioblastoma Microenvironment and Cellular Interactions" Cancers 14, no. 4: 1092. https://doi.org/10.3390/cancers14041092
APA StyleCrivii, C. -B., Boșca, A. B., Melincovici, C. S., Constantin, A. -M., Mărginean, M., Dronca, E., Suflețel, R., Gonciar, D., Bungărdean, M., & Șovrea, A. (2022). Glioblastoma Microenvironment and Cellular Interactions. Cancers, 14(4), 1092. https://doi.org/10.3390/cancers14041092