
MMP9: A Tough Target for Targeted Therapy for Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
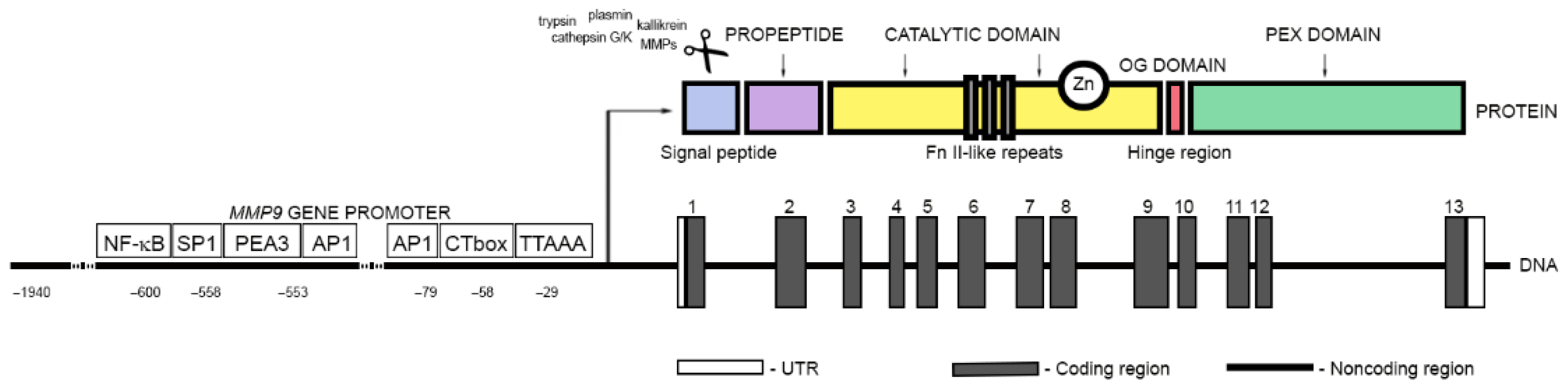
2. Matrix Metalloproteinase 9: A Secreted Member of the Zinc Metalloendopeptidase Family
2.1. Biochemical Characteristics of MMP9
2.2. Transcriptional and Post-Transcriptional Regulation of MMP9 Gene Expression
2.3. Extracellular Signal-Induced Upregulation of MMP9
2.4. MMP9 Synthesis and Activation
2.5. Substrate Specificity of MMP9
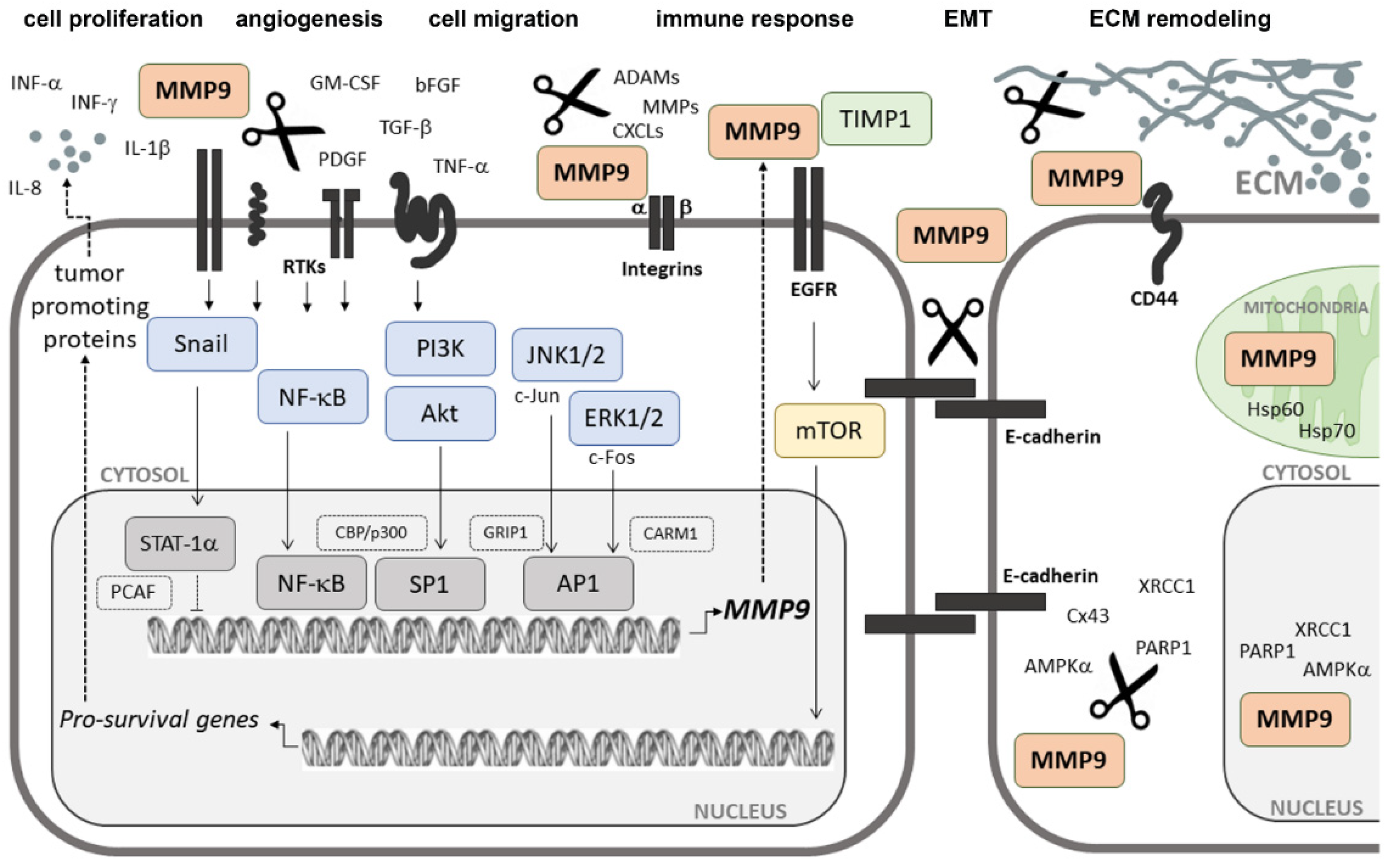
3. MMP9 and Cancer
3.1. MMP9 in Cancer Cell Adhesion and Migration
3.2. MMP9 in Cancer-Related Inflammation
3.3. MMP9 in Tumor Microenvironment Formation
3.4. Intracellular Activities of MMP9
3.5. Anticancer Effect of MMP9
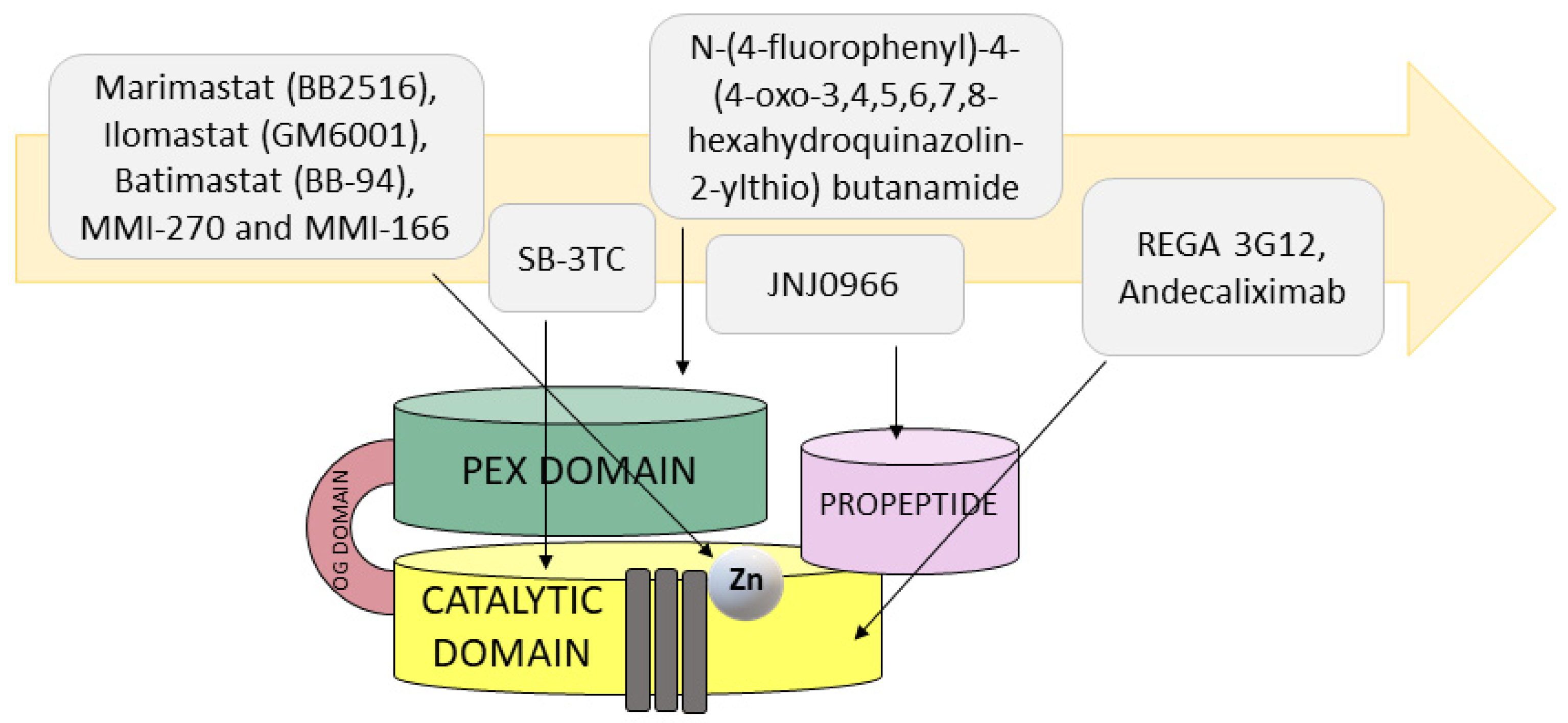
4. MMP9 as an Anticancer Drug Target
4.1. Small-Molecule Inhibitors of MMP9
4.2. Inhibitory Antibodies for MMP9
4.3. Natural Products with Anti-MMP9 Activity
4.4. RNAi Therapeutics
5. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R.; Czapla, L.; Szelachowski, P.; Wierzbicki, J.; Grabowski, K.; Sikorski, A.F. Upregulated expression and activation of membrane? Associated proteases in esophageal squamous cell carcinoma. Oncol. Rep. 2014, 31, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- Martins, L.M.; Dourado, C.S.D.M.E.; Campos-Verdes, L.M.; Sampaio, F.A.; Revoredo, C.M.S.; Costa-Silva, D.R.; Barros-Oliveira, M.D.C.; Junior, E.D.J.N.; Rego-Medeiros, L.M.D.; Gebrim, L.H.; et al. Expression of matrix metalloproteinase 2 and 9 in breast cancer and breast fibroadenoma: A randomized, double-blind study. Oncotarget 2019, 10, 6879–6884. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, R.; Hagström, J.; Tervahartiala, T.; Sorsa, T.; Haglund, C.; Isoniemi, H. High Expression of MMP-9 in Primary Tumors and High Preoperative MPO in Serum Predict Improved Prognosis in Colorectal Cancer with Operable Liver Metastases. Oncology 2021, 99, 144–160. [Google Scholar] [CrossRef]
- Jiang, H.; Li, H. Prognostic values of tumoral MMP2 and MMP9 overexpression in breast cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 149. [Google Scholar] [CrossRef]
- Shao, W.; Wang, W.; Xiong, X.-G.; Cao, C.; Yan, T.D.; Chen, G.; Chen, H.; Yin, W.; Liu, J.; Gu, Y.; et al. Prognostic impact of MMP-2 and MMP-9 expression in pathologic stage IA non-small cell lung cancer. J. Surg. Oncol. 2011, 104, 841–846. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Collier, I.E.; Marmer, B.L.; Eisen, A.Z.; Grant, G.A.; Goldberg, G.I. SV40-transformed human lung fibroblasts secrete a 92-kDa type IV collagenase which is identical to that secreted by normal human macrophages. J. Biol. Chem. 1989, 264, 17213–17221, Erratum in: J. Biol. Chem. 1990, 265, 22570.. [Google Scholar] [CrossRef]
- Christensen, J.; Shastri, V.P. Matrix-metalloproteinase-9 is cleaved and activated by cathepsin K. BMC Res. Notes 2015, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, G.; Meroueh, S.; Toth, M.; Fisher, J.F.; Fridman, R.; Mobashery, S.; Sagi, I. Molecular Structures and Dynamics of the Stepwise Activation Mechanism of a Matrix Metalloproteinase Zymogen: Challenging the Cysteine Switch Dogma. J. Am. Chem. Soc. 2007, 129, 13566–13574. [Google Scholar] [CrossRef]
- Tallant, C.; Marrero, A.; Gomis-Rüth, F. Matrix metalloproteinases: Fold and function of their catalytic domains. Biochim. Biophys. Acta 2010, 1803, 20–28. [Google Scholar] [CrossRef]
- Woessner, J.F., Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wart, E.H.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeb, E.; Schleinkofer, K.; Kernebeck, T.; Pötsch, S.; Jansen, B.; Behrmann, I.; Matern, S.; Grötzinger, J. The Matrix Metalloproteinase 9 (MMP-9) Hemopexin Domain Is a Novel Gelatin Binding Domain and Acts as an Antagonist. J. Biol. Chem. 2002, 277, 50326–50332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Steen, P.E.; Van Aelst, I.; Hvidberg, V.; Piccard, H.; Fiten, P.; Jacobsen, C.; Moestrup, S.K.; Fry, S.; Royle, L.; Wormald, M.R.; et al. The Hemopexin and O-Glycosylated Domains Tune Gelatinase B/MMP-9 Bioavailability via Inhibition and Binding to Cargo Receptors. J. Biol. Chem. 2006, 281, 18626–18637. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.; Kopetzki, E.; Huber, R.; Lanzendörfer, M.; Brandstetter, H. Structural Basis of the Adaptive Molecular Recognition by MMP9. J. Mol. Biol. 2002, 320, 1065–1079. [Google Scholar] [CrossRef]
- Kotra, L.P.; Zhang, L.; Fridman, R.; Orlando, R.; Mobashery, S. N-Glycosylation pattern of the zymogenic form of human matrix metalloproteinase-9. Bioorganic Chem. 2002, 30, 356–370. [Google Scholar] [CrossRef]
- Duellman, T.; Burnett, J.; Yang, J. Functional Roles of N-Linked Glycosylation of Human Matrix Metalloproteinase 9. Traffic 2015, 16, 1108–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Cieplak, P. Role of N-glycosylation in activation of proMMP-9. A molecular dynamics simulations study. PLoS ONE 2018, 13, e0191157. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.W.; Bernardo, M.M.; Pietila, M.; Gervasi, D.C.; Toth, M.; Kotra, L.P.; Massova, I.; Mobashery, S.; Fridman, R. Characterization of the Monomeric and Dimeric Forms of Latent and Active Matrix Metalloproteinase-9. J. Biol. Chem. 2000, 275, 2661–2668. [Google Scholar] [CrossRef] [Green Version]
- Labrie, M.; St-Pierre, Y. Epigenetic regulation of mmp-9 gene expression. Cell. Mol. Life Sci. 2012, 70, 3109–3124. [Google Scholar] [CrossRef]
- Liu, P.; Wilson, M.J. miR-520c and miR-373 upregulate MMP9 expression by targeting mTOR and SIRT1, and activate the Ras/Raf/MEK/Erk signaling pathway and NF-κB factor in human fibrosarcoma cells. J. Cell. Physiol. 2011, 227, 867–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huhtala, P.; Tuuttila, A.; Chow, L.; Lohi, J.; Keski-Oja, J.; Tryggvason, K. Complete structure of the human gene for 92-kDa type IV collagenase. Divergent regulation of expression for the 92- and 72-kilodalton enzyme genes in HT-1080 cells. J. Biol. Chem. 1991, 266, 16485–16490. [Google Scholar] [CrossRef]
- He, C. Molecular mechanism of transcriptional activation of human gelatinase B by proximal promoter. Cancer Lett. 1996, 106, 185–191. [Google Scholar] [CrossRef]
- Han, Y.-P.; Tuan, T.-L.; Hughes, M.; Wu, H.; Garner, W.L. Transforming Growth Factor-β- and Tumor Necrosis Factor-α-mediated Induction and Proteolytic Activation of MMP-9 in Human Skin. J. Biol. Chem. 2001, 276, 22341–22350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Seiki, M. Regulatory mechanism of 92 kDa type IV collagenase gene expression which is associated with invasiveness of tumor cells. Oncogene 1993, 8, 395–405. [Google Scholar] [PubMed]
- Bond, M.; Fabunmi, R.P.; Baker, A.H.; Newby, A.C. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: An absolute requirement for transcription factor NF-κB. FEBS Lett. 1998, 435, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Benveniste, E.N. Transcriptional Activation of Human Matrix Metalloproteinase-9 Gene Expression by Multiple Co-activators. J. Mol. Biol. 2008, 383, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Fanjul-Fernández, M.; Folgueras, A.R.; Cabrera, S.; López-Otín, C. Matrix metalloproteinases: Evolution, gene regulation and functional analysis in mouse models. Biochim. Biophys. Acta 2010, 1803, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Chang, M.J.; Shah, R.C.; Benveniste, E.N. Interferon-γ-activated STAT-1α suppresses MMP-9 gene transcription by sequestration of the coactivators CBP/p300. J. Leukoc. Biol. 2005, 78, 515–523. [Google Scholar] [CrossRef]
- Zaremba-Czogalla, M.; Hryniewicz-Jankowska, A.; Tabola, R.; Nienartowicz, M.; Stach, K.; Wierzbicki, J.; Cirocchi, R.; Ziolkowski, P.; Tabaczar, S.; Augoff, K. A novel regulatory function of CDKN1A/p21 in TNFα-induced matrix metalloproteinase 9-dependent migration and invasion of triple-negative breast cancer cells. Cell Signal. 2018, 47, 27–36. [Google Scholar] [CrossRef]
- Snowden, A.W.; Anderson, L.A.; Webster, G.A.; Perkins, N.D. A Novel Transcriptional Repression Domain Mediates p21 WAF1/CIP1 Induction of p300 Transactivation. Mol. Cell. Biol. 2000, 20, 2676–2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opdenakker, G.; Steen, P.E.V.D.; Dubois, B.; Nelissen, I.; Van Coillie, E.; Masure, S.; Proost, P.; Van Damme, J. Gelatinase B functions as regulator and effector in leukocyte biology. J. Leukoc. Biol. 2001, 69, 851–859. [Google Scholar] [PubMed]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2010, 41, 271–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, J.; Knapnougel, P.; Lesavre, P.; Bauvois, B. Inhibition of matrix metalloproteinase-9 by interferons and TGF-β1 through distinct signalings accounts for reduced monocyte invasiveness. FEBS Lett. 2005, 579, 5487–5493. [Google Scholar] [CrossRef] [Green Version]
- Nee, L.E.; McMorrow, T.; Campbell, E.; Slattery, C.; Ryan, M.P. TNF-α and IL-1β–mediated regulation of MMP-9 and TIMP-1 in renal proximal tubular cells. Kidney Int. 2004, 66, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Kuo, C.-T.; Cheng, C.-Y.; Wu, C.-Y.; Lee, C.-W.; Hsieh, H.-L.; Lee, I.-T.; Yang, C.-M. IL-1β promotes A549 cell migration via MAPKs/AP-1- and NF-κB-dependent matrix metalloproteinase-9 expression. Cell Signal. 2009, 21, 1652–1662. [Google Scholar] [CrossRef]
- Wolczyk, D.; Zaremba-Czogalla, M.; Hryniewicz-Jankowska, A.; Tabola, R.; Grabowski, K.; Sikorski, A.F.; Augoff, K. TNF-α promotes breast cancer cell migration and enhances the concentration of membrane-associated proteases in lipid rafts. Cell. Oncol. 2016, 39, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-C.; Wei, X.-T.; Guan, J.-H.; Shu, H.; Chen, D. EGF stimulates glioblastoma metastasis by induction of matrix metalloproteinase-9 in an EGFR-dependent mechanism. Oncotarget 2017, 8, 65969–65982. [Google Scholar] [CrossRef] [Green Version]
- Bond, M.; Chase, A.J.; Baker, A.H.; Newby, A.C. Inhibition of transcription factor NF-κB reduces matrix metalloproteinase-1, -3 and -9 production by vascular smooth muscle cells. Cardiovasc. Res. 2001, 50, 556–565. [Google Scholar] [CrossRef]
- Zhou, M.; Zhang, Y.; Ardans, J.A.; Wahl, L.M. Interferon-γ Differentially Regulates Monocyte Matrix Metalloproteinase-1 and -9 through Tumor Necrosis Factor-α and Caspase 8. J. Biol. Chem. 2003, 278, 45406–45413. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.E.; Fernandez-Vilaseca, M.; Elkington, P.T.G.; Horncastle, D.E.; Graeber, M.B.; Friedland, J.S. IFN synergizes with IL-1α to up-regulate MMP-9 secretion in a cellular model of central nervous system tuberculosis. FASEB J. 2006, 21, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Park, K.-K.; Magae, J.; Ando, K.; Lee, T.-S.; Kwon, T.K.; Kwak, J.-Y.; Kim, C.-H.; Chang, Y.-C. Ascochlorin Inhibits Matrix Metalloproteinase-9 Expression by Suppressing Activator Protein-1-mediated Gene Expression through the ERK1/2 Signaling Pathway: Inhibitory effects of ascochlorin on the invasion of renal carcinoma cells. J. Biol. Chem. 2005, 280, 25202–25209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cáceres, L.C.; Bonacci, G.R.; Sánchez, M.C.; Chiabrando, G.A. Activated α2 macroglobulin induces matrix metalloproteinase 9 expression by low-density lipoprotein receptor-related protein 1 through MAPK-ERK1/2 and NF-κB activation in macrophage-derived cell lines. J. Cell. Biochem. 2010, 111, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Lian, S.; Khoi, P.N.; Yoon, H.J.; Joo, Y.E.; Chay, K.O.; Kim, K.K.; Jung, Y.D. Chrysin Inhibits Tumor Promoter-Induced MMP-9 Expression by Blocking AP-1 via Suppression of ERK and JNK Pathways in Gastric Cancer Cells. PLoS ONE 2015, 10, e0124007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, K.-Y.; Shieh, Y.-S.; Lee, C.-S.; Shiah, S.-G.; Wu, C.-W. Axl promotes cell invasion by inducing MMP-9 activity through activation of NF-κB and Brg-1. Oncogene 2008, 27, 4044–4055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakka, S.S.; Jasti, S.L.; Gondi, C.; Boyd, D.; Chandrasekar, N.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Downregulation of MMP-9 in ERK-mutated stable transfectants inhibits glioma invasion in vitro. Oncogene 2002, 21, 5601–5608. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.-Y.; Hsieh, H.-L.; Hsiao, L.-D.; Yang, C.-M. PI3-K/Akt/JNK/NF-κB is essential for MMP-9 expression and outgrowth in human limbal epithelial cells on intact amniotic membrane. Stem Cell Res. 2012, 9, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.T.; Sie, S.S.; Kuan, T.C.; Lin, C.S. Identifying the Regulative Role of NF-κB Binding Sites Within Promoter Region of Human Matrix Metalloproteinase 9 (mmp-9) by TNF-α Induction. Appl. Biochem. Biotechnol. 2012, 169, 438–449. [Google Scholar] [CrossRef]
- Ogawa, K.; Chen, F.; Kuang, C.; Chen, Y. Suppression of matrix metalloproteinase-9 transcription by transforming growth factor-β is mediated by a nuclear factor-κB site. Biochem. J. 2004, 381, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Hanania, R.; Sun, H.S.; Xu, K.; Pustylnik, S.; Jeganathan, S.; Harrison, R.E. Classically Activated Macrophages Use Stable Microtubules for Matrix Metalloproteinase-9 (MMP-9) Secretion. J. Biol. Chem. 2012, 287, 8468–8483. [Google Scholar] [CrossRef] [Green Version]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roderfeld, M.; Graf, J.; Giese, B.; Salguero-Palacios, R.; Tschuschner, A.; Müller-Newen, G.; Roeb, E. Latent MMP-9 is bound to TIMP-1 before secretion. Biol. Chem. 2007, 388, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Nagase, H. Preferential Inactivation of Tissue Inhibitor of Metalloproteinases-1 That Is Bound to the Precursor of Matrix Metalloproteinase 9 (Progelatinase B) by Human Neutrophil Elastase. J. Biol. Chem. 1995, 270, 16518–16521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjeldsen, L.; Johnsen, A.H.; Sengeløv, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [CrossRef]
- Tschesche, H.; Zölzer, V.; Triebel, S.; Bartsch, S. The human neutrophil lipocalin supports the allosteric activation of matrix metalloproteinases. JBIC J. Biol. Inorg. Chem. 2001, 268, 1918–1928. [Google Scholar] [CrossRef]
- Yan, L.; Borregaard, N.; Kjeldsen, L.; Moses, M.A. The High Molecular Weight Urinary Matrix Metalloproteinase (MMP) Activity is a Complex of Gelatinase B/MMP-9 and Neutrophil Gelatinase-associated Lipocalin (NGAL): Modulation of MMP-9 activity by NGAL. J. Biol. Chem. 2001, 276, 37258–37265. [Google Scholar] [CrossRef] [Green Version]
- Ogata, Y.; Enghild, J.; Nagase, H. Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J. Biol. Chem. 1992, 267, 3581–3584. [Google Scholar] [CrossRef]
- Ramos-DeSimone, N.; Hahn-Dantona, E.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of Matrix Metalloproteinase-9 (MMP-9) via a Converging Plasmin/Stromelysin-1 Cascade Enhances Tumor Cell Invasion. J. Biol. Chem. 1999, 274, 13066–13076. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, S.D.; Fliszar, C.J.; Broekelmann, T.J.; Mecham, R.P.; Senior, R.M.; Welgus, H.G. Activation of the 92-kDa Gelatinase by Stromelysin and 4-Aminophenylmercuric Acetate. Differential processing and stabilization of the carboxyl-terminal domain by tissue inhibitor of metalloproteinases (TIMP). J. Biol. Chem. 1995, 270, 6351–6356. [Google Scholar] [CrossRef] [Green Version]
- Bannikov, G.A.; Karelina, T.V.; Collier, I.E.; Marmer, B.L.; Goldberg, G.I. Substrate Binding of Gelatinase B Induces Its Enzymatic Activity in the Presence of Intact Propeptide. J. Biol. Chem. 2002, 277, 16022–16027. [Google Scholar] [CrossRef] [Green Version]
- Chelladurai, P.; Seeger, W.; Pullamsetti, S.S. Matrix metalloproteinases and their inhibitors in pulmonary hypertension. Eur. Respir. J. 2012, 40, 766–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigg, H.F.; Rowan, A.D.; Barker, M.D.; Cawston, T.E. Activity of matrix metalloproteinase-9 against native collagen types I and III. FEBS J. 2007, 274, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, G.; Steen, P.E.V.D.; Cohen, S.R.; Bitler, A.; Brand, D.D.; Opdenakker, G.; Sagi, I. Direct Visualization of Protease Action on Collagen Triple Helical Structure. PLoS ONE 2010, 5, e11043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kridel, S.J.; Chen, E.; Kotra, L.P.; Howard, E.W.; Mobashery, S.; Smith, J.W. Substrate Hydrolysis by Matrix Metalloproteinase-9. J. Biol. Chem. 2001, 276, 20572–20578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouw, J.K.; Ou, G.; Weaver, V.M. Extracellular matrix assembly: A multiscale deconstruction. Nat. Rev. Mol. Cell Biol. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, R.; Yamada, K.M. Basement Membranes in Development and Disease. Curr. Top. Dev. Biol. 2018, 130, 143–191. [Google Scholar] [CrossRef] [PubMed]
- Kyriakides, T.R.; Wulsin, D.; Skokos, E.A.; Fleckman, P.; Pirrone, A.; Shipley, J.M.; Senior, R.M.; Bornstein, P. Mice that lack matrix metalloproteinase-9 display delayed wound healing associated with delayed reepithelization and disordered collagen fibrillogenesis. Matrix Biol. 2009, 28, 65–73. [Google Scholar] [CrossRef] [Green Version]
- McMillan, S.J.; Kearley, J.; Campbell, J.D.; Zhu, X.-W.; Larbi, K.Y.; Shipley, J.M.; Senior, R.M.; Nourshargh, S.; Lloyd, C. Matrix Metalloproteinase-9 Deficiency Results in Enhanced Allergen-Induced Airway Inflammation. J. Immunol. 2004, 172, 2586–2594. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.H.; Shipley, J.; Bergers, G.; E Berger, J.; A Helms, J.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/Gelatinase B Is a Key Regulator of Growth Plate Angiogenesis and Apoptosis of Hypertrophic Chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Cho, A.; Reidy, M.A. Matrix Metalloproteinase-9 Is Necessary for the Regulation of Smooth Muscle Cell Replication and Migration After Arterial Injury. Circ. Res. 2002, 91, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Liu, L.; Jiang, C.; Pan, K.; Deng, J.; Wan, C. MMP9 protects against LPS-induced inflammation in osteoblasts. Innate Immun. 2019, 26, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Shchors, K.; Nozawa, H.; Xu, J.; Rostker, F.; Swigart-Brown, L.; Evan, G.; Hanahan, D. Increased invasiveness of MMP-9-deficient tumors in two mouse models of neuroendocrine tumorigenesis. Oncogene 2012, 32, 502–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlichenko, L.S.; Radisky, D.C. Matrix metalloproteinases stimulate epithelial-mesenchymal transition during tumor development. Clin. Exp. Metastasis 2008, 25, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.D.C.; Symowicz, J.; Ning, Y.; Gutierrez, E.; Fishman, D.A.; Adley, B.P.; Stack, M.S.; Hudson, L.G. Matrix Metalloproteinase 9 Is a Mediator of Epidermal Growth Factor–Dependent E-Cadherin Loss in Ovarian Carcinoma Cells. Cancer Res. 2008, 68, 4606–4613. [Google Scholar] [CrossRef] [Green Version]
- Symowicz, J.; Adley, B.P.; Gleason, K.J.; Johnson, J.J.; Ghosh, S.; Fishman, D.A.; Hudson, L.G.; Stack, M.S. Engagement of Collagen-Binding Integrins Promotes Matrix Metalloproteinase-9–Dependent E-Cadherin Ectodomain Shedding in Ovarian Carcinoma Cells. Cancer Res. 2007, 67, 2030–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, F.; Shimada, Y.; Watanabe, G.; Uchida, S.; Makino, T.; Imamura, M. Expression of vascular endothelial growth factor, matrix metalloproteinase-9 and E-cadherin in the process of lymph node metastasis in oesophageal cancer. Br. J. Cancer 1999, 80, 1366–1372. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Lan, X.; Li, S.; Xue, Y. Relationships of MMP-9, E-cadherin, and VEGF expression with clinicopathological features and response to chemosensitivity in gastric cancer. Tumor Biol. 2017, 39, 1010428317698368. [Google Scholar] [CrossRef] [Green Version]
- Heikenwalder, M.; Lorentzen, A. The role of polarisation of circulating tumour cells in cancer metastasis. Cell. Mol. Life Sci. 2019, 76, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Kaszak, I.; Witkowska-Piłaszewicz, O.; Niewiadomska, Z.; Dworecka-Kaszak, B.; Toka, F.N.; Jurka, P. Role of Cadherins in Cancer—A Review. Int. J. Mol. Sci. 2020, 21, 7624. [Google Scholar] [CrossRef]
- Thomas, G.J.; Lewis, M.P.; Hart, I.R.; Marshall, J.F.; Speight, P.M. AlphaVbeta6 integrin promotes invasion of squamous carcinoma cells through up-regulation of matrix metalloproteinase-9. Int. J. Cancer 2001, 92, 641–650. [Google Scholar] [CrossRef]
- Huhtala, P.; Humphries, M.; McCarthy, J.B.; Tremble, P.M.; Werb, Z.; Damsky, C.H. Cooperative signaling by alpha 5 beta 1 and alpha 4 beta 1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J. Cell Biol. 1995, 129, 867–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, X.; Niu, J.; Dorahy, D.J.; Scott, R.; Agrez, M.V. Integrin αvβ6-associated ERK2 mediates MMP-9 secretion in colon cancer cells. Br. J. Cancer 2002, 87, 348–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noe, V.; Fingleton, B.; Jacobs, K.; Crawford, H.; Vermeulen, S.; Steelant, W.; Bruyneel, E.; Matrisian, L.; Mareel, M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 2001, 114, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.K.; Ip, P.P.; Wong, A.S. New insights into the role of soluble E-cadherin in tumor angiogenesis. Cell Stress 2018, 2, 236–238. [Google Scholar] [CrossRef]
- Chan, A.O.-O.; Chu, K.-M.; Lam, S.-K.; Wong, B.C.-Y.; Kwok, K.-F.; Law, S.; Ko, S.; Hui, W.-M.; Yueng, Y.-H.; Wong, J. Soluble E-Cadherin is an Independent Pretherapeutic Factor for Long-Term Survival in Gastric Cancer. J. Clin. Oncol. 2003, 21, 2288–2293. [Google Scholar] [CrossRef] [Green Version]
- Kuefer, R.; Hofer, M.D.; Gschwend, J.E.; Pienta, K.J.; Sanda, M.G.; Chinnaiyan, A.M.; A Rubin, M.; Day, M.L. The role of an 80 kDa fragment of E-cadherin in the metastatic progression of prostate cancer. Clin. Cancer Res. 2003, 9, 6447–6552. [Google Scholar]
- Brouxhon, S.M.; Kyrkanides, S.; Teng, X.; Athar, M.; Ghazizadeh, S.; Simon, M.; O’Banion, M.K.; Ma, L. Soluble E-cadherin: A critical oncogene modulating receptor tyrosine kinases, MAPK and PI3K/Akt/mTOR signaling. Oncogene 2013, 33, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Kim, H.-J.; Park, K.-G.; Kim, Y.-N.; Kwon, T.-K.; Park, J.-Y.; Lee, K.-U.; Kim, J.-G.; Lee, I.-K. α-Lipoic acid inhibits matrix metalloproteinase-9 expression by inhibiting NF-κB transcriptional activity. Exp. Mol. Med. 2007, 39, 106–113. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Nakamura, K.; Iwai, S.; Murakami, M.; Itoh, T.; Kijima, H.; Shipley, J.; Senior, R.M.; Shibuya, M. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2002, 2, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Mañes, S.; Llorente, M.; Lacalle, R.A.; Gómez, M.L.; Kremer, L.; Mira, E.; Martínez-A, C. The Matrix Metalloproteinase-9 Regulates the Insulin-like Growth Factor-triggered Autocrine Response in DU-145 Carcinoma Cells. J. Biol. Chem. 1999, 274, 6935–6945. [Google Scholar] [CrossRef] [Green Version]
- Ellerbroek, S.M.; Halbleib, J.M.; Benavidez, M.; Warmka, J.K.; Wattenberg, E.V.; Stack, M.S.; Hudson, L.G. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix metalloproteinase-9 production and cell surface association. Cancer Res. 2001, 61, 1855–1861. [Google Scholar] [PubMed]
- Al-Sadi, R.; Youssef, M.; Rawat, M.; Guo, S.; Dokladny, K.; Haque, M.; Watterson, M.D.; Ma, T.Y. MMP-9-induced increase in intestinal epithelial tight permeability is mediated by p38 kinase signaling pathway activation of MLCK gene. Am. J. Physiol. Liver Physiol. 2019, 316, G278–G290. [Google Scholar] [CrossRef] [PubMed]
- Chiu, P.-S.; Lai, S.-C. Matrix Metalloproteinase-9 Leads to Claudin-5 Degradation via the NF-κB Pathway in BALB/c Mice with Eosinophilic Meningoencephalitis Caused by Angiostrongylus cantonensis. PLoS ONE 2013, 8, e53370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999, 13, 35–48. [Google Scholar] [CrossRef] [Green Version]
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of Matrix Metalloproteinase-9 Dimers in Cell Migration: Design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956. [Google Scholar] [CrossRef] [Green Version]
- Abécassis, I.; Olofsson, B.; Schmid, M.; Zalcman, G.; Karniguian, A. RhoA induces MMP-9 expression at CD44 lamellipodial focal complexes and promotes HMEC-1 cell invasion. Exp. Cell Res. 2003, 291, 363–376. [Google Scholar] [CrossRef]
- Lagarrigue, F.; Dupuis-Coronas, S.; Ramel, D.; Delsol, G.; Tronchère, H.; Payrastre, B.; Gaits-Iacovoni, F. Matrix Metalloproteinase-9 Is Upregulated in Nucleophosmin-Anaplastic Lymphoma Kinase–Positive Anaplastic Lymphomas and Activated at the Cell Surface by the Chaperone Heat Shock Protein 90 to Promote Cell Invasion. Cancer Res. 2010, 70, 6978–6987. [Google Scholar] [CrossRef] [Green Version]
- Foger, N.; Marhaba, R.; Zoller, M. Involvement of CD44 in cytoskeleton rearrangement and raft reorganization in T cells. J. Cell Sci. 2001, 114, 1169–1178. [Google Scholar] [CrossRef]
- Dayer, C.; Stamenkovic, I. Recruitment of Matrix Metalloproteinase-9 (MMP-9) to the Fibroblast Cell Surface by Lysyl Hydroxylase 3 (LH3) Triggers Transforming Growth Factor-β (TGF-β) Activation and Fibroblast Differentiation. J. Biol. Chem. 2015, 290, 13763–13778. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Kim, H.; Liu, X.; Sugiura, H.; Kohyama, T.; Fang, Q.; Wen, F.-Q.; Abe, S.; Wang, X.; Atkinson, J.J.; et al. Matrix metalloproteinase-9 activates TGF-β and stimulates fibroblast contraction of collagen gels. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L1006–L1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Li, Y.-Y.; Zhang, H.-Y.; Wang, F.; He, H.-L.; Yao, J.-C.; Liu, L.; Li, S.-S. Role of matrix metalloproteinase-9 in transforming growth factor-β1-induced epithelial–mesenchymal transition in esophageal squamous cell carcinoma. OncoTargets Ther. 2017, ume 10, 2837–2847. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; He, J.; Wang, F.; Wang, X.; Yang, F.; Zhao, C.; Feng, C.; Li, T. Role of MMP-9 in epithelial-mesenchymal transition of thyroid cancer. World J. Surg. Oncol. 2020, 18, 181. [Google Scholar] [CrossRef] [PubMed]
- Midgley, A.C.; Rogers, M.; Hallett, M.; Clayton, A.; Bowen, T.; Phillips, A.; Steadman, R. Transforming Growth Factor-β1 (TGF-β1)-stimulated Fibroblast to Myofibroblast Differentiation Is Mediated by Hyaluronan (HA)-facilitated Epidermal Growth Factor Receptor (EGFR) and CD44 Co-localization in Lipid Rafts. J. Biol. Chem. 2013, 288, 14824–14838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Diamond, M.E.; Ottaviano, A.J.; Joseph, M.J.; Ananthanarayan, V.; Munshi, H.G. Transforming Growth Factor-β1 Promotes Matrix Metalloproteinase-9–Mediated Oral Cancer Invasion through Snail Expression. Mol. Cancer Res. 2008, 6, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulkhalek, S.; Amith, S.R.; Franchuk, S.L.; Jayanth, P.; Guo, M.; Finlay, T.; Gilmour, A.; Guzzo, C.; Gee, K.; Beyaert, R.; et al. Neu1 Sialidase and Matrix Metalloproteinase-9 Cross-talk Is Essential for Toll-like Receptor Activation and Cellular Signaling. J. Biol. Chem. 2011, 286, 36532–36549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayanth, P.; Amith, S.R.; Gee, K.; Szewczuk, M.R. Neu1 sialidase and matrix metalloproteinase-9 cross-talk is essential for neurotrophin activation of Trk receptors and cellular signaling. Cell Signal. 2010, 22, 1193–1205. [Google Scholar] [CrossRef]
- Gilmour, A.M.; Abdulkhalek, S.; Cheng, T.S.; Alghamdi, F.; Jayanth, P.; O’Shea, L.K.; Geen, O.; Arvizu, L.A.; Szewczuk, M.R. A novel epidermal growth factor receptor-signaling platform and its targeted translation in pancreatic cancer. Cell Signal. 2013, 25, 2587–2603. [Google Scholar] [CrossRef] [Green Version]
- Alghamdi, F.; Guo, M.; Abdulkhalek, S.; Crawford, N.; Amith, S.R.; Szewczuk, M.R. A novel insulin receptor-signaling platform and its link to insulin resistance and type 2 diabetes. Cell Signal. 2014, 26, 1355–1368. [Google Scholar] [CrossRef] [Green Version]
- Sindhu, S.; Al-Roub, A.; Koshy, M.; Thomas, R.; Ahmad, R. Palmitate-Induced MMP-9 Expression in the Human Monocytic Cells is Mediated through the TLR4-MyD88 Dependent Mechanism. Cell. Physiol. Biochem. 2016, 39, 889–900. [Google Scholar] [CrossRef]
- Kashani, B.; Zandi, Z.; Pourbagheri-Sigaroodi, A.; Bashash, D.; Ghaffari, S.H. The role of toll-like receptor 4 (TLR4) in cancer progression: A possible therapeutic target? J. Cell. Physiol. 2020, 236, 4121–4137. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, F.; Wei, F.; Ren, X. The role of toll-like receptor 4 in tumor microenvironment. Oncotarget 2017, 8, 66656–66667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Van den Steen, P.E.; Proost, P.; Wuyts, A.; Van Damme, J.; Opdenakker, G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood 2000, 96, 2673–2681. [Google Scholar] [CrossRef]
- Palena, C.; Hamilton, D.H.; I Fernando, R. Influence of IL-8 on the epithelial–mesenchymal transition and the tumor microenvironment. Futur. Oncol. 2012, 8, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Investig. 2010, 120, 485–497. [Google Scholar] [CrossRef]
- Inoue, K.; Slaton, J.W.; Perrotte, P.; Eve, B.Y.; Dinney, C.P.N. Interleukin-8 (IL-8) expression regulates tumorigenicity and metastasis in human bladder cancer. J. Urol. 1999, 60, 2290–2299. [Google Scholar] [CrossRef]
- Bekes, E.M.; Schweighofer, B.; Kupriyanova, T.A.; Zajac, E.; Ardi, V.C.; Quigley, J.P.; Deryugina, E.I. Tumor-Recruited Neutrophils and Neutrophil TIMP-Free MMP-9 Regulate Coordinately the Levels of Tumor Angiogenesis and Efficiency of Malignant Cell Intravasation. Am. J. Pathol. 2011, 179, 1455–1470. [Google Scholar] [CrossRef]
- Steen, P.E.V.D.; Wuyts, A.; Husson, S.J.; Proost, P.; Van Damme, J.; Opdenakker, G. Gelatinase B/MMP-9 and neutrophil collagenase/MMP-8 process the chemokines human GCP-2/CXCL6, ENA-78/CXCL5 and mouse GCP-2/LIX and modulate their physiological activities. JBIC J. Biol. Inorg. Chem. 2003, 270, 3739–3749. [Google Scholar] [CrossRef]
- Steen, P.E.V.D.; Husson, S.J.; Proost, P.; Van Damme, J.; Opdenakker, G. Carboxyterminal cleavage of the chemokines MIG and IP-10 by gelatinase B and neutrophil collagenase. Biochem. Biophys. Res. Commun. 2003, 310, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.H.; Dean, R.A.; Roberts, C.R.; Overall, C.M. Matrix Metalloproteinase Processing of CXCL11/I-TAC Results in Loss of Chemoattractant Activity and Altered Glycosaminoglycan Binding. J. Biol. Chem. 2008, 283, 19389–19399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuibban, G.A.; Butler, G.S.; Gong, J.-H.; Bendall, L.; Power, C.; Clark-Lewis, I.; Overall, C.M. Matrix Metalloproteinase Activity Inactivates the CXC Chemokine Stromal Cell-derived Factor-1. J. Biol. Chem. 2001, 276, 43503–43508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, W.-Z.; Zhang, H.-B.; Xia, W.-X.; Ke, L.-R.; Yang, J.; Yu, Y.-H.; Liang, H.; Huang, X.-J.; Liu, G.-Y.; Li, W.-Z.; et al. The CXCL5/CXCR2 axis contributes to the epithelial-mesenchymal transition of nasopharyngeal carcinoma cells by activating ERK/GSK-3β/snail signalling. J. Exp. Clin. Cancer Res. 2018, 37, 85. [Google Scholar] [CrossRef] [Green Version]
- Mao, Z.; Zhang, J.; Shi, Y.; Li, W.; Shi, H.; Ji, R.; Mao, F.; Qian, H.; Xu, W.; Zhang, X. CXCL5 promotes gastric cancer metastasis by inducing epithelial-mesenchymal transition and activating neutrophils. Oncogenesis 2020, 9, 63. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Gong, J.-H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood 2002, 100, 1160–1167. [Google Scholar] [CrossRef]
- Horiuchi, T.; Mitoma, H.; Harashima, S.-I.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Gearing, A.J.H.; Beckett, P.; Christodoulou, M.; Churchill, M.; Clements, J.; Davidson, A.H.; Drummond, A.H.; Galloway, W.A.; Gilbert, R.; Gordon, J.L.; et al. Processing of tumour necrosis factor-α precursor by metalloproteinases. Nature 1994, 370, 555–557. [Google Scholar] [CrossRef]
- Schönbeck, U.; Mach, F.; Libby, P. Generation of biologically active IL-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of IL-1 beta processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar]
- Dreschers, S.; Platen, C.; Ludwig, A.; Gille, C.; Köstlin, N.; Orlikowsky, T.W. Metalloproteinases TACE and MMP-9 Differentially Regulate Death Factors on Adult and Neonatal Monocytes After Infection with Escherichia coli. Int. J. Mol. Sci. 2019, 20, 1399. [Google Scholar] [CrossRef] [Green Version]
- Sheu, B.-C.; Hsu, S.M.; Ho, H.-N.; Lien, H.-C.; Huang, S.C.; Lin, R.H. A novel role of metalloproteinase in cancer-mediated immunosuppression. Cancer Res. 2001, 61, 237–242. [Google Scholar] [PubMed]
- Gomez, I.G.; Tang, J.; Wilson, C.L.; Yan, W.; Heinecke, J.W.; Harlan, J.M.; Raines, E.W. Metalloproteinase-mediated Shedding of Integrin β2 Promotes Macrophage Efflux from Inflammatory Sites. J. Biol. Chem. 2012, 287, 4581–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisar, T.; Kassim, S.Y.; Gomez, I.G.; Green, P.S.; Hargarten, S.; Gough, P.J.; Parks, W.C.; Wilson, C.L.; Raines, E.W.; Heinecke, J.W. MMP-9 Sheds the β2 Integrin Subunit (CD18) from Macrophages. Mol. Cell. Proteom. 2009, 8, 1044–1060. [Google Scholar] [CrossRef] [Green Version]
- Leifler, K.S.; Svensson, S.; Abrahamsson, A.; Bendrik, C.; Robertson, J.; Gauldie, J.; Olsson, A.-K.; Dabrosin, C. Inflammation Induced by MMP-9 Enhances Tumor Regression of Experimental Breast Cancer. J. Immunol. 2013, 190, 4420–4430. [Google Scholar] [CrossRef] [PubMed]
- Coticchia, C.M.; Curatolo, A.S.; Zurakowski, D.; Yang, J.; Daniels, K.E.; Matulonis, U.A.; Moses, M.A. Urinary MMP-2 and MMP-9 predict the presence of ovarian cancer in women with normal CA125 levels. Gynecol. Oncol. 2011, 123, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.; Damery, S.; Stocken, D.D.; Dowswell, G.; Holder, R.; Ward, S.T.; Redman, V.; Wakelam, M.J.; James, J.; Hobbs, F.D.R.; et al. Serum matrix metalloproteinase 9 and colorectal neoplasia: A community-based evaluation of a potential diagnostic test. Br. J. Cancer 2012, 106, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Prieto, S.; Barcia-Castro, L.; De La Cadena, M.P.; Rodríguez-Berrocal, F.J.; Vázquez-Iglesias, L.; Botana-Rial, M.I.; Fernández-Villar, A.; De Chiara, L. Relevance of matrix metalloproteases in non-small cell lung cancer diagnosis. BMC Cancer 2017, 17, 823. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, Y.; Bai, J.; Xue, Y.; Peng, Q. MMP1 and MMP9 are potential prognostic biomarkers and targets for uveal melanoma. BMC Cancer 2021, 21, 1068. [Google Scholar] [CrossRef]
- Thieringer, F.R.; Maass, T.; Anthon, B.; Meyer, E.; Schirmacher, P.; Longerich, T.; Galle, P.R.; Kanzler, S.; Teufel, A. Liver-specific overexpression of matrix metalloproteinase 9 (MMP-9) in transgenic mice accelerates development of hepatocellular carcinoma. Mol. Carcinog. 2011, 51, 439–448. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef] [Green Version]
- Belotti, D.; Paganoni, P.; Manenti, L.; Garofalo, A.; Marchini, S.; Taraboletti, G.; Giavazzi, R. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: Implications for ascites formation. Cancer Res. 2003, 63, 5224–5229. [Google Scholar] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Owyong, M.; Chou, J.; Bijgaart, R.J.V.D.; Kong, N.; Efe, G.; Maynard, C.; Talmi-Frank, D.; Solomonov, I.; Koopman, C.; Hadler-Olsen, E.; et al. MMP9 modulates the metastatic cascade and immune landscape for breast cancer anti-metastatic therapy. Life Sci. Alliance 2019, 14, e201800226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Kwon, H.-J.; Kim, D.-S. Matrix Metalloproteinase 9 (MMP-9)-dependent Processing of βig-h3 Protein Regulates Cell Migration, Invasion, and Adhesion. J. Biol. Chem. 2012, 287, 38957–38969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendrik, C.; Robertson, J.; Gauldie, J.; Dabrosin, C. Gene Transfer of Matrix Metalloproteinase-9 Induces Tumor Regression of Breast Cancer In vivo. Cancer Res. 2008, 68, 3405–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamano, Y.; Zeisberg, M.; Sugimoto, H.; Lively, J.C.; Maeshima, Y.; Yang, C.; O Hynes, R.; Werb, Z.; Sudhakar, A.; Kalluri, R. Physiological levels of tumstatin, a fragment of collagen IV α3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via αVβ3 integrin. Cancer Cell 2003, 3, 589–601. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.; Sarma, D.; Jeppsson, S.; Patel, N.R.; Gewirtz, A.T.; Merlin, D.; Sitaraman, S.V. Matrix Metalloproteinase-9 Functions as a Tumor Suppressor in Colitis-Associated Cancer. Cancer Res. 2010, 70, 792–801. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Pujada, A.; Bhatnagar, N.; Bialkowska, A.B.; Yang, V.W.; Laroui, H.; Garg, P. Epithelial derived-matrix metalloproteinase (MMP9) exhibits a novel defensive role of tumor suppressor in colitis associated cancer by activating MMP9-Notch1-ARF-p53 axis. Oncotarget 2016, 8, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Sans-Fons, M.G.; Sole, S.; Sanfeliu, C.; Planas, A.M. Matrix Metalloproteinase-9 and Cell Division in Neuroblastoma Cells and Bone Marrow Macrophages. Am. J. Pathol. 2010, 177, 2870–2885. [Google Scholar] [CrossRef] [Green Version]
- Jobin, P.G.; Butler, G.S.; Overall, C.M. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim. Biophys. Acta 2017, 1864, 2043–2055. [Google Scholar] [CrossRef] [PubMed]
- Bassiouni, W.; Ali, M.A.M.; Schulz, R. Multifunctional intracellular matrix metalloproteinases: Implications in disease. FEBS J. 2021, 288, 7162–7182. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akaike, T.; Sawa, T.; Miyamoto, Y.; van der Vliet, A.; Maeda, H. Activation of Matrix Metalloproteinases by Peroxynitrite-induced Protein S-Glutathiolation via Disulfide S-Oxide Formation. J. Biol. Chem. 2001, 276, 29596–29602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, M.A.M.; Garcia-Vilas, J.A.; Cromwell, C.R.; Hubbard, B.P.; Hendzel, M.J.; Schulz, R. Matrix metalloproteinase-2 mediates ribosomal RNA transcription by cleaving nucleolar histones. FEBS J. 2021, 288, 6736–6751. [Google Scholar] [CrossRef]
- Kim, K.; Punj, V.; Kim, J.-M.; Lee, S.; Ulmer, T.S.; Lu, W.; Rice, J.C.; An, W. MMP-9 facilitates selective proteolysis of the histone H3 tail at genes necessary for proficient osteoclastogenesis. Genes Dev. 2016, 30, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Amorosa, L.F.; Coyle, S.M.; Macor, M.A.; Lubitz, S.; Carson, J.L.; Birnbaum, M.; Lee, L.Y.; Haimovich, B. Proteolytic Cleavage of AMPKα and Intracellular MMP9 Expression Are Both Required for TLR4-Mediated mTORC1 Activation and HIF-1α Expression in Leukocytes. J. Immunol. 2015, 195, 2452–2460. [Google Scholar] [CrossRef] [PubMed]
- Cao, J. MMP Inhibitors Past present and future. Front. Biosci. 2015, 20, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.; Schnell, C.; Cozens, R.; O’Reilly, T.; Cox, D. CGS 27023A, a potent and orally active matrix metalloprotease inhibitor with antitumor activity. Proc. Am. Assoc. Cancer Res. 1998, 39, 83a. [Google Scholar]
- Nakabayashi, H.; Yawata, T.; Shimizu, K. Anti-invasive and antiangiogenic effects of MMI-166 on malignant glioma cells. BMC Cancer 2010, 10, 339. [Google Scholar] [CrossRef] [Green Version]
- Eatock, M.; Cassidy, J.; Johnson, J.; Morrison, R.; Devlin, M.; Blackey, R.; Owen, S.; Choi, L.; Twelves, C. A dose-finding and pharmacokinetic study of the matrix metalloproteinase inhibitor MMI270 (previously termed CGS27023A) with 5-FU and folinic acid. Cancer Chemother. Pharmacol. 2004, 55, 39–46. [Google Scholar] [CrossRef]
- Agrawal, A.; Romero-Perez, D.; Jacobsen, J.; Villarreal, F.J.; Cohen, S.M. Zinc-Binding Groups Modulate Selective Inhibition of MMPs. ChemMedChem 2008, 3, 812–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeDour, G.; Moroy, G.; Rouffet, M.; Bourguet, E.; Guillaume, D.; Decarme, M.; ElMourabit, H.; Augé, F.; Alix, A.J.; Laronze, J.-Y.; et al. Introduction of the 4-(4-bromophenyl)benzenesulfonyl group to hydrazide analogs of Ilomastat leads to potent gelatinase B (MMP-9) inhibitors with improved selectivity. Bioorganic Med. Chem. 2008, 16, 8745–8759. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Bernardo, M.M.; Meroueh, S.O.; Brown, S.; Fridman, A.R.; Mobashery, S. Synthesis of Chiral 2-(4-Phenoxyphenylsulfonylmethyl)thiiranes as Selective Gelatinase Inhibitors. Org. Lett. 2005, 7, 4463–4465. [Google Scholar] [CrossRef]
- Krüger, A.; Arlt, M.J.; Gerg, M.; Kopitz, C.; Bernardo, M.M.; Chang, M.; Mobashery, S.; Fridman, R. Antimetastatic Activity of a Novel Mechanism-Based Gelatinase Inhibitor. Cancer Res. 2005, 65, 3523–3526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfil, R.D.; Sabbota, A.; Nabha, S.; Bernardo, M.M.; Dong, Z.; Meng, H.; Yamamoto, H.; Chinni, S.R.; Lim, I.T.; Chang, M.; et al. Inhibition of human prostate cancer growth, osteolysis and angiogenesis in a bone metastasis model by a novel mechanism-based selective gelatinase inhibitor. Int. J. Cancer 2006, 118, 2721–2726. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Kuang, X.; Xie, Z.; Liang, L.; Zhang, Z.; Zhang, Y.; Ma, F.; Gao, Q.; Chang, R.; Lee, H.-H.; et al. Small-molecule MMP2/MMP9 inhibitor SB-3CT modulates tumor immune surveillance by regulating PD-L1. Genome Med. 2020, 83, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.-M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [Green Version]
- Dufour, A.; Sampson, N.; Li, J.; Kuscu, C.; Rizzo, R.C.; DeLeon, J.L.; Zhi, J.; Jaber, N.; Liu, E.; Zucker, S.; et al. Small-Molecule Anticancer Compounds Selectively Target the Hemopexin Domain of Matrix Metalloproteinase-9. Cancer Res. 2011, 71, 4977–4988. [Google Scholar] [CrossRef] [Green Version]
- Alford, V.M.; Kamath, A.; Ren, X.; Kumar, K.; Gan, Q.; Awwa, M.; Tong, M.; Seeliger, M.A.; Cao, J.; Ojima, I.; et al. Targeting the Hemopexin-like Domain of Latent Matrix Metalloproteinase-9 (proMMP-9) with a Small Molecule Inhibitor Prevents the Formation of Focal Adhesion Junctions. ACS Chem. Biol. 2017, 12, 2788–2803. [Google Scholar] [CrossRef]
- Yosef, G.; Hayun, H.; Papo, N. Simultaneous targeting of CD44 and MMP9 catalytic and hemopexin domains as a therapeutic strategy. Biochem. J. 2021, 478, 1139–1157. [Google Scholar] [CrossRef]
- Laronha, H.; Carpinteiro, I.; Portugal, J.; Azul, A.C.; Polido, M.; Petrova, K.T.; Salema-Oom, M.; Caldeira, J. Challenges in Matrix Metalloproteinases Inhibition. Biomolecules 2020, 10, 717. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.E.; Chang, M. Selective small-molecule inhibitors as chemical tools to define the roles of matrix metalloproteinases in disease. Biochim. Biophys. Acta 2017, 1864, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Paemen, L.; Martens, E.; Masure, S.; Opdenakker, G. Monoclonal Antibodies Specific for Natural Human Neutrophil Gelatinase B Used for Affinity Purification, Quantitation by Two-Site ELISA and Inhibition of Enzymatic Activity. JBIC J. Biol. Inorg. Chem. 1995, 234, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.; Leyssen, A.; Van Aelst, I.; Fiten, P.; Piccard, H.; Hu, J.; Descamps, F.J.; Van den Steen, P.E.; Proost, P.; Van Damme, J.; et al. A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim. Biophys. Acta 2007, 1770, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.C.; Lyman, S.K.; McCauley, S.; Kovalenko, M.; Spangler, R.; Liu, C.; Lee, M.; O’Sullivan, C.; Barry-Hamilton, V.; Ghermazien, H.; et al. Selective Allosteric Inhibition of MMP9 Is Efficacious in Preclinical Models of Ulcerative Colitis and Colorectal Cancer. PLoS ONE 2015, 10, e0127063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleby, T.C.; Greenstein, A.; Hung, M.; Liclican, A.; Velasquez, M.; Villaseñor, A.G.; Wang, R.; Wong, M.H.; Liu, X.; Papalia, G.A.; et al. Biochemical characterization and structure determination of a potent, selective antibody inhibitor of human MMP9. J. Biol. Chem. 2017, 292, 6810–6820. [Google Scholar] [CrossRef] [Green Version]
- Bendell, J.C.; Starodub, A.; Huang, X.; Maltzman, J.D.; Wainberg, Z.A.; Shah, M.A. A phase 3 randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of GS-5745 combined with mFOLFOX6 as first-line treatment in patients with advanced gastric or gastroesophageal junction adenocarcinoma. J. Clin. Oncol. 2017, 35, TPS4139. [Google Scholar] [CrossRef]
- Shah, M.A.; Starodub, A.N.; Sharma, S.; Berlin, J.; Patel, M.R.; Wainberg, Z.A.; Chaves, J.; Gordon, M.S.; Windsor, K.; Brachmann, C.B.; et al. Andecaliximab/GS-5745 Alone and Combined with mFOLFOX6 in Advanced Gastric and Gastroesophageal Junction Adenocarcinoma: Results from a Phase I Study. Clin. Cancer Res. 2018, 24, 3829–3837. [Google Scholar] [CrossRef] [Green Version]
- Zuk, J.; Ozernov-Palchik, O.; Kim, H.; Lakshminarayanan, K.; Gabrieli, J.D.E.; Tallal, P.; Gaab, N. Enhanced Syllable Discrimination Thresholds in Musicians. PLoS ONE 2013, 8, e80546. [Google Scholar] [CrossRef]
- Awasthi, N.; Mikels-Vigdal, A.J.; Stefanutti, E.; Schwarz, M.A.; Monahan, S.; Smith, V.; Schwarz, R.E. Therapeutic efficacy of anti-MMP9 antibody in combination with nab-paclitaxel-based chemotherapy in pre-clinical models of pancreatic cancer. J. Cell. Mol. Med. 2019, 23, 3878–3887. [Google Scholar] [CrossRef]
- Kim, S.; Kim, S.H.; Hur, S.M.; Lee, S.-K.; Kim, W.W.; Kim, J.S.; Kim, J.-H.; Choe, J.-H.; Nam, S.J.; Lee, J.E.; et al. Silibinin prevents TPA-induced MMP-9 expression by down-regulation of COX-2 in human breast cancer cells. J. Ethnopharmacol. 2009, 126, 252–257. [Google Scholar] [CrossRef]
- Verma, S.; Singh, A.; Mishra, A. Gallic acid: Molecular rival of cancer. Environ. Toxicol. Pharmacol. 2013, 35, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Chang, L.-S. Gallic acid downregulates matrix metalloproteinase-2 (MMP-2) and MMP-9 in human leukemia cells with expressed Bcr/Abl. Mol. Nutr. Food Res. 2012, 56, 1398–1412. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Li, Y.; Teruya, K.; Katakura, Y.; Ichikawa, A.; Eto, H.; Hosoi, M.; Hosoi, M.; Nishimoto, S.; Shirahata, S. Enzyme-digested Fucoidan Extracts Derived from Seaweed Mozuku of Cladosiphon novae-caledoniae kylin Inhibit Invasion and Angiogenesis of Tumor Cells. Cytotechnology 2005, 47, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalva, S.; Singam, E.A.; Rajapandian, V.; Saleena, L.M.; Subramanian, V. Discovery of potent inhibitor for matrix metalloproteinase-9 by pharmacophore based modeling and dynamics simulation studies. J. Mol. Graph. Model. 2014, 49, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.K.; Whittaker, P.A. RNA interference: From gene silencing to gene-specific therapeutics. Pharmacol. Ther. 2005, 107, 222–239. [Google Scholar] [CrossRef] [PubMed]
- De Fougerolles, A.; Vornlocher, H.-P.; Maraganore, J.; Lieberman, J. Interfering with disease: A progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Tang, Z.-Y.; Liu, Y.; Liu, L.-X.; Ding, X.-Y.; Zhang, H.; Fang, L.-Q. RNAi-mediated MMP-9 silencing inhibits mouse melanoma cell invasion and migration in vitro and in vivo. Cell Biol. Int. 2013, 37, 849–854. [Google Scholar] [CrossRef]
- Lakka, S.S.; Gondi, C.S.; Yanamandra, N.; Olivero, W.C.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene 2004, 23, 4681–4689. [Google Scholar] [CrossRef] [Green Version]
- Nalla, A.K.; Gorantla, B.; Gondi, C.S.; Lakka, S.S.; Rao, J.S. Targeting MMP-9, uPAR, and cathepsin B inhibits invasion, migration and activates apoptosis in prostate cancer cells. Cancer Gene Ther. 2010, 17, 599–613. [Google Scholar] [CrossRef] [Green Version]
- Grijalvo, S.; Puras, G.; Zarate, J.; Sainz-Ramos, M.; Al Qtaish, N.; Lopez-Mendez, T.B.; Mashal, M.; Attia, N.; Díaz, D.D.; Pons, R.; et al. Cationic Niosomes as Non-Viral Vehicles for Nucleic Acids: Challenges and Opportunities in Gene Delivery. Pharmaceutics 2019, 11, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Zhang, H.; Yu, J.; Liufu, C.; Chen, Z. Ultrasound-mediated microbubble destruction: A new method in cancer immunotherapy. OncoTargets Ther. 2018, 11, 5763–5775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darband, S.G.; Mirza-Aghazadeh-Attari, M.; Kaviani, M.; Mihanfar, A.; Sadighparvar, S.; Yousefi, B.; Majidinia, M. Exosomes: Natural nanoparticles as bio shuttles for RNAi delivery. J. Control. Release 2018, 289, 158–170. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Target | Effect of MMP9 | Biological Consequences in Cancer | References |
|---|---|---|---|
| E-cadherin | Releasing the sE-cad fragment | Disruption of tight junction (TJ) integrity; cell dissociation; promotion of the EMT process; activation of EGFR, HER, and IGF-1R-dependent signaling pathways (MAPK, PI3K/Akt, and mTOR) | [75,83,84] |
| claudin-5 | Degradation and loss of function | Disturbances of cellular polarity and epithelial barrier function, disruption of tight junction (TJ) integrity through the NF-κB signaling pathway | [93] |
| CD44 | Formation of the CD44-MMP9 complex | Increasing the concentration and proteolytic activity of MMP9 against: (1) type IV of collagen to direct the migration of cancer cells, (2) TGF-β to promote cancer-associated fibroblast (CAF) differentiation and stimulate FN expression; initiating cross-talk between CD44 and HER-1 and triggering the activation of downstream effectors for cell migration; regulation of migratory potential and invasiveness of cancer cells | [94,95,97,98,100,101,102,103,104] |
| TLR-4, TrkA, EGFR/HER, and IR | Formation of a signaling platform | Induction of inflammation triggered by endogenous danger-associated molecular pattern (DAMP) molecules, increasing the tumorigenic potential of cancer cells, promoting immune evasion | [106,107,108,109,110,111,112] |
| IL-8/CXCL8 | Releasing the truncated IL-8(7-77) form | Increasing the migration and activity of immune cells by activating the FAK/Akt/FOXO3A pathway, promoting the trafficking of neutrophils and MDSCs into the tumor stroma, inducing the EMT process, increasing the expression of MMP9 in cancer cells, increasing metastatic potential, promoting neutrophil degranulation, enhancement of angiogenic activity | [114,115,118,119] |
| CXCL5/ENA-78 | Releasing truncated fragments | Activation of the EMT process by the ERK/GSK-3β/Snail pathway | [120] |
| CXCL11/I-TAC | Releasing the CXCL11/I-TAC(5-73) fragment | Inhibiting the antitumor immune response by acting as a natural antagonist of CXCR3 | [122] |
| CXCL1/GRO-α, CXCL4/PF-4, CXCL7/CTAP-III, and CXCL12/SDF-1 | Degradation and loss of function | Loss of chemotactic capacity and inhibition of the antitumor immune response | [120,121,122,123,126] |
| TNF-α and IL-1β | Releasing mature forms of TNF-α and IL-1β | Activation of signaling pathways that enhance cell proliferation and survival | [114,128,129] |
| TNFR1 and FAS/APO-1/TNFRSF6 death receptor | Cleavage of extracellular regions | Reduction of chronic inflammation by downregulation of cell-contact-related phagocytosis-induced cell death (PICD) in monocytes | [130] |
| IL-2Rα | Generating the soluble IL-2Rα/DC25 form | Abrogation of the efficacy of tumor-reactive cytotoxic lymphocytes antagonized with wild-type IL-2Rα | [131] |
| ITGB2 | Releasing soluble fragments | Reducing local inflammation by maintaining the ability to bind ligands, such as ICAM-1, fibrin, or collagen, and acting as receptor antagonists | [80,81,82,132,133] |
| ECM proteins (i.e., laminins, collagens, and FN) | Degradation and releasing signaling fragments | Facilitating the spread of invading cancer cells and migration of immune cells, stimulation of angiogenic activators including VEGF and bFGF, promoting the differentiation of normal fibroblasts to CAFs, creating metastatic niches in secondary sites, promoting invasion by activating the FAK-Src-related signaling pathways due to the binding MMP9-degraded FN to αvβ6 and α5β1 integrins | [80,81,82,138,140,141,142,144] |
| βig-h3 | Degradation and loss of function | Increasing the invasive potential of cancer cells | [145] |
| Group | Name/Description | Target | Clinical Outcome | References |
|---|---|---|---|---|
| Small inhibitors | Marimastat (BB2516, (2S,3R)-N4-[(1S)-2,2-dimethyl-1-[(methylamino)carbonyl] propyl]-N1,2-dihydroxy-3-(2-methylpropyl)butanediamide) | Catalytic domain (zinc chelator) | Cancelled in phase III clinical trials | [157] |
| Ilomastat (GM6001, N-[(2R)-2-(hydroxamidocarbonylmethyl)-4-methylpentanoyl]-L-tryptophan methylamide, also known as galardin) | Catalytic domain (zinc chelator) | Cancelled in phase II clinical trials | [157] | |
| Batimastat (BB-94, (2R,3S)-N4-hydroxy-N1-[(1S)-2-(methylamino)-2-oxo-1-(phenylmethyl)ethyl]-2-(2-methylpropyl)-3-[(2-thienylthio)methyl]butanediamide) | Catalytic domain (zinc chelator) | Cancelled in phase III clinical trials | [157] | |
| MMI-270 (N-hydroxy-2(R)-[(4-methoxysulfonyl)(3-picolyl)-amino]-3-methylbutaneamide hydrochloride monohydrate, also known as CGS-27023A) | Catalytic domain (zinc chelator) | Cancelled in phase I clinical trials | [157,158] | |
| MMI-166 (Nα-[4-(2- phenyl-2H-tetrazole-5-yl) phenyl sulfonyl]-D-tryptophan) | Catalytic domain (zinc chelator) | Preclinical studies | [158,159] | |
| SB-3CT (2-[[(4-phenoxyphenyl)sulfonyl]methyl]thiirane) | Catalytic domain (zinc chelator) | Preclinical studies | [163] | |
| JNJ0966 (N-[2-[(2-methoxyphenyl)amino]-4′-methyl[4,5′-bithiazol]-2′-yl]acetamide) | Zymogen activation | Preclinical studies | [167] | |
| N-[4-(difluoromethoxy)phenyl]-2-[(4-oxo-6-propyl-1H-pyrimidin-2-yl)sulfanyl]-acetamide | PEX domain | Preclinical studies | [168] | |
| N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio) butanamide | PEX domain | Preclinical studies | [168] | |
| C9-PEX | Catalytic domain and PEX domain | Preclinical studies | [170] | |
| Inhibitory antibodies | REGA 3G12 | N-terminal region of catalytic domain but not the Zn2+-binding site | Preclinical studies | [174] |
| B0041 | Zymogen activation and catalytic domain distal to active site | Preclinical studies | [175] | |
| B0046 | Zymogen activation and catalytic domain distal to active site | Preclinical studies | [175] | |
| GS-5745 (andecaliximab) | Zymogen activation and catalytic domain distal to active site | Phase I, II, and III clinical trial solid tumors and phase III gastric adenocarcinoma (combined therapy of GS-5745 with mFOLFOX6) and phase II clinical trials gastric and gastroesophageal junction adenocarcinomas (GS-5745 coupled with nivolumab) | [176,177,178,179] | |
| Naturally occurring inhibitors | Silibinin A, a compound isolated from milk thistle seeds | N/A | Preclinical studies | [181] |
| Gallic acid (GA), also known as 3,4,5-trihydroxybenzoic acid | N/A | Preclinical studies | [182] | |
| Methanolic extracts from the marine red alga Corallina pilulifera | N/A | Preclinical studies | [184] | |
| Fucoidan extracts from the seaweed Cladosiphon novae-caledoniae | N/A | Preclinical studies | [184] | |
| Hinokiflavone from Juniperus communis | N/A | Preclinical studies | [185] | |
| RNAi therapeutics | RNAi-mediated MMP9 gene silencing | Preclinical studies | [186,187,188,189,190] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Augoff, K.; Hryniewicz-Jankowska, A.; Tabola, R.; Stach, K. MMP9: A Tough Target for Targeted Therapy for Cancer. Cancers 2022, 14, 1847. https://doi.org/10.3390/cancers14071847
Augoff K, Hryniewicz-Jankowska A, Tabola R, Stach K. MMP9: A Tough Target for Targeted Therapy for Cancer. Cancers. 2022; 14(7):1847. https://doi.org/10.3390/cancers14071847
Chicago/Turabian StyleAugoff, Katarzyna, Anita Hryniewicz-Jankowska, Renata Tabola, and Kamilla Stach. 2022. "MMP9: A Tough Target for Targeted Therapy for Cancer" Cancers 14, no. 7: 1847. https://doi.org/10.3390/cancers14071847
APA StyleAugoff, K., Hryniewicz-Jankowska, A., Tabola, R., & Stach, K. (2022). MMP9: A Tough Target for Targeted Therapy for Cancer. Cancers, 14(7), 1847. https://doi.org/10.3390/cancers14071847