
Imaging Kv1.3 Expressing Memory T Cells as a Marker of Immunotherapy Response

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. [18F]AlF-NOTA-KCNA3P Radiochemistry
2.3. Electrophysiology Studies
2.4. Animal Procedures
2.5. PET-CT Imaging
2.6. Flow Cytometry
2.7. Dimension Reduction Analysis
2.8. Statistical Analysis
3. Results
3.1. Evaluation of Treatment Efficacy Using Tumour Volumes
3.2. [19F]AlF-Electrophysiological Characterization of NOTA-KCNA3P
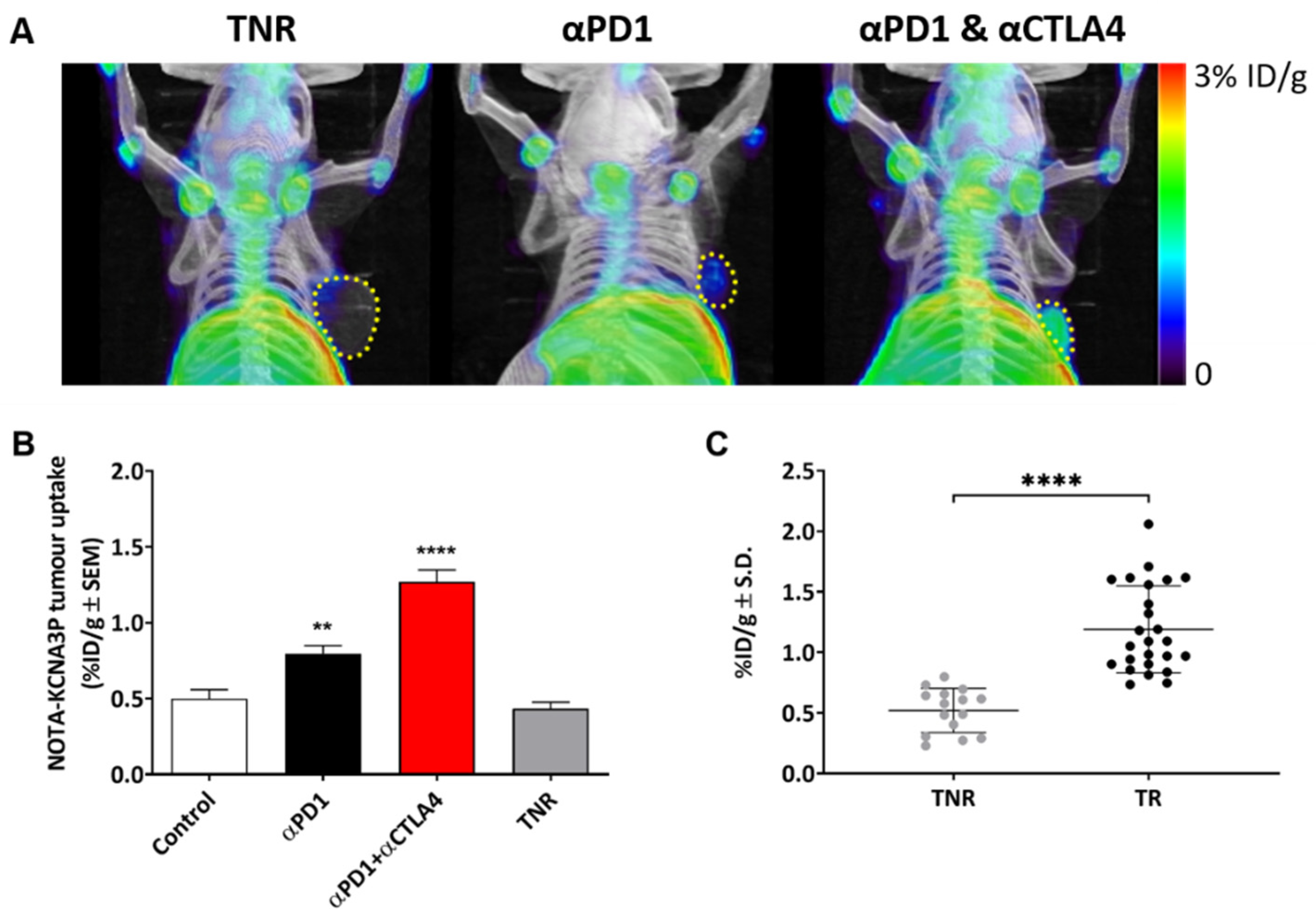
3.3. [18F]AlF-NOTA-KCNA3P In Vivo PET Imaging
3.4. Tumour Uptake of [18F]AlF-NOTA-KCNA3P Is Linked to Infiltration of Kv1.3 Expressing TEM Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brahmer, J.R.; Pardoll, D.M. Immune checkpoint inhibitors: Making immunotherapy a reality for the treatment of lung cancer. Cancer Immunol. Res. 2013, 1, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llosa, N.J.; Luber, B.; Siegel, N.; Awan, A.H.; Oke, T.; Zhu, Q.; Bartlett, B.R.; Aulakh, L.K.; Thompson, E.D.; Jaffee, E.M.; et al. Immunopathologic Stratification of Colorectal Cancer for Checkpoint Blockade Immunotherapy. Cancer Immunol. Res. 2019, 7, 1574–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y. Cancer immunotherapy: Harnessing the immune system to battle cancer. J. Clin. Investig. 2015, 125, 3335–3337. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.W.; Tavare, R.; Mahakian, L.M.; Silvestrini, M.T.; Tam, S.; Ingham, E.S.; Salazar, F.B.; Borowsky, A.D.; Wu, A.M.; Ferrara, K.W. CD8(+) T-Cell Density Imaging with (64)Cu-Labeled Cys-Diabody Informs Immunotherapy Protocols. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4976–4987. [Google Scholar] [CrossRef] [Green Version]
- Tavare, R.; Escuin-Ordinas, H.; Mok, S.; McCracken, M.N.; Zettlitz, K.A.; Salazar, F.B.; Witte, O.N.; Ribas, A.; Wu, A.M. An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy. Cancer Res. 2016, 76, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavare, R.; McCracken, M.N.; Zettlitz, K.A.; Knowles, S.M.; Salazar, F.B.; Olafsen, T.; Witte, O.N.; Wu, A.M. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 1108–1113. [Google Scholar] [CrossRef] [Green Version]
- Tavare, R.; McCracken, M.N.; Zettlitz, K.A.; Salazar, F.B.; Olafsen, T.; Witte, O.N.; Wu, A.M. Immuno-PET of Murine T Cell Reconstitution Postadoptive Stem Cell Transplantation Using Anti-CD4 and Anti-CD8 Cys-Diabodies. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2015, 56, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Goggi, J.L.; Tan, Y.X.; Hartimath, S.V.; Jieu, B.; Hwang, Y.Y.; Jiang, L.; Boominathan, R.; Cheng, P.; Yuen, T.Y.; Chin, H.X.; et al. Granzyme B PET Imaging of Immune Checkpoint Inhibitor Combinations in Colon Cancer Phenotypes. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2020, 22, 1392–1402. [Google Scholar] [CrossRef] [PubMed]
- Pandit-Taskar, N.; Postow, M.A.; Hellmann, M.D.; Harding, J.J.; Barker, C.A.; O’Donoghue, J.A.; Ziolkowska, M.; Ruan, S.; Lyashchenko, S.K.; Tsai, F.; et al. First-in-Humans Imaging with (89)Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2020, 61, 512–519. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.; Lim, L.H.K.; Cheung, F.S.G. The role of the tumour microenvironment in immunotherapy. Endocr.-Relat. Cancer 2017, 24, T283–T295. [Google Scholar] [CrossRef] [PubMed]
- Goggi, J.L.; Hartimath, S.V.; Hwang, Y.; Tan, Y.X.; Khanapur, S.; Ramasamy, B.; Jiang, L.; Yong, F.F.; Cheng, P.; Tan, P.W.; et al. Examining Immunotherapy Response Using Multiple Radiotracers. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2020, 22, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, M.D.; Chandy, K.G. The functional network of ion channels in T lymphocytes. Immunol. Rev. 2009, 231, 59–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feske, S.; Skolnik, E.Y.; Prakriya, M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012, 12, 532–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.T.; Bajaj, S.; Tanner, M.R.; Chang, S.C.; Krishnarjuna, B.; Ng, X.R.; Morales, R.A.V.; Chen, M.W.; Luo, D.; Patel, D.; et al. Modulation of Lymphocyte Potassium Channel KV1.3 by Membrane-Penetrating, Joint-Targeting Immunomodulatory Plant Defensin. ACS Pharm. Transl. Sci. 2020, 3, 720–736. [Google Scholar] [CrossRef] [PubMed]
- Newton, H.S.; Gawali, V.S.; Chimote, A.A.; Lehn, M.A.; Palackdharry, S.M.; Hinrichs, B.H.; Jandarov, R.; Hildeman, D.; Janssen, E.M.; Wise-Draper, T.M.; et al. PD1 blockade enhances K+ channel activity, Ca2+ signaling, and migratory ability in cytotoxic T lymphocytes of patients with head and neck cancer. J. Immunother. Cancer 2020, 8, e000844. [Google Scholar] [CrossRef]
- Ribas, A.; Shin, D.S.; Zaretsky, J.; Frederiksen, J.; Cornish, A.; Avramis, E.; Seja, E.; Kivork, C.; Siebert, J.; Kaplan-Lefko, P.; et al. PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol. Res. 2016, 4, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Grissmer, S.; Nguyen, A.N.; Aiyar, J.; Hanson, D.C.; Mather, R.J.; Gutman, G.A.; Karmilowicz, M.J.; Auperin, D.D.; Chandy, K.G. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol. Pharm. 1994, 45, 1227–1234. [Google Scholar]
- Goggi, J.L.; Hartimath, S.V.; Xuan, T.Y.; Khanapur, S.; Jieu, B.; Chin, H.X.; Ramasamy, B.; Cheng, P.; Rong, T.J.; Fong, Y.F.; et al. Granzyme B PET Imaging of Combined Chemotherapy and Immune Checkpoint Inhibitor Therapy in Colon Cancer. Mol. Imaging Biol. MIB Off. Publ. Acad. Mol. Imaging 2021, 23, 714–723. [Google Scholar] [CrossRef]
- Principe, N.; Kidman, J.; Goh, S.; Tilsed, C.M.; Fisher, S.A.; Fear, V.S.; Forbes, C.A.; Zemek, R.M.; Chopra, A.; Watson, M.; et al. Tumor Infiltrating Effector Memory Antigen-Specific CD8(+) T Cells Predict Response to Immune Checkpoint Therapy. Front. Immunol. 2020, 11, 584423. [Google Scholar] [CrossRef]
- Beeton, C.; Wulff, H.; Standifer, N.E.; Azam, P.; Mullen, K.M.; Pennington, M.W.; Kolski-Andreaco, A.; Wei, E.; Grino, A.; Counts, D.R.; et al. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc. Natl. Acad. Sci. USA 2006, 103, 17414–17419. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.; Wulff, H. The Lymphocyte Potassium Channels Kv1.3 and KCa3.1 as Targets for Immunosuppression. Drug Dev. Res. 2011, 72, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; Calabresi, P.A.; Allie, R.; Yun, S.; Pennington, M.; Beeton, C.; Chandy, K.G. The voltage-gated Kv1.3 K(+) channel in effector memory T cells as new target for MS. J. Clin. Investig. 2003, 111, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teisseyre, A.; Gasiorowska, J.; Michalak, K. Voltage-Gated Potassium Channels Kv1.3--Potentially New Molecular Target in Cancer Diagnostics and Therapy. Adv. Clin. Exp. Med. 2015, 24, 517–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul, M.; Hoosein, N. Reduced Kv1.3 potassium channel expression in human prostate cancer. J. Membr. Biol. 2006, 214, 99–102. [Google Scholar] [CrossRef]
- Bielanska, J.; Hernandez-Losa, J.; Perez-Verdaguer, M.; Moline, T.; Somoza, R.; Ramon, Y.C.S.; Condom, E.; Ferreres, J.C.; Felipe, A. Voltage-dependent potassium channels Kv1.3 and Kv1.5 in human cancer. Curr. Cancer Drug Targets 2009, 9, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Brevet, M.; Fucks, D.; Chatelain, D.; Regimbeau, J.M.; Delcenserie, R.; Sevestre, H.; Ouadid-Ahidouch, H. Deregulation of 2 potassium channels in pancreas adenocarcinomas: Implication of KV1.3 gene promoter methylation. Pancreas 2009, 38, 649–654. [Google Scholar] [CrossRef]
- Brevet, M.; Haren, N.; Sevestre, H.; Merviel, P.; Ouadid-Ahidouch, H. DNA methylation of K(v)1.3 potassium channel gene promoter is associated with poorly differentiated breast adenocarcinoma. Cell. Physiol. Biochem. 2009, 24, 25–32. [Google Scholar] [CrossRef]
- Felipe, A.; Bielanska, J.; Comes, N.; Vallejo, A.; Roig, S.; Ramon, Y.C.S.; Condom, E.; Hernandez-Losa, J.; Ferreres, J.C. Targeting the voltage-dependent K(+) channels Kv1.3 and Kv1.5 as tumor biomarkers for cancer detection and prevention. Curr. Med. Chem. 2012, 19, 661–674. [Google Scholar] [CrossRef]
- Felipe, A.; Vicente, R.; Villalonga, N.; Roura-Ferrer, M.; Martinez-Marmol, R.; Sole, L.; Ferreres, J.C.; Condom, E. Potassium channels: New targets in cancer therapy. Cancer Detect. Prev. 2006, 30, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Gutman, G.A.; Chandy, K.G.; Adelman, J.P.; Aiyar, J.; Bayliss, D.A.; Clapham, D.E.; Covarriubias, M.; Desir, G.V.; Furuichi, K.; Ganetzky, B.; et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: Potassium channels. Pharm. Rev. 2003, 55, 583–586. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Group | [18F]AlF-NOTA-KCNA3P Tumour Uptake (%ID/g ± SD) |
|---|---|
| Control | 0.51 ± 0.19 |
| Treatment Responsive tumours (TR) αPD1 | 0.87 ± 0.15 * |
| αPD1 + αCTLA4 | 1.30 ± 0.36 ** |
| Treatment Non-Responders (TNR) | 0.53 ± 0.20 |
| CT26 Tumour-Infiltrating Immune Cells | ||||
|---|---|---|---|---|
| Treatment Group | CD8+ % of CD3+ | GZB+ CD8+ % of CD8+ | CD8+ TEM % of CD8+ | CD4+ TEM % of CD4+ |
| Control | 14.74 ± 5.38 | 34.02 ± 8.15 | 7.32 ± 4.53 | 7.25 ± 5.75 |
| TR αPD1 | 21.35 ± 2.78 * | 58.46 ± 7.96 * | 54.61 ± 8.96 ** | 42.68 ± 9.65 * |
| αPD1 + αCTLA4 | 42.13 ± 7.35 *** | 76.06 ± 9.20 ** | 77.67 ± 21.50 *** | 66.81 ± 18.09 ** |
| TNR | 16.43 ± 4.61 | 40.66 ± 3.20 | 20.69 ± 8.26 | 23.09 ± 10.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goggi, J.L.; Khanapur, S.; Ramasamy, B.; Hartimath, S.V.; Rong, T.J.; Cheng, P.; Tan, Y.X.; Yeo, X.Y.; Jung, S.; Goay, S.S.M.; et al. Imaging Kv1.3 Expressing Memory T Cells as a Marker of Immunotherapy Response. Cancers 2022, 14, 1217. https://doi.org/10.3390/cancers14051217
Goggi JL, Khanapur S, Ramasamy B, Hartimath SV, Rong TJ, Cheng P, Tan YX, Yeo XY, Jung S, Goay SSM, et al. Imaging Kv1.3 Expressing Memory T Cells as a Marker of Immunotherapy Response. Cancers. 2022; 14(5):1217. https://doi.org/10.3390/cancers14051217
Chicago/Turabian StyleGoggi, Julian L., Shivashankar Khanapur, Boominathan Ramasamy, Siddesh V. Hartimath, Tang Jun Rong, Peter Cheng, Yun Xuan Tan, Xin Yi Yeo, Sangyong Jung, Stephanie Shee Min Goay, and et al. 2022. "Imaging Kv1.3 Expressing Memory T Cells as a Marker of Immunotherapy Response" Cancers 14, no. 5: 1217. https://doi.org/10.3390/cancers14051217
APA StyleGoggi, J. L., Khanapur, S., Ramasamy, B., Hartimath, S. V., Rong, T. J., Cheng, P., Tan, Y. X., Yeo, X. Y., Jung, S., Goay, S. S. M., Ong, S. T., Hwang, Y. Y., Chandy, K. G., & Robins, E. G. (2022). Imaging Kv1.3 Expressing Memory T Cells as a Marker of Immunotherapy Response. Cancers, 14(5), 1217. https://doi.org/10.3390/cancers14051217