
At the Intersection of Cardiology and Oncology: TGFβ as a Clinically Translatable Therapy for TNBC Treatment and as a Major Regulator of Post-Chemotherapy Cardiomyopathy

Abstract
:Simple Summary
Abstract
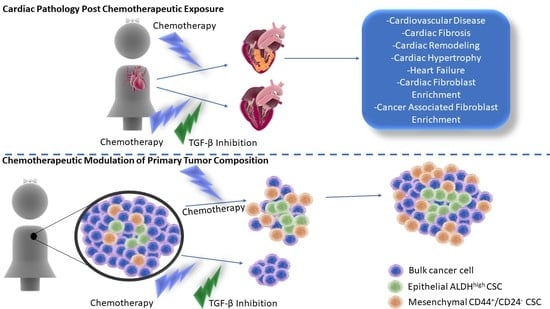
1. Introduction
2. Post-Chemotherapeutic Cardiomyopathy
3. Anthracycline and Taxane Mechanisms of Cardiotoxicity
4. Clinical Assessment of Cardiotoxicity
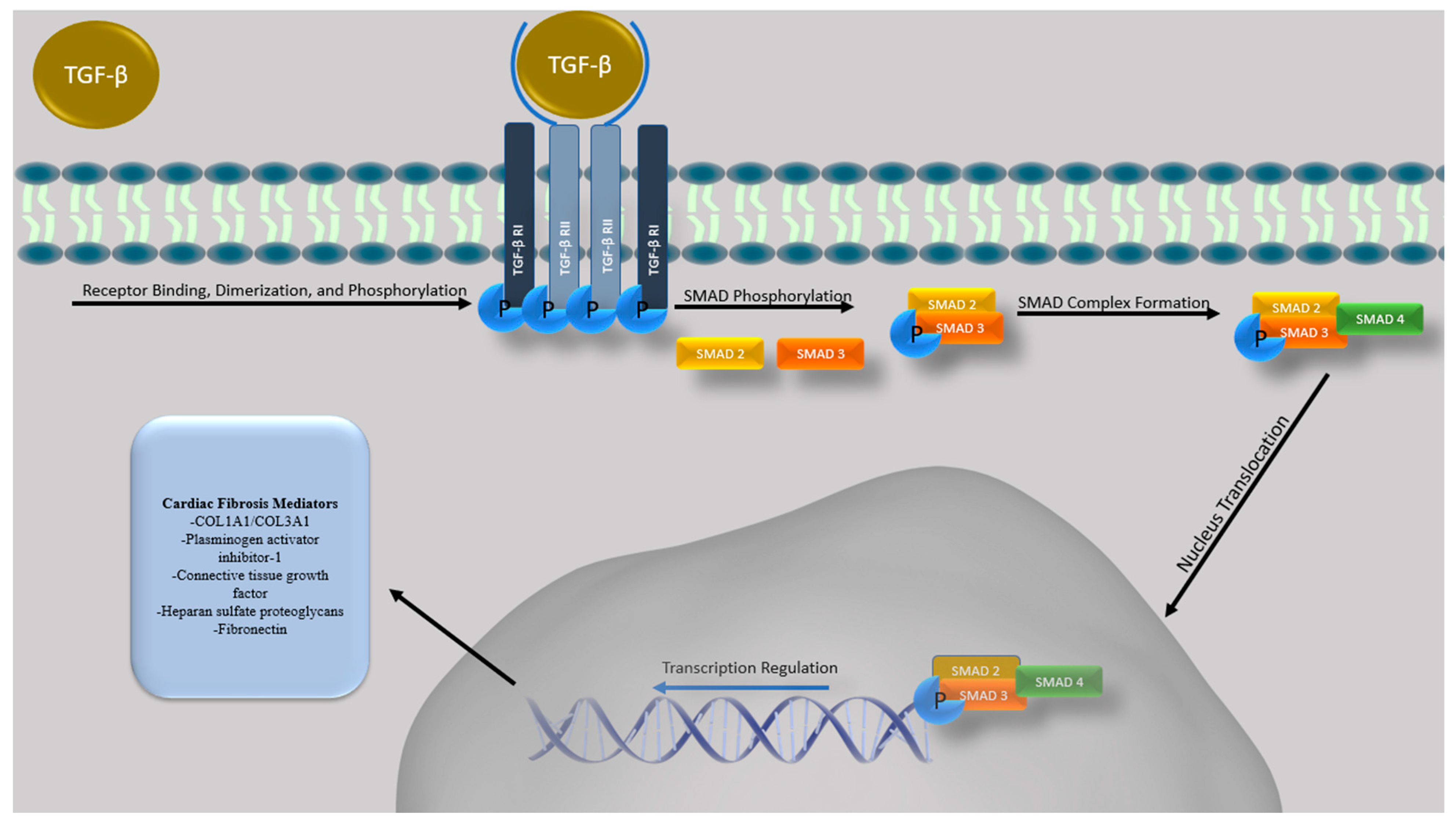
5. TGF-β Overview
6. The Role of TGFB in Cardiac Fibrosis, Remodeling and Regulation of Cardiac Fibrocytes
7. TGF-β Inhibition to Prevent Cardiomyopathy

{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Clinical Trial Number | Mechanism | References |
|---|---|---|---|
| Enalapril | NCT01968200 | ACEI with antifibrotic activity via inhibition of TGFB1 and p-SMAD2/3 expression | [129,130] |
| Carvedilol | NCT02177175 NCT01347970 | Suppression of myocardial fibrosis by inhibiting TGFB1 mRNA expression | [131,132] |
| Simvastatin | NCT02096588 | Downregulates TGFb1-mediated phosphorylation of Smad2/3 via activation of PP2A and PP2C/PPM1A phosphatases | [133,134] |
| Rivaroxaban | NCT02303795 NCT01776424 NCT02066662 | Downregulates mRNA expression of TGFB in the infarcted area following an MI, potentially via suppression of PAR-1 and PAR-2 pathways | [135] |
| Clopidogrel | NCT02044250 NCT02317198 | Platelet blocker that inhibits the expression of TGFB mRNA and the protein levels preventing cardiac fibrosis | [136] |
| Rituximab | NCT03072199 | Monoclonal antibody against CD20 inhibits fibrotic signaling of TGF-β1 and p-Smad2/3 | [137] |
| LCZ696 | NCT02816736 NCT03190304 NCT02468232 NCT02924727 | Angiotensin receptor–neprilysin inhibitor that improves cardiac function by downregulating cardiac fibrosis via suppression of TGF-β expression, primarily through its specific inhibition of neprilysin | [138,139] |
| Spironolactone | NCT03409627 NCT02673463 | SP prevents cardiac fibrosis by inhibiting the production of TGFβ1 and phosphorylation of Smad2/3 | [140,141] |
| Macitentan | NCT03153111 | Dual endothelin receptor antagonist (ETA and ETB) that suppresses expression of TGFβ, especially in DM patients in whom TGFβ is upregulated | [142,143] |
| Ivabradine | NCT04448899 NCT04308031 | Hyperpolarization-activated pacemaker current (If) channel inhibitor ivabradine inhibits the expression of TGFb1 and Smad2 post-MI, suppressing collagen synthesis and pro-fibrotic activity | [144,145] |
| Empagliflozin | NCT03128528 NCT03030222 NCT03057977 NCT03057951 NCT03485092 NCT02998970 | Inhibits the fibrotic activity of TGFb in the heart by suppressing the expression of TGFb1, p-Smad2/3 and upregulating TGFb inhibitor Smad7, further resulting in decreased expression of collagen I and II mediated by the TGFb/Smad pathway | [146,147] |
| Pirfenidone | NCT02932566 | Inhibits Ang II-induced expression of TGFb1 and suppresses myocardial interstitial fibrosis | [148,149] |
| Atorvastatin | NCT02679261 | Suppresses cardiac fibrosis by attenuating TGFb1-mediated phosphorylation of Smad3, PI-3 kinase, Akt, collagen I and endoglin expression | [150] |
| Eplerenone | NCT01857856 | Inhibits the expression of TGFb1 and collagen I, resulting in downregulation of cardiac remodeling induced by cardiomyopathy | [151] |
| Olmesartan | NCT04174456 | Angiotensin II type 1 receptor blocker which reduces the expression of TGFb in pressure-overloaded, diabetic, obese patients, preventing cardiovascular injury | [152,153] |
| Tadalafil | NCT03049540 | cGMP-mediated inhibition of TGFb1 expression | [154] |
| Berberine | NCT04434365 | Antifibrotic activity by inhibition of TGFb1 secretion, potentially by upregulation of AMPK phosphorylation and downregulation of mTOR and p70S6K phosphorylation | [155] |
| Melatonin | NCT02099331 | Antifibrotic activity via suppression of TGFb1 expression | [156] |
| N-Acetylcysteine (NAC) | NCT02750319 w/Amiodarone NCT01878669 NCT01878344 | Antioxidant that inhibits the TGFb1-mediated signaling involved in fibrosis, potentially by suppressing its interaction with TGB1R, downregulating phosphorylation of Smad2/3 and upregulating Smad7 mRNA | [157,158] |
| Colchicine | NCT02594111 NCT01709981 NCT02624180 NCT04382443 | Antifibrotic via inhibition of expression of TGFb1 mRNA | [159] |
| Ticagrelor | NCT02539160 NCT03437044 NCT01944800 | Antifibrotic activity via inhibition of the expression of TGFb | [160] |
| Valsartan | NCT01912534 | Inhibition of Ang II type I (AT 1) receptors, resulting in suppression of AT 1-mediated action of the TGFb/Smad pathway | [161] |
| Metformin | NCT03629340 | Suppression of cardiac fibrosis via inhibition of TGFb1 production and phosphorylation of Smad3 | [162] |
| Nitrite | NCT03015402 NCT02980068 | Downregulation of cardiac remodeling via suppression of AT II and AT 1R, inhibiting TGFb1 | [163] |
| Nebivolol | NCT02053246 NCT01648634 | Attenuated profibrotic activity and prevention of vascular remodeling by downregulating the expression of TGFb1and MMP-2/9 | [164] |
| Riociguat | NCT01065454 | Guyanalate cyclase stimulant with antifibrotic activity via inhibition of TGFb1-mediated collagen synthesis | [165] |
8. TGF-β as a Therapeutic Target in TNBC
9. Conclusions and Future Directions
10. Materials and Methods
Clinical Database Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9, S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Long, J.B.; Hurria, A.; Owusu, C.; Steingart, R.M.; Gross, C.P. Incidence of Heart Failure or Cardiomyopathy After Adjuvant Trastuzumab Therapy for Breast Cancer. J. Am. Coll. Cardiol. 2012, 60, 2504–2512. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.; Denlinger, C. Cardiovascular toxicity in cancer survivors: Current guidelines and future directions. Am. Coll. Cardiol. Expert Anal. 2018, 29. [Google Scholar]
- Patnaik, J.L.; Byers, T.; DiGuiseppi, C.; Dabelea, D.; Denberg, T.D. Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: A retrospective cohort study. Breast Cancer Res. 2011, 13, R64. [Google Scholar] [CrossRef] [Green Version]
- Bardia, A.; Arieas, E.T.; Zhang, Z.; DeFilippis, A.; Tarpinian, K.; Jeter, S.; Nguyen, A.; Henry, N.L.; Flockhart, D.A.; Hayes, D.F.; et al. Comparison of breast cancer recurrence risk and cardiovascular disease incidence risk among postmenopausal women with breast cancer. Breast Cancer Res. Treat. 2012, 131, 907–914. [Google Scholar] [CrossRef] [Green Version]
- Sturgeon, K.M.; Deng, L.; Bluethmann, S.M.; Zhou, S.; Trifiletti, D.M.; Jiang, C.; Kelly, S.P.; Zaorsky, N.G. A population-based study of cardiovascular disease mortality risk in US cancer patients. Eur. Heart J. 2019, 40, 3889–3897. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.; Coates, A.S.; Darby, S.C.; Davies, C.; Gelber, R.D.; Godwin, J.; Goldhirsch, A.; Gray, R.; Peto, R.; Pritchard, K.I.; et al. Adjuvant chemotherapy in oestrogen-receptor-poor breast cancer: Patient-level meta-analysis of randomised trials. Lancet 2008, 371, 29–40. [Google Scholar] [CrossRef]
- Ozkan, M.; Berk, V.; Kaplan, M.A.; Benekli, M.; Coskun, U.; Bilici, A.; Gumus, M.; Alkis, N.; Dane, F.; Ozdemir, N.Y.; et al. Gemcitabine and cisplatin combination chemotherapy in triple negative metastatic breast cancer previously treated with a taxane/anthracycline chemotherapy; multicenter experience. Neoplasma 2012, 59, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Singal, P.K.; Iliskovic, N. Doxorubicin-Induced Cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Mercuro, G.; Cadeddu, C.; Piras, A.; Dessì, M.; Madeddu, C.; Deidda, M.; Serpe, R.; Massa, E.; Mantovani, G. Early Epirubicin-Induced Myocardial Dysfunction Revealed by Serial Tissue Doppler Echocardiography: Correlation with Inflammatory and Oxidative Stress Markers. Oncologist 2007, 12, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Pennesi, G.; Donatelli, F.; Cammarota, R.; De Flora, S.; Noonan, D.M. Cardiotoxicity of Anticancer Drugs: The Need for Cardio-Oncology and Cardio-Oncological Prevention. JNCI J. Natl. Cancer Inst. 2010, 102, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, V.B.; Nahata, M.C. Cardiotoxicity of Chemotherapeutic Agents: Incidence, treatment and prevention. Drug Saf. 2000, 22, 263–302. [Google Scholar] [CrossRef]
- Ewer, M.S.; Lippman, S.M. Type II Chemotherapy-Related Cardiac Dysfunction: Time to Recognize a New Entity. J. Clin. Oncol. 2005, 23, 2900–2902. [Google Scholar] [CrossRef]
- Giordano, S.H.; Lin, Y.-L.; Kuo, Y.F.; Hortobagyi, G.N.; Goodwin, J.S. Decline in the Use of Anthracyclines for Breast Cancer. J. Clin. Oncol. 2012, 30, 2232–2239. [Google Scholar] [CrossRef] [Green Version]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Wallace, K.B. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007, 23, 15–25. [Google Scholar] [CrossRef]
- Barth, E.; Stämmler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Šimůnek, T.; Štěrba, M.; Popelová, O.; Adamcova, M.; Hrdina, R.; Geršl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Thomas, C.E.; Aust, S.D. Release of iron from ferritin by cardiotoxic anthracycline antibiotics. Arch. Biochem. Biophys. 1986, 248, 684–689. [Google Scholar] [CrossRef]
- Wang, S.; Leonard, S.S.; Ye, J.; Ding, M.; Shi, X. The role of hydroxyl radical as a messenger in Cr(VI)-induced p53 activation. Am. J. Physiol. Cell Physiol. 2000, 279, C868–C875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Capranico, G.; Tinelli, S.; Austin, C.A.; Fisher, M.L.; Zunino, F. Different patterns of gene expression of topoisomerase II isoforms in differentiated tissues during murine development. Biochim. Biophys. Acta 1992, 1132, 43–48. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Gehl, J.; Boesgaard, M.; Paaske, T.; Vittrup Jensen, B.; Dombernowsky, P. Combined doxorubicin and paclitaxel in advanced breast cancer: Effective and cardiotoxic. Ann. Oncol. 1996, 7, 687–693. [Google Scholar] [CrossRef]
- Chan, S.; Friedrichs, K.; Noel, D.; Pintér, T.; Van Belle, S.; Vorobiof, D.; Duarte, R.; Gil Gil, M.; Bodrogi, I.; Murray, E.; et al. Prospective Randomized Trial of Docetaxel Versus Doxorubicin in Patients with Metastatic Breast Cancer. J. Clin. Oncol. 1999, 17, 2341–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowinsky, E.K.; McGuire, W.P.; Guarnieri, T.; Fisherman, J.S.; Christian, M.C.; Donehower, R.C. Cardiac disturbances during the administration of taxol. J. Clin. Oncol. 1991, 9, 1704–1712. [Google Scholar] [CrossRef]
- McGuire, W.P.; Rowinsky, E.K.; Rosenshein, N.B.; Grumbine, F.C.; Ettinger, D.S.; Armstrong, D.K.; Donehower, R.C. Taxol: A Unique Antineoplastic Agent with Significant Activity in Advanced Ovarian Epithelial Neoplasms. Ann. Intern. Med. 1989, 111, 273–279. [Google Scholar] [CrossRef]
- Bristow, M.R.; Sageman, W.S.; Scott, R.H.; Billingham, M.E.; Bowden, R.E.; Kernoff, R.S.; Snidow, G.H.; Daniels, J.R. Acute and Chronic Cardiovascular Effects of Doxorubicin in the Dog: The cardiovascular pharmacology of drug-induced histamine release. J. Cardiovasc. Pharmacol. 1980, 2, 487–515. [Google Scholar] [CrossRef]
- Gianni, L.; Munzone, E.; Capri, G.; Fulfaro, F.; Tarenzi, E.; Villani, F.; Spreafico, C.; Laffranchi, A.; Caraceni, A.; Martini, C. Paclitaxel by 3-hour infusion in combination with bolus doxorubicin in women with untreated metastatic breast cancer: High antitumor efficacy and cardiac effects in a dose-finding and sequence-finding study. J. Clin. Oncol. 1995, 13, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Viganò, L.; Locatelli, A.; Capri, G.; Giani, A.; Tarenzi, E.; Bonadonna, G. Human pharmacokinetic characterization and in vitro study of the interaction between doxorubicin and paclitaxel in patients with breast cancer. J. Clin. Oncol. 1997, 15, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Burridge, P.W.; Li, Y.F.; Matsa, E.; Wu, H.; Ong, S.-G.; Sharma, A.; Holmström, A.; Chang, A.C.; Coronado, M.J.; Ebert, A.D.; et al. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016, 22, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piver, M.S.; Marchetti, D.L.; Parthasarathy, K.L.; Bakshi, S.; Reese, P. Doxorubicin hydrochloride (adriamycin) cardiotoxicity evaluated by sequential radionuclide angiocardiography. Cancer 1985, 56, 76–80. [Google Scholar] [CrossRef]
- van Royen, N.; Jaffe, C.C.; Krumholz, H.M.; Johnson, K.M.; Lynch, P.J.; Natale, D.; Atkinson, P.; Demon, P.; Wackers, F.J. Comparison and reproducibility of visual echocardiographic and quantitative radionyclide left ventricular ejection fractions. Am. J. Cardiol. 1996, 77, 843–850. [Google Scholar] [CrossRef]
- Seidman, A.; Hudis, C.; Pierri, M.K.; Shak, S.; Paton, V.; Ashby, M.; Murphy, M.; Stewart, S.J.; Keefe, D. Cardiac Dysfunction in the Trastuzumab Clinical Trials Experience. J. Clin. Oncol. 2002, 20, 1215–1221. [Google Scholar] [CrossRef]
- Martín, M.; Esteva, F.J.; Alba, E.; Khandheria, B.; Pérez-Isla, L.; García-Sáenz, J.; Márquez, A.; Sengupta, P.; Zamorano, J. Minimizing Cardiotoxicity While Optimizing Treatment Efficacy with Trastuzumab: Review and Expert Recommendations. Oncologist 2009, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Suter, T.M.; Procter, M.; van Veldhuisen, D.J.; Muscholl, M.; Bergh, J.; Carlomagno, C.; Perren, T.; Passalacqua, R.; Bighin, C.; Klijn, J.G.; et al. Trastuzumab-Associated Cardiac Adverse Effects in the Herceptin Adjuvant Trial. J. Clin. Oncol. 2007, 25, 3859–3865. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef]
- Hoffmann, R.; Von Bardeleben, S.; Cate, F.T.; Borges, A.C.; Kasprzak, J.; Firschke, C.; Lafitte, S.; Al-Saadi, N.; Kuntz-Hehner, S.; Engelhardt, M.; et al. Assessment of systolic left ventricular function: A multi-centre comparison of cineventriculography, cardiac magnetic resonance imaging, unenhanced and contrast-enhanced echocardiography. Eur. Heart J. 2004, 26, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Jiji, R.S.; Kramer, C.M.; Salerno, M. Non-invasive imaging and monitoring cardiotoxicity of cancer therapeutic drugs. J. Nucl. Cardiol. 2012, 19, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitani, I.; Jain, D.; Joska, T.M.; Burtness, B.; Zaret, B.L. Doxorubicin cardiotoxicity: Prevention of congestive heart failure with serial cardiac function monitoring with equilibrium radionuclide angiocardiography in the current era. J. Nucl. Cardiol. 2003, 10, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Hendel, R.C.; Patel, M.R.; Kramer, C.M.; Poon, M.; Hendel, R.C.; Carr, J.C.; Gerstad, N.A.; Gillam, L.D.; Hodgson, J.M.; Kim, R.J.; et al. ACCF/ACR/SCCT/SCMR/ASNC/NASCI/SCAI/SIR 2006 appropriateness criteria for cardiac computed tomography and cardiac magnetic resonance imaging: A report of the American College of Cardiology Foundation Quality Strategic Directions Committee Appropriateness Criteria Working Group, American College of Radiology, Society of Cardiovascular Computed Tomography, Society for Cardiovascular Magnetic Resonance, American Society of Nuclear Cardiology, North American Society for Cardiac Imaging, Society for Cardiovascular Angiography and Interventions, and Society of Interventional Radiology. J. Am. Coll. Cardiol. 2006, 48, 1475–1497. [Google Scholar] [CrossRef] [Green Version]
- Horacek, J.; Jakl, M.; Horackova, J.; Pudil, R.; Jebavy, L.; Malý, J. Assessment of anthracycline-induced cardiotoxicity with electrocardiography. Exp. Oncol. 2009, 31, 115–117. [Google Scholar] [PubMed]
- Fukumi, D.; Uchikoba, Y.; Maeda, M.; Ogawa, S. Longitudinal evaluation of anthracycline cardiotoxicity by signal-averaged electrocardiography in children with cancer. Pediatr. Int. 2002, 44, 134–140. [Google Scholar] [CrossRef]
- Adamcova, M.; Šimůnek, T.; Kaiserová, H.; Popelová, O.; Štěrba, M.; Potáčová, A.; Vávrová, J.; Maláková, J.; Geršl, V. In vitro and in vivo examination of cardiac troponins as biochemical markers of drug-induced cardiotoxicity. Toxicology 2007, 237, 218–228. [Google Scholar] [CrossRef]
- Cardinale, D.; Sandri, M.T.; Colombo, A.; Colombo, N.; Boeri, M.; Lamantia, G.; Civelli, M.; Peccatori, F.; Martinelli, G.; Fiorentini, C.; et al. Prognostic Value of Troponin I in Cardiac Risk Stratification of Cancer Patients Undergoing High-Dose Chemotherapy. Circulation 2004, 109, 2749–2754. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, D.; Sandri, M.T.; Martinoni, A.; Tricca, A.; Civelli, M.; Lamantia, G.; Cinieri, S.; Martinelli, G.; Cipolla, C.M.; Fiorentini, C. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J. Am. Coll. Cardiol. 2000, 36, 517–522. [Google Scholar] [CrossRef]
- Cao, L.; Zhu, W.; Wagar, E.A.; Meng, Q.H. Biomarkers for monitoring chemotherapy-induced cardiotoxicity. Crit. Rev. Clin. Lab. Sci. 2016, 54, 87–101. [Google Scholar] [CrossRef]
- Suga, S.; Nakao, K.; Hosoda, K.; Mukoyama, M.; Ogawa, Y.; Shirakami, G.; Arai, H.; Saito, Y.; Kambayashi, Y.; Inouye, K. Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology 1992, 130, 229–239. [Google Scholar] [CrossRef]
- Babuin, L.; Jaffe, A.S. Troponin: The biomarker of choice for the detection of cardiac injury. Can. Med. Assoc. J. 2005, 173, 1191–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, S.; Fratini, S.; Ricevuto, E.; Procaccini, V.; Stifano, G.; Mancini, M.; Di Mauro, M.; Ficorella, C.; Penco, M. Serial measurements of NT-proBNP are predictive of not-high-dose anthracycline cardiotoxicity in breast cancer patients. Br. J. Cancer 2011, 105, 1663–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katus, H.A.; Remppis, A.; Looser, S.; Hallermeier, K.; Scheffold, T.; Kübler, W. Enzyme linked immuno assay of cardiac troponin T for the detection of acute myocardial infarction in patients. J. Mol. Cell. Cardiol. 1989, 21, 1349–1353. [Google Scholar] [CrossRef]
- Lenihan, D.J.; Stevens, P.L.; Massey, M.; Plana, J.C.; Araujo, D.M.; Fanale, M.A.; Fayad, L.E.; Fisch, M.J.; Yeh, E.T. The Utility of Point-of-Care Biomarkers to Detect Cardiotoxicity During Anthracycline Chemotherapy: A Feasibility Study. J. Card. Fail. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Feola, M.; Garrone, O.; Occelli, M.; Francini, A.; Biggi, A.; Visconti, G.; Albrile, F.; Bobbio, M.; Merlano, M. Cardiotoxicity after anthracycline chemotherapy in breast carcinoma: Effects on left ventricular ejection fraction, troponin I and brain natriuretic peptide. Int. J. Cardiol. 2011, 148, 194–198. [Google Scholar] [CrossRef]
- Toba, H.; Lindsey, M.L. Extracellular matrix roles in cardiorenal fibrosis: Potential therapeutic targets for CVD and CKD in the elderly. Pharmacol. Ther. 2018, 193, 99–120. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Sarrazy, V.; Koehler, A.; Chow, M.L.; Zimina, E.; Li, C.X.; Kato, H.; Caldarone, C.A.; Hinz, B. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc. Res. 2014, 102, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hagood, J.S.; Lu, B.; Merryman, W.D.; Murphy-Ullrich, J.E. Thy-1-Integrin alphav beta5 Interactions Inhibit Lung Fibroblast Contraction-induced Latent Transforming Growth Factor-β1 Activation and Myofibroblast Differentiation. J. Biol. Chem. 2010, 285, 22382–22393. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flevaris, P.; Vaughan, D. The Role of Plasminogen Activator Inhibitor Type-1 in Fibrosis. Semin. Thromb. Hemost. 2017, 43, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenes. Tissue Repair 2012, 5, S24. [Google Scholar] [CrossRef] [Green Version]
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: Mechanisms and targets. Semin. Cell Dev. Biol. 2020, 101, 115–122. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Bansal, T.; Chatterjee, E.; Singh, J.; Ray, A.; Kundu, B.; Thankamani, V.; Sengupta, S.; Sarkar, S. Arjunolic acid, a peroxisome proliferator-activated receptor α agonist, regresses cardiac fibrosis by inhibiting non-canonical TGF-β signaling. J. Biol. Chem. 2017, 292, 16440–16462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological Ventricular Remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Holweg, C.T.; Baan, C.C.; Niesters, H.G.; Vantrimpont, P.J.; Mulder, P.G.; Maat, A.P.; Weimar, W.; Balk, A.H. TGF-β1 Gene polymorphisms in patients with end-stage heart failure. J. Heart Lung Transplant. 2001, 20, 979–984. [Google Scholar] [CrossRef]
- Cho, N.; Razipour, S.E.; McCain, M.L. Featured Article: TGF-β1 dominates extracellular matrix rigidity for inducing differentiation of human cardiac fibroblasts to myofibroblasts. Exp. Biol. Med. 2018, 243, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Liguori, T.T.A.; Liguori, G.R.; Moreira, L.F.P.; Harmsen, M.C. Fibroblast growth factor-2, but not the adipose tissue-derived stromal cells secretome, inhibits TGF-β1-induced differentiation of human cardiac fibroblasts into myofibroblasts. Sci. Rep. 2018, 8, 16633. [Google Scholar] [CrossRef] [PubMed]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Dobaczewski, M.; Bujak, M.; Li, N.; Gonzalez-Quesada, C.; Mendoza, L.H.; Wang, X.-F.; Frangogiannis, N.G. Smad3 Signaling Critically Regulates Fibroblast Phenotype and Function in Healing Myocardial Infarction. Circ. Res. 2010, 107, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Gerarduzzi, C.; Di Battista, J.A. Myofibroblast repair mechanisms post-inflammatory response: A fibrotic perspective. Inflamm. Res. 2016, 66, 451–465. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Ren, G.; Kweon, H.J.; Dobaczewski, M.; Reddy, A.; Taffet, G.; Wang, X.-F.; Frangogiannis, N. Essential Role of Smad3 in Infarct Healing and in the Pathogenesis of Cardiac Remodeling. Circulation 2007, 116, 2127–2138. [Google Scholar] [CrossRef] [Green Version]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.-J.; et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Gao, X.; He, X.; Luo, B.; Peng, L.; Lin, J.; Zuo, Z. Angiotensin II increases collagen I expression via transforming growth factor-beta1 and extracellular signal-regulated kinase in cardiac fibroblasts. Eur. J. Pharmacol. 2009, 606, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.K.S.; Falkenham, A.; Myers, T.; Légaré, J.-F. Connective tissue growth factor expression after angiotensin II exposure is dependent on transforming growth factor-β signaling via the canonical Smad-dependent pathway in hypertensive induced myocardial fibrosis. J. Renin Angiotensin Aldosterone Syst. 2018, 19, 1470320318759358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Deng, C.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential Role of Smad3 in Angiotensin II–Induced Vascular Fibrosis. Circ. Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Zhang, Y.Y.; Huang, X.R.; Wu, Y.; Chung, A.C.; Wu, E.X.; Szalai, A.J.; Wong, B.C.; Lau, C.-P.; Lan, H.Y. C-Reactive Protein Promotes Cardiac Fibrosis and Inflammation in Angiotensin II–Induced Hypertensive Cardiac Disease. Hypertension 2010, 55, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563. [Google Scholar] [CrossRef]
- Huang, H.; Tang, Q.-Z.; Wang, A.-B.; Chen, M.; Yan, L.; Liu, C.; Jiang, H.; Yang, Q.; Bian, Z.-Y.; Bai, X.; et al. Tumor Suppressor A20 Protects Against Cardiac Hypertrophy and Fibrosis by Blocking Transforming Growth Factor-β–Activated Kinase 1–Dependent Signaling. Hypertension 2010, 56, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Laviades, C.; Varo, N.; Díez, J. Transforming Growth Factor β in Hypertensives With Cardiorenal Damage. Hypertension 2000, 36, 517–522. [Google Scholar] [CrossRef] [Green Version]
- Vliegen, H.W.; Van Der Laarse, A.; Cornelisse, C.J.; Eulderink, F. Myocardial changes in pressure overload-induced left ventricular hypertrophy. A study on tissue composition, polyploidization and multinucleation. Eur. Heart J. 1991, 12, 488–494. [Google Scholar] [CrossRef]
- Caulfield, J.B.; Borg, T.K. The collagen network of the heart. Lab. Investig. 1979, 40, 364–372. [Google Scholar] [PubMed]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2013, 70, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Border, W.A.; Noble, N.A. Transforming Growth Factor β in Tissue Fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Furtado, M.B.; Costa, M.W.; Pranoto, E.A.; Salimova, E.; Pinto, A.R.; Lam, N.; Park, A.; Snider, P.; Chandran, A.; Harvey, R.; et al. Cardiogenic Genes Expressed in Cardiac Fibroblasts Contribute to Heart Development and Repair. Circ. Res. 2014, 114, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Takefuji, M.; Ngai, C.Y.; Carvalho, J.; Bayer, J.; Wietelmann, A.; Poetsch, A.; Hoelper, S.; Conway, S.J.; Möllmann, H.; et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ. Res. 2016, 118, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- van Putten, S.; Shafieyan, Y.; Hinz, B. Mechanical control of cardiac myofibroblasts. J. Mol. Cell. Cardiol. 2016, 93, 133–142. [Google Scholar] [CrossRef]
- Stempien-Otero, A.; Kim, D.-H.; Davis, J. Molecular networks underlying myofibroblast fate and fibrosis. J. Mol. Cell. Cardiol. 2016, 97, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Gourdie, R.G.; Dimmeler, S.; Kohl, P. Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nat. Rev. Drug Discov. 2016, 15, 620–638. [Google Scholar] [CrossRef] [Green Version]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef]
- Olson, R.D.; Mushlin, P.S. Doxorubicin cardiotoxicity: Analysis of prevailing hypotheses. FASEB J. 1990, 4, 3076–3086. [Google Scholar] [CrossRef]
- Krstić, J.; Trivanović, D.; Mojsilović, S.; Santibanez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxid. Med. Cell. Longev. 2015, 2015, 654594. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming Growth Factor-β Function Blocking Prevents Myocardial Fibrosis and Diastolic Dysfunction in Pressure-Overloaded Rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Turaga, R.C.; Yuan, Y.; Satyanarayana, G.; Mishra, F.; Bian, Z.; Liu, W.; Sun, L.; Yang, J.; Liu, Z.-R. Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J. Exp. Med. 2021, 218, e20200712. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- De Wever, O.; Nguyen, Q.-D.; Van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent proinvasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Surowiak, P.; Murawa, D.; Materna, V.; Maciejczyk, A.; Pudelko, M.; Ciesla, S.; Breborowicz, J.; Murawa, P.; Zabel, M.; Dietel, M.; et al. Occurence of stromal myofibroblasts in the invasive ductal breast cancer tissue is an unfavourable prognostic factor. Anticancer Res. 2007, 27, 2917–2924. [Google Scholar]
- Neve, A.; Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Extracellular Matrix Modulates Angiogenesis in Physiological and Pathological Conditions. BioMed Res. Int. 2014, 2014, 756078. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Rowe, R.G.; Hiraoka, N.; George, J.P.; Wirtz, D.; Mosher, D.F.; Virtanen, I.; Chernousov, M.A.; Weiss, S.J. Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. 2008, 22, 1231–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beacham, D.A.; Cukierman, E. Stromagenesis: The changing face of fibroblastic microenvironments during tumor progression. Semin. Cancer Biol. 2005, 15, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; DeVecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabek, J.; Kułach, A.; Monastyrska-Cup, B.; Gasior, Z. Transforming growth factor beta and cardiovascular diseases: The other facet of the ‘protective cytokine’. Pharmacol. Rep. 2006, 58, 799–805. [Google Scholar] [PubMed]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti-inflammatory and pro-inflammatory roles of TGF-β, IL-10, and IL-22 in immunity and autoimmunity. Curr. Opin. Pharmacol. 2009, 9, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Hogan, B.L.M.; Blessing, M.; Winnier, G.E.; Suzuki, N.; Jones, C.M. Growth factors in development: The role of TGF-β related polypeptide signalling molecules in embryogenesis. Development 1994, 1994, 53–60. [Google Scholar] [CrossRef]
- De Oliveira, F.L.; Araújo-Jorge, T.C.; De Souza, E.M.; De Oliveira, G.M.; Degrave, W.M.; Feige, J.-J.; Bailly, S.; Waghabi, M.C. Oral Administration of GW788388, an Inhibitor of Transforming Growth Factor Beta Signaling, Prevents Heart Fibrosis in Chagas Disease. PLoS Negl. Trop. Dis. 2012, 6, e1696. [Google Scholar] [CrossRef]
- Ferreira, R.R.; Abreu, R.d.S.; Vilar-Pereira, G.; Degrave, W.; Meuser-Batista, M.; Ferreira, N.V.C.; da Cruz Moreira, O.; da Silva Gomes, N.L.; Mello de Souza, E.; Ramos, I.P.; et al. TGF-β inhibitor therapy decreases fibrosis and stimulates cardiac improvement in a pre-clinical study of chronic Chagas’ heart disease. PLoS Negl. Trop. Dis. 2019, 13, e0007602. [Google Scholar] [CrossRef] [Green Version]
- Deten, A.; Hölzl, A.; Leicht, M.; Barth, W.; Zimmer, H.-G. Changes in Extracellular Matrix and in Transforming Growth Factor Beta Isoforms After Coronary Artery Ligation in Rats. J. Mol. Cell. Cardiol. 2001, 33, 1191–1207. [Google Scholar] [CrossRef]
- Dewald, O.; Ren, G.; Duerr, G.D.; Zoerlein, M.; Klemm, C.; Gersch, C.; Tincey, S.; Michael, L.H.; Entman, M.L.; Frangogiannis, N.G. Of Mice and Dogs: Species-Specific Differences in the Inflammatory Response Following Myocardial Infarction. Am. J. Pathol. 2004, 164, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Heymans, S.; Schroen, B.; Vermeersch, P.; Milting, H.; Gao, F.; Kassner, A.; Gillijns, H.; Herijgers, P.; Flameng, W.; Carmeliet, P.; et al. Increased Cardiac Expression of Tissue Inhibitor of Metalloproteinase-1 and Tissue Inhibitor of Metalloproteinase-2 Is Related to Cardiac Fibrosis and Dysfunction in the Chronic Pressure-Overloaded Human Heart. Circulation 2005, 112, 1136–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto-Ida, M.; Takimoto, Y.; Aoyama, T.; Akao, M.; Takeda, T.; Kita, T. Activation of TGF-β1-TAK1-p38 MAPK pathway in spared cardiomyocytes is involved in left ventricular remodeling after myocardial infarction in rats. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H709–H715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellmers, L.J.; Scott, N.J.A.; Medicherla, S.; Pilbrow, A.; Bridgman, P.G.; Yandle, T.G.; Richards, A.M.; Protter, A.A.; Cameron, V.A. Transforming Growth Factor-β Blockade Down-Regulates the Renin-Angiotensin System and Modifies Cardiac Remodeling after Myocardial Infarction. Endocrinology 2008, 149, 5828–5834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotfy, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic complications of diabetes mellitus: A mini review. Curr. Diabetes Rev. 2017, 13, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cui, L.; Guan, G.; Wang, J.; Qiu, C.; Yang, T.; Guo, Y.; Liu, Z. Matrine suppresses cardiac fibrosis by inhibiting the TGF-β/Smad pathway in experimental diabetic cardiomyopathy. Mol. Med. Rep. 2018, 17, 1775–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumantra, I.G.; Astuti, A.; Martanto, E.; Sihite, T.A.; Maman Abdurahman, R.; Iqbal, M.; Rizki Akbar, M.; Martha, J.W. ORThe Effect of Carvedilol on Subclinical Left Ventricular Dysfunction after Chemotherapy of FAC Regimen in Breast Cancer Patients: An Experimental Study. Eur. Heart J. Suppl. 2021, 23, suab122-064. [Google Scholar]
- Akpek, M.; Ozdogru, I.; Sahin, O.; Inanc, M.; Dogan, A.; Yazici, C.; Berk, V.; Karaca, H.; Kalay, N.; Oguzhan, A.; et al. Protective effects of spironolactone against anthracycline-induced cardiomyopathy. Eur. J. Heart Fail. 2015, 17, 81–89. [Google Scholar] [CrossRef]
- Calvillo-Argüelles, O.; Abdel-Qadir, H.; Michalowska, M.; Billia, F.; Suntheralingam, S.; Amir, E.; Thavendiranathan, P. Cardioprotective Effect of Statins in Patients With HER2-Positive Breast Cancer Receiving Trastuzumab Therapy. Can. J. Cardiol. 2018, 35, 153–159. [Google Scholar] [CrossRef]
- Chen, J.L.; Shang, Q.H.; Hu, W.; Liu, C.; Mao, W.H.; Liu, H.Q. Role of TGF-β1/Smads pathway in carotid artery remodeling in renovascular hypertensive rats and prevention by Enalapril and Amlodipine. J. Geriatr. Cardiol. 2012, 9, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Peters, H.; Border, W.A.; Noble, N.A. Targeting TGF-β overexpression in renal disease: Maximizing the antifibrotic action of angiotensin II blockade. Kidney Int. 1998, 54, 1570–1580. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, E.; Marui, A.; Tsukashita, M.; Nishina, T.; Wang, J.; Muranaka, H.; Ikeda, T.; Komeda, M. Carvedilol may alleviate late cardiac remodelling following surgical ventricular restoration. Eur. J. Cardio-Thorac. Surg. 2009, 37, 362–367. [Google Scholar] [CrossRef] [PubMed]
- El-Wakeel, S.A.; Rahmo, R.M.; El-Abhar, H.S. Anti-fibrotic impact of Carvedilol in a CCl-4 model of liver fibrosis via serum microRNA-200a/SMAD7 enhancement to bridle TGF-β1/EMT track. Sci. Rep. 2018, 8, 14327. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, F.; Siddiqui, R.; DeFranco, A.; Homar, P.; Emelyanova, L.; Holmuhamedov, E.; Ross, G.; Tajik, A.J.; Jahangir, A. Simvastatin reduces TGF-β1-induced SMAD2/3-dependent human ventricular fibroblasts differentiation: Role of protein phosphatase activation. Int. J. Cardiol. 2018, 270, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Chang, G.; Liu, J.; Sun, G.; Liu, L.; Qin, S.; Zhang, D. Simvastatin ameliorates ventricular remodeling via the TGF-β1 signaling pathway in rats following myocardial infarction. Mol. Med. Rep. 2016, 13, 5093–5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, N.; Kaikita, K.; Ishii, M.; Oimatsu, Y.; Mitsuse, T.; Ito, M.; Yamanaga, K.; Fujisue, K.; Kanazawa, H.; Sueta, D.; et al. Cardioprotective Effects of Rivaroxaban on Cardiac Remodeling After Experimental Myocardial Infarction in Mice. Circ. Rep. 2020, 2, 158–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.-X.; Qi, G.-M.; Liu, O.; Li, T.-T.; Yang, M.; Cui, W.; Zhang, W.-M.; Qi, Y.-F.; Du, J. Inhibition of Platelet Activation by Clopidogrel Prevents Hypertension-Induced Cardiac Inflammation and Fibrosis. Cardiovasc. Drugs Ther. 2013, 27, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-L.; Lin, Q.-Y.; Wang, L.; Xie, X.; Zhang, Y.-L.; Li, H.-H. Rituximab prevents and reverses cardiac remodeling by depressing B cell function in mice. Biomed. Pharmacother. 2019, 114, 108804. [Google Scholar] [CrossRef]
- Malek, V.; Gaikwad, A.B. Neprilysin inhibitors: A new hope to halt the diabetic cardiovascular and renal complications? Biomed. Pharmacother. 2017, 90, 752–759. [Google Scholar] [CrossRef]
- Suematsu, Y.; Miura, S.-I.; Goto, M.; Matsuo, Y.; Arimura, T.; Kuwano, T.; Imaizumi, S.; Iwata, A.; Yahiro, E.; Saku, K. LCZ696, an angiotensin receptor-neprilysin inhibitor, improves cardiac function with the attenuation of fibrosis in heart failure with reduced ejection fraction in streptozotocin-induced diabetic mice. Eur. J. Heart Fail. 2016, 18, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Liu, Y.; Wang, R.; Hou, T.; Chen, C.; Zheng, S.; Dong, Z. Spironolactone Attenuates Doxorubicin-induced Cardiotoxicity in Rats. Cardiovasc. Ther. 2016, 34, 216–224. [Google Scholar] [CrossRef]
- Luo, J.; Gao, X.; Peng, L.; Sun, H.; Dai, G. Effects of hydrochlorothiazide on cardiac remodeling in a rat model of myocardial infarction-induced congestive heart failure. Eur. J. Pharmacol. 2011, 667, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Bellaye, P.-S.; Yanagihara, T.; Granton, E.; Sato, S.; Shimbori, C.; Upagupta, C.; Imani, J.; Hambly, N.; Ask, K.; Gauldie, J.; et al. Macitentan reduces progression of TGF-β1-induced pulmonary fibrosis and pulmonary hypertension. Eur. Respir. J. 2018, 52, 1701857. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Chen, S.; Feng, B.; Iglarz, M.; Chakrabarti, S. Renal, retinal and cardiac changes in type 2 diabetes are attenuated by macitentan, a dual endothelin receptor antagonist. Life Sci. 2012, 91, 658–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, L.P.; Zhang, R.-L.; Zheng, W.; Campanelli, J.J.; Dedkov, E.I.; Weiss, R.M.; Tomanek, R.J. Postmyocardial infarction remodeling and coronary reserve: Effects of ivabradine and beta blockade therapy. Am. J. Physiol. Circ. Physiol. 2009, 297, H322–H330. [Google Scholar] [CrossRef]
- Dias, P.; Navaratnarajah, M.; Alayoubi, S.; Cartledge, J.E.; Jayaratne, N.; Starke, R.; Sarathchandra, P.; Latif, N.; Randi, A.M.; Yacoub, M.H.; et al. 9 Ivabradine Alters Fibroblast Number and Transforming Growth Factor beta 1 Expression in Heart Failure. Heart 2014, 100, A4. [Google Scholar] [CrossRef]
- Kang, S.; Verma, S.; Hassanabad, A.F.; Teng, G.; Belke, D.D.; Dundas, J.A.; Guzzardi, D.G.; Svystonyuk, D.A.; Pattar, S.S.; Park, D.S.; et al. Direct Effects of Empagliflozin on Extracellular Matrix Remodelling in Human Cardiac Myofibroblasts: Novel Translational Clues to Explain EMPA-REG OUTCOME Results. Can. J. Cardiol. 2020, 36, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, X.; Bai, Y.; Cui, C.; Li, J.; Li, Y.; Hu, S.; Wei, Y. In Vitro Effects of Pirfenidone on Cardiac Fibroblasts: Proliferation, Myofibroblast Differentiation, Migration and Cytokine Secretion. PLoS ONE 2011, 6, e28134. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Yamashita, N.; Izumi, Y.; Nakamura, Y.; Shiota, M.; Hanatani, A.; Shimada, K.; Muro, T.; Iwao, H.; Yoshiyama, M. The antifibrotic agent pirfenidone inhibits angiotensin II-induced cardiac hypertrophy in mice. Hypertens. Res. 2011, 35, 34–40. [Google Scholar] [CrossRef]
- Shyu, K.-G.; Wang, B.-W.; Chen, W.-J.; Kuan, P.; Hung, C.-R. Mechanism of the inhibitory effect of atorvastatin on endoglin expression induced by transforming growth factor-β1 in cultured cardiac fibroblasts. Eur. J. Heart Fail. 2010, 12, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Yoshida, K.; Nakano, S.; Ohno, T.; Honda, T.; Tsubokou, Y.; Matsuoka, H. Cardioprotective Mechanisms of Eplerenone on Cardiac Performance and Remodeling in Failing Rat Hearts. Hypertension 2006, 47, 671–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, M.; Hirohata, S.; Ogawa, H.; Ohtsuki, T.; Shinohata, R.; Miyoshi, T.; Hatipoglu, F.O.; Kusachi, S.; Yamamoto, K.; Ninomiya, Y. Connective tissue growth factor induction in a pressure-overloaded heart ameliorated by the angiotensin II type 1 receptor blocker olmesartan. Hypertens. Res. 2010, 33, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, E.; Dong, Y.-F.; Kataoka, K.; Yamashita, T.; Tokutomi, Y.; Matsuba, S.; Ichijo, H.; Ogawa, H.; Kim-Mitsuyama, S. Olmesartan Prevents Cardiovascular Injury and Hepatic Steatosis in Obesity and Diabetes, Accompanied by Apoptosis Signal Regulating Kinase-1 Inhibition. Hypertension 2008, 52, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Zhang, J.; Zhi, H.; Hong, B.; Zhang, S.; Guo, H.; Li, L. The beneficial effects of tadalafil on left ventricular dysfunction in doxorubicin-induced cardiomyopathy. J. Cardiol. 2013, 62, 110–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, F.; Chen, M.; Yu, B.; Yang, Y.; Xu, G.; Gui, F.; Liu, Z.; Bai, X.; Chen, Z. Berberine regulates proliferation, collagen synthesis and cytokine secretion of cardiac fibroblasts via AMPK-mTOR-p70S6K signaling pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 12509–12516. [Google Scholar]
- Salam, T.; El-Bakly, W.; Badawy, A.; Hasanin, A.; Raafat, M. Melatonin combination with perindopril alleviated doxorubicin cardiac toxicity in L-NAME hypertensive rats: Comparative study with perindopril. Ain Shams J. Forensic Med. Clin. Toxicol. 2020, 34, 69–81. [Google Scholar] [CrossRef]
- Meurer, S.K.; Lahme, B.; Tihaa, L.; Weiskirchen, R.; Gressner, A.M. N-Acetyl-l-cysteine suppresses TGF-β signaling at distinct molecular steps: The biochemical and biological efficacy of a multifunctional, antifibrotic drug. Biochem. Pharmacol. 2005, 70, 1026–1034. [Google Scholar] [CrossRef]
- Talasaz, A.H.; Khalili, H.; Jenab, Y.; Salarifar, M.; Broumand, M.A.; Darabi, F. N-Acetylcysteine Effects on Transforming Growth Factor-β and Tumor Necrosis Factor-α Serum Levels as Pro-Fibrotic and Inflammatory Biomarkers in Patients Following ST-Segment Elevation Myocardial Infarction. Drugs R&D 2013, 13, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine—Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lu, Y.; Qu, C.; Zhang, E.; Tang, Y. GW26-e0768 Ticagrelor Prevents Cardiac Inflammation and Fibrosis of Hyertension Rats. J. Am. Coll. Cardiol. 2015, 66, C268. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Wei, H.; Wang, D. Novel mechanism of cardiac protection by valsartan: Synergetic roles of TGF -β1 and HIF -1α in Ang II-mediated fibrosis after myocardial infarction. J. Cell. Mol. Med. 2015, 19, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Ma, X.; Feng, W.; Fu, Y.; Lu, Z.; Xu, M.; Shen, Q.; Zhu, Y.; Zhang, Y. Metformin attenuates cardiac fibrosis by inhibiting the TGFβ1–Smad3 signalling pathway. Cardiovasc. Res. 2010, 87, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoda, K.; Ohtake, K.; Uchida, H.; Ito, J.; Uchida, M.; Natsume, H.; Tamada, H.; Kobayashi, J. Dietary nitrite supplementation attenuates cardiac remodeling in l -NAME-induced hypertensive rats. Nitric Oxide 2017, 67, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ceron, C.S.; Rizzi, E.; Guimarães, D.A.; Martins-Oliveira, A.; Gerlach, R.F.; Tanus-Santos, J.E. Nebivolol attenuates prooxidant and profibrotic mechanisms involving TGF-β and MMPs, and decreases vascular remodeling in renovascular hypertension. Free Radic. Biol. Med. 2013, 65, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Rai, N.; Veeroju, S.; Schymura, Y.; Janssen, W.; Wietelmann, A.; Kojonazarov, B.; Weissmann, N.; Stasch, J.-P.; Ghofrani, H.A.; Seeger, W.; et al. Effect of Riociguat and Sildenafil on Right Heart Remodeling and Function in Pressure Overload Induced Model of Pulmonary Arterial Banding. BioMed Res. Int. 2018, 2018, 3293584. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.-J.; Su, K.; Cui, G.-Z.; Yang, W.-H.; Chen, L.; Yang, M.; Liu, Y.-Q.; Dai, D.-L. Association between transforming growth factor-β1 expression and the clinical features of triple negative breast cancer. Oncol. Lett. 2016, 11, 4040–4044. [Google Scholar] [CrossRef] [Green Version]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of Colorectal Cancer on a TGF-β-Driven Program in Stromal Cells for Metastasis Initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Yokouchi, H.; Nishihara, H.; Harada, T.; Ishida, T.; Yamazaki, S.; Kikuchi, H.; Oizumi, S.; Uramoto, H.; Tanaka, F.; Harada, M.; et al. Immunohistochemical profiling of receptor tyrosine kinases, MED12, and TGF-βRII of surgically resected small cell lung cancer, and the potential of c-kit as a prognostic marker. Oncotarget 2017, 8, 39711–39726. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, A.; McGarry, S.; Chilumula, S.C.; Kandunuri, R.; Vinod, V. Clinically Translatable Approaches of Inhibiting TGF-β to Target Cancer Stem Cells in TNBC. Biomedicines 2021, 9, 1386. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.; Georgoudaki, A.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.; Nelson, C.M.; et al. TGF-β1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Wang, C.; Zhou, Z.; Chen, C.; Liu, R.; Wang, D. Transforming growth factor-beta increases breast cancer stem cell population partially through upregulating PMEPA1 expression. Acta Biochim. Biophys. Sin. 2016, 48, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Buck, M.B.; Fritz, P.; Dippon, J.; Zugmaier, G.; Knabbe, C. Prognostic Significance of Transforming Growth Factor β Receptor II in Estrogen Receptor-Negative Breast Cancer Patients. Clin. Cancer Res. 2004, 10, 491–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleksakhina, S.; Kashyap, A.; Imyanitov, E.N. Mechanisms of acquired tumor drug resistance. Biochim. Biophys. Acta 2019, 1872, 188310. [Google Scholar] [CrossRef]
- Murayama, T.; Gotoh, N. Drug resistance mechanisms of cancer stem-like cells and their therapeutic potential as drug targets. Cancer Drug Resist 2019, 2, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, A.; McGarry, S.; Li, L.; Jia, D.; Ooi, S.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; Nessim, C.; Yao, Z.; et al. Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 2018, 12, 423–440. [Google Scholar] [CrossRef]
- Sulaiman, A.; Yao, Z.; Wang, L. Re-evaluating the role of epithelial-mesenchymal-transition in cancer progression. J. Biomed. Res. 2018, 32, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Sommariva, M.; Limonta, P.; Gagliano, N. Epithelial-To-Mesenchymal Transition Markers and CD44 Isoforms Are Differently Expressed in 2D and 3D Cell Cultures of Prostate Cancer Cells. Cells 2019, 8, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Vikram, R.; Chou, W.C.; Hung, S.-C.; Shen, C.-Y. Tumorigenic and Metastatic Role of CD44−/low/CD24−/low Cells in Luminal Breast Cancer. Cancers 2020, 12, 1239. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D.; et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFβ/TNFα-mediated epithelial–mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wu, J.; Mao, K.; Deng, H.; Yang, Y.; Zhou, E.; Liu, J. Role of transforming growth factor-β1 in triple negative breast cancer patients. Int. J. Surg. 2017, 45, 72–76. [Google Scholar] [CrossRef]
- Lu, S.; Dong, Z. Characterization of TGF-β-regulated interleukin-8 expression in human prostate cancer cells. Prostate 2006, 66, 996–1004. [Google Scholar] [CrossRef]
- Bates, R.C.; De Leo, M.J., III; Mercurio, A.M. The epithelial–mesenchymal transition of colon carcinoma involves expression of IL-8 and CXCR-1-mediated chemotaxis. Exp. Cell Res. 2004, 299, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Tan, Y.; Liu, H.; Ooi, S.; Li, L.; Wright, K.; Bennett, S.; Addison, C.L.; Wang, L. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget 2015, 7, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Wang, J. Correlations of MRP1 gene with serum TGF-beta 1 and IL-8 in breast cancer patients during chemotherapy. Off. J. Balk. Union Oncol. 2018, 23, 1302–1308. [Google Scholar]
- Ignacio, R.M.C.; Gibbs, C.R.; Lee, E.-S.; Son, D.-S. The TGFα-EGFR-Akt signaling axis plays a role in enhancing proinflammatory chemokines in triple-negative breast cancer cells. Oncotarget 2018, 9, 29286–29303. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.N.; Burton, L.J.; Henderson, V.; Randle, D.D.; Morton, D.J.; Smith, B.A.; Taliaferro-Smith, L.; Nagappan, P.; Yates, C.; Zayzafoon, M.; et al. Snail Promotes Epithelial Mesenchymal Transition in Breast Cancer Cells in Part via Activation of Nuclear ERK. PLoS ONE 2014, 9, e104987. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gu, L.-N.; Shan, B.-E.; Geng, C.-Z.; Sang, M.-X. Biomarkers for EMT and MET in breast cancer: An update. Oncol. Lett. 2016, 12, 4869–4876. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Kim, M.-J.; Park, S.-A.; Kim, J.-S.; Min, K.-N.; Kim, D.-K.; Lim, W.; Nam, J.-S.; Sheen, Y.Y. Combinatorial TGF-β attenuation with paclitaxel inhibits the epithelial-to-mesenchymal transition and breast cancer stem-like cells. Oncotarget 2015, 6, 37526–37543. [Google Scholar] [CrossRef] [Green Version]
- Wardhani, B.W.; Puteri, M.U.; Watanabe, Y.; Louisa, M.; Setiabudy, R.; Kato, M. TGF-β-Induced TMEPAI Attenuates the Response of Triple-Negative Breast Cancer Cells to Doxorubicin and Paclitaxel. J. Exp. Pharmacol. 2020, 12, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Singha, P.K.; Yeh, I.-T.; Venkatachalam, M.A.; Saikumar, P. Transforming growth factor-β (TGF-β)–inducible gene TMEPAI converts TGF-β from a tumor suppressor to a tumor promoter in breast cancer. Cancer Res. 2010, 70, 6377–6383. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, Y.-L.; Ji, G.; Fang, W.; Gao, Z.; Liu, Y.; Wang, J.; Ding, X.; Gao, F. Sorafenib Inhibits Epithelial-Mesenchymal Transition through an Epigenetic-Based Mechanism in Human Lung Epithelial Cells. PLoS ONE 2013, 8, e64954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yu, T.; Yuan, Y.; Zhuang, H.; Wang, Z.; Liu, X.; Feng, M. Sorafenib reduces hepatic infiltrated regulatory T cells in hepatocellular carcinoma patients by suppressing TGF-beta signal. J. Surg. Oncol. 2013, 107, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ozawa, Y.; Kimura, T.; Sato, Y.; Kuznetsov, G.; Xu, S.; Uesugi, M.; Agoulnik, S.; Taylor, N.P.; Funahashi, Y.; et al. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial–mesenchymal transition (EMT) to mesenchymal–epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Schöffski, P.; Littlefield, B.A. Multiple modes of action of eribulin mesylate: Emerging data and clinical implications. Cancer Treat. Rev. 2018, 70, 190–198. [Google Scholar] [CrossRef] [Green Version]
- Kudo, M. Pembrolizumab for the Treatment of Hepatocellular Carcinoma. Liver Cancer 2019, 8, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, X.; He, X.; Xu, Y.; Wang, X. Complete response to the combination of Lenvatinib and Pembrolizumab in an advanced hepatocellular carcinoma patient: A case report. BMC Cancer 2019, 19, 1062. [Google Scholar] [CrossRef]
- Qiu, H.; Li, J.; Liu, Q.; Tang, M.; Wang, Y. Apatinib, a novel tyrosine kinase inhibitor, suppresses tumor growth in cervical cancer and synergizes with Paclitaxel. Cell Cycle 2018, 17, 1235–1244. [Google Scholar] [CrossRef]
- Liu, B.; An, T.; Li, M.; Yi, Z.; Li, C.; Sun, X.; Guan, X.; Li, L.; Wang, Y.; Zhang, Y.; et al. The association between early-onset cardiac events caused by neoadjuvant or adjuvant chemotherapy in triple-negative breast cancer patients and some novel autophagy-related polymorphisms in their genomic DNA: A real-world study. Cancer Commun. 2018, 38, 71. [Google Scholar] [CrossRef] [Green Version]
- Avalyan, A.; Shitov, V.; Saidova, M.; Bolotova, M.; Oshchepkova, E.; Chazova, I. Cardiotoxicity in Patients with Triple Negative Breast Cancer Undergoing Anthracycline Chemotherapy Depending on Blood Pressure Level. J. Hypertens. 2018, 36, e56. [Google Scholar] [CrossRef]
- Strongman, H.; Gadd, S.; Matthews, A.; Mansfield, K.E.; Stanway, S.; Lyon, A.R.; Dos-Santos-Silva, I.; Smeeth, L.; Bhaskaran, K. Medium and long-term risks of specific cardiovascular diseases in survivors of 20 adult cancers: A population-based cohort study using multiple linked UK electronic health records databases. Lancet 2019, 394, 1041–1054. [Google Scholar] [CrossRef] [Green Version]


| Inhibitor | Clinical Trial Number | Mechanism | References |
|---|---|---|---|
| Sorafenib | NCT02624700—w/Pemetrexed | -Suppression of TGFb1-mediated EMT via epigenetic modification of TGFb1 and Smad2/3 promoters through loss of active histone markers (H3K4me3 and/or H3K9ac) -Has also been shown to disrupt the phosphorylation of Smad2/3 -Suppression of TGFb signaling in hepatocellular carcinoma | [201,202] |
| Halaven (eribulin mesylate) | NCT01372579—w/Carboplatin NCT02120469 | Suppresses metastasis by inhibiting TGFb-mediated phosphorylation of Smad2/3 (potentially by altering the interactions between Smad proteins and microtubules following erlubin binding) | [203,204] |
| Pembrolizumab (MK-3475) | NCT02644369 NCT02730130 NCT02734290 NCT03036488 NCT02555657 NCT02819518 NCT02981303—w/Imprime PGG NCT03567720 NCT02657889—w/Niraparib NCT02971761—w/Enobosarm NCT01676753—w/Dinaciclib NCT02178722 | Decreased the production of TGFb in the tumor microenvironment | [205,206] |
| Apatinib | NCT03075462 NCT03394287 | Downregulates the TGFb1 pathway | [207] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulaiman, A.; Chambers, J.; Chilumula, S.C.; Vinod, V.; Kandunuri, R.; McGarry, S.; Kim, S. At the Intersection of Cardiology and Oncology: TGFβ as a Clinically Translatable Therapy for TNBC Treatment and as a Major Regulator of Post-Chemotherapy Cardiomyopathy. Cancers 2022, 14, 1577. https://doi.org/10.3390/cancers14061577
Sulaiman A, Chambers J, Chilumula SC, Vinod V, Kandunuri R, McGarry S, Kim S. At the Intersection of Cardiology and Oncology: TGFβ as a Clinically Translatable Therapy for TNBC Treatment and as a Major Regulator of Post-Chemotherapy Cardiomyopathy. Cancers. 2022; 14(6):1577. https://doi.org/10.3390/cancers14061577
Chicago/Turabian StyleSulaiman, Andrew, Jason Chambers, Sai Charan Chilumula, Vishak Vinod, Rohith Kandunuri, Sarah McGarry, and Sung Kim. 2022. "At the Intersection of Cardiology and Oncology: TGFβ as a Clinically Translatable Therapy for TNBC Treatment and as a Major Regulator of Post-Chemotherapy Cardiomyopathy" Cancers 14, no. 6: 1577. https://doi.org/10.3390/cancers14061577
APA StyleSulaiman, A., Chambers, J., Chilumula, S. C., Vinod, V., Kandunuri, R., McGarry, S., & Kim, S. (2022). At the Intersection of Cardiology and Oncology: TGFβ as a Clinically Translatable Therapy for TNBC Treatment and as a Major Regulator of Post-Chemotherapy Cardiomyopathy. Cancers, 14(6), 1577. https://doi.org/10.3390/cancers14061577