
Development and Functional Characterization of a Versatile Radio-/Immunotheranostic Tool for Prostate Cancer Management

 ,
,  , , , , ,
, , , , ,  and add
Show full author list
and add
Show full author list
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines
2.3. Generation and Cultivation of UniCAR T Cells
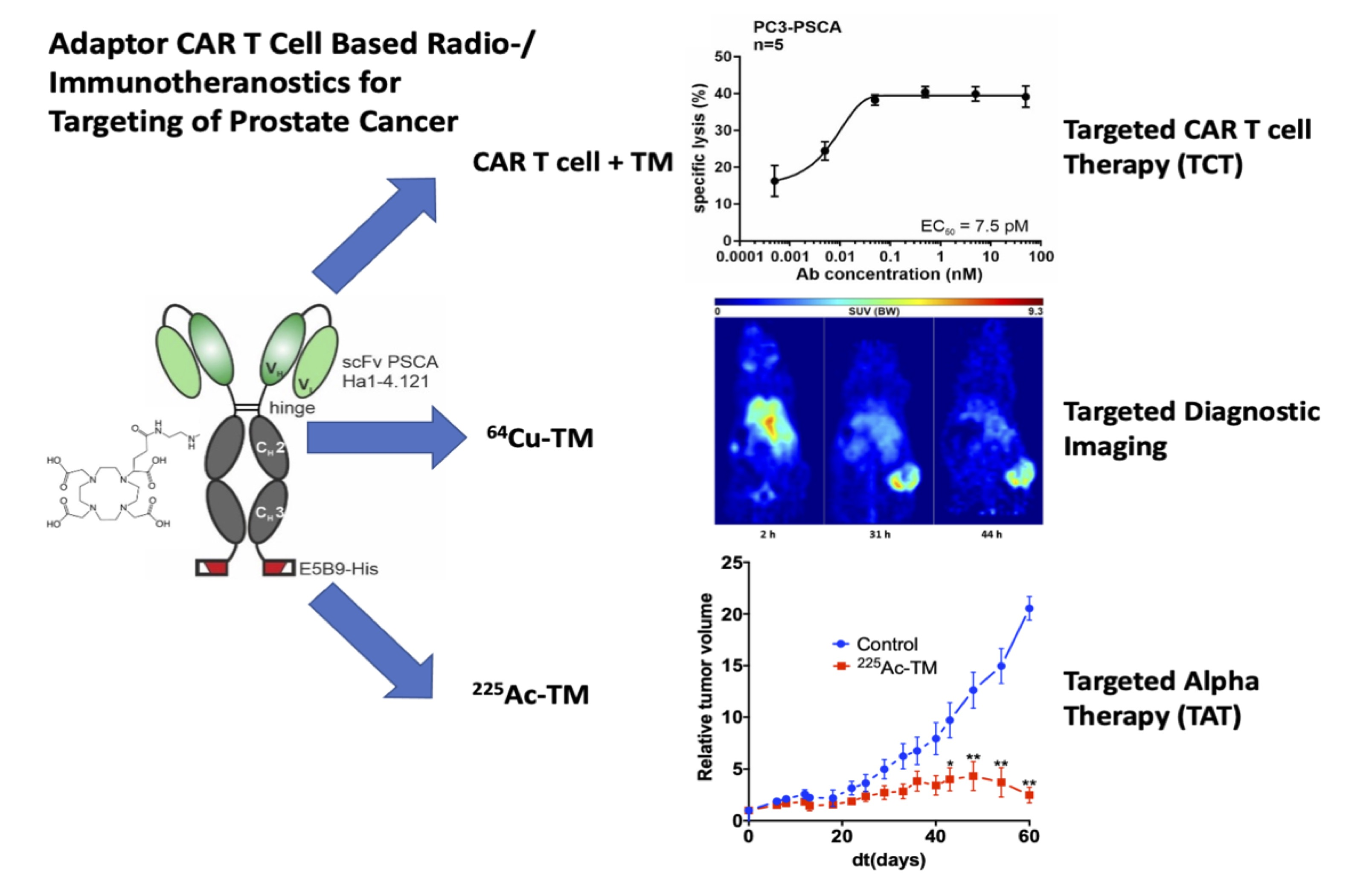
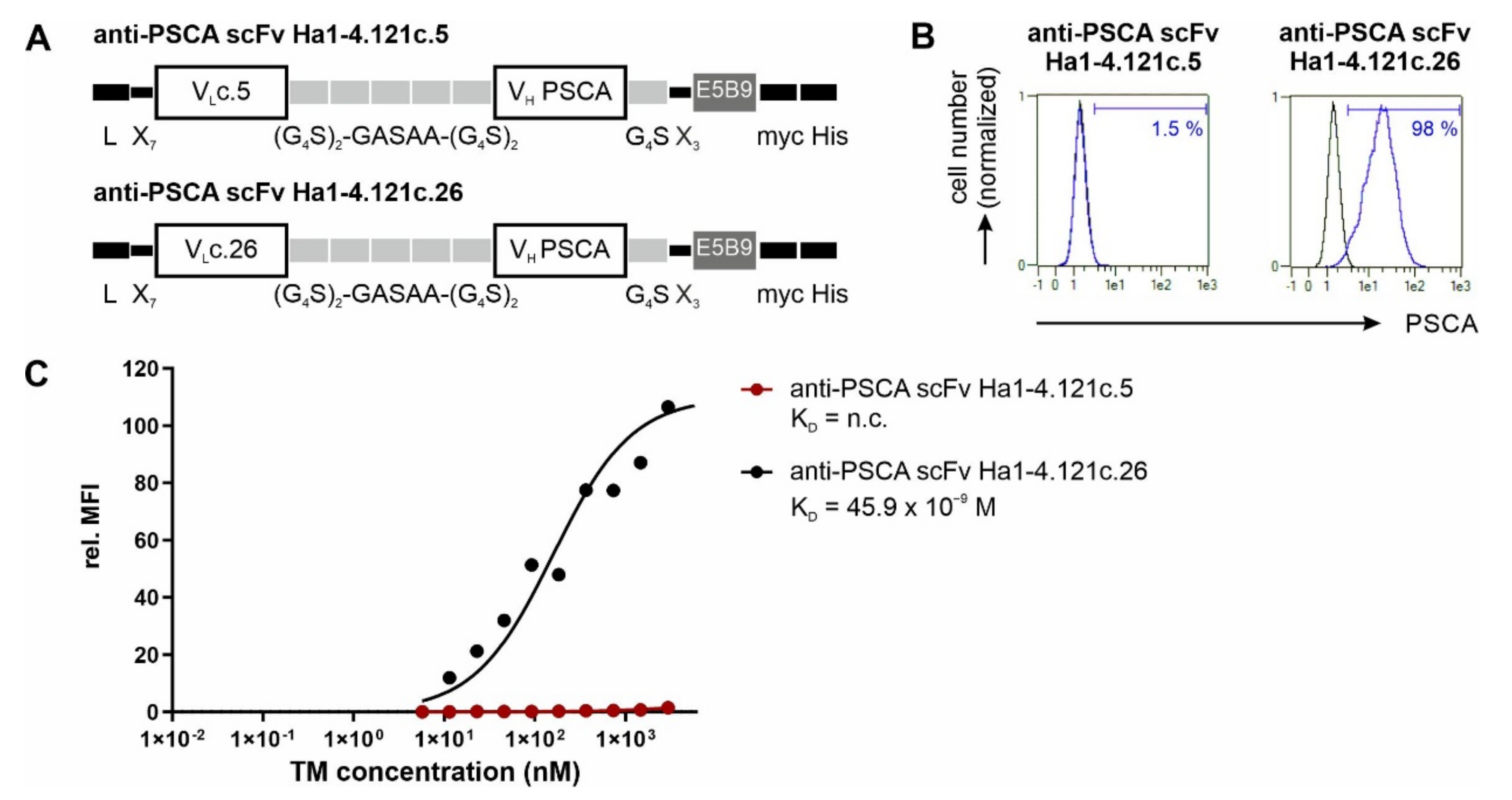
2.4. Cloning of Recombinant Antibodies
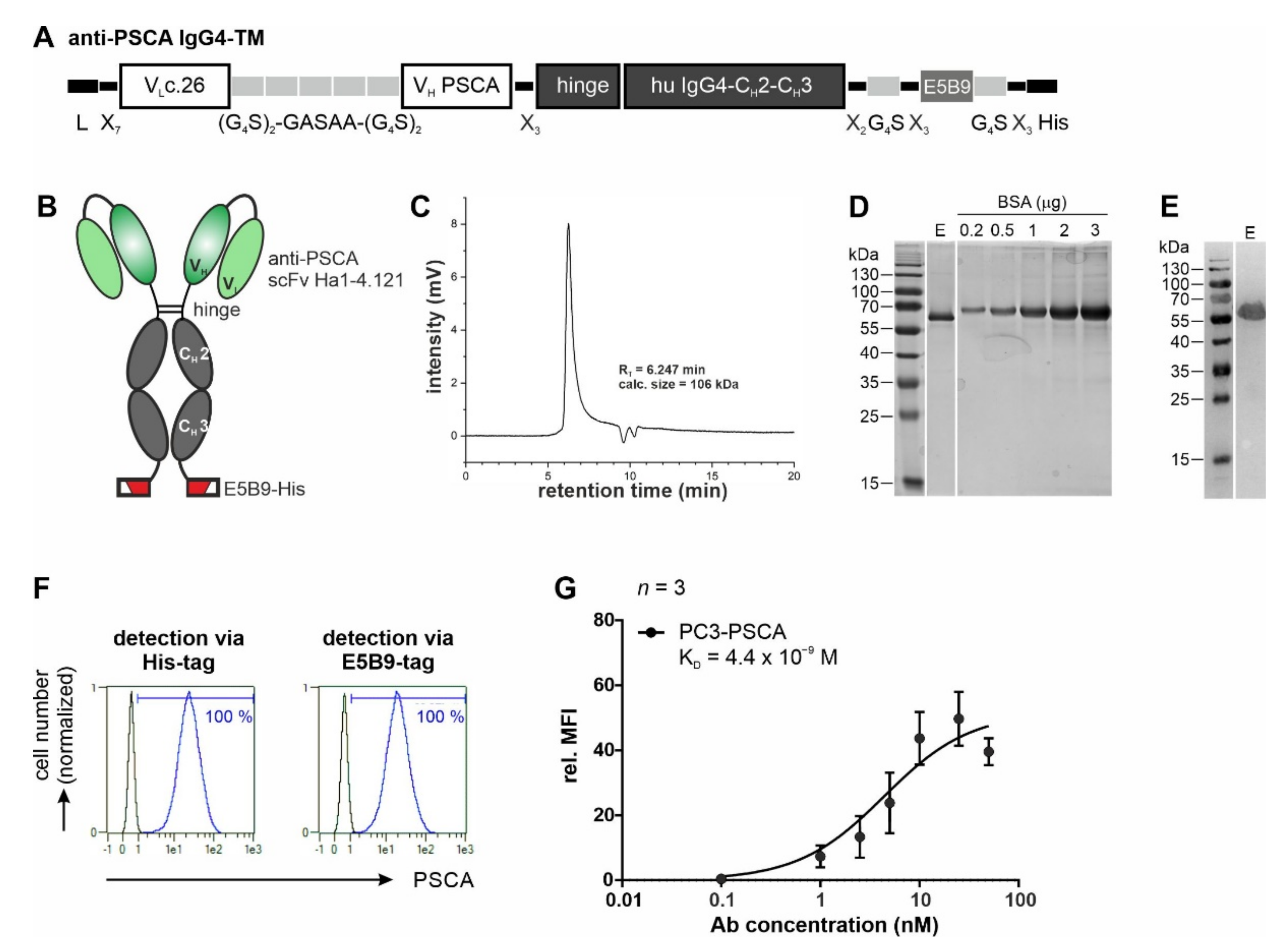
2.5. Expression, Purification, and Biochemical Characterization of Ab Constructs
2.6. Flow Cytometric Binding Analysis
2.7. Activation Assay and Enzyme-Linked Immunosorbent Assay (ELISA)
2.8. Chromium Release Assay
2.9. Radiolabeling of the TM with 64Cu and 225Ac
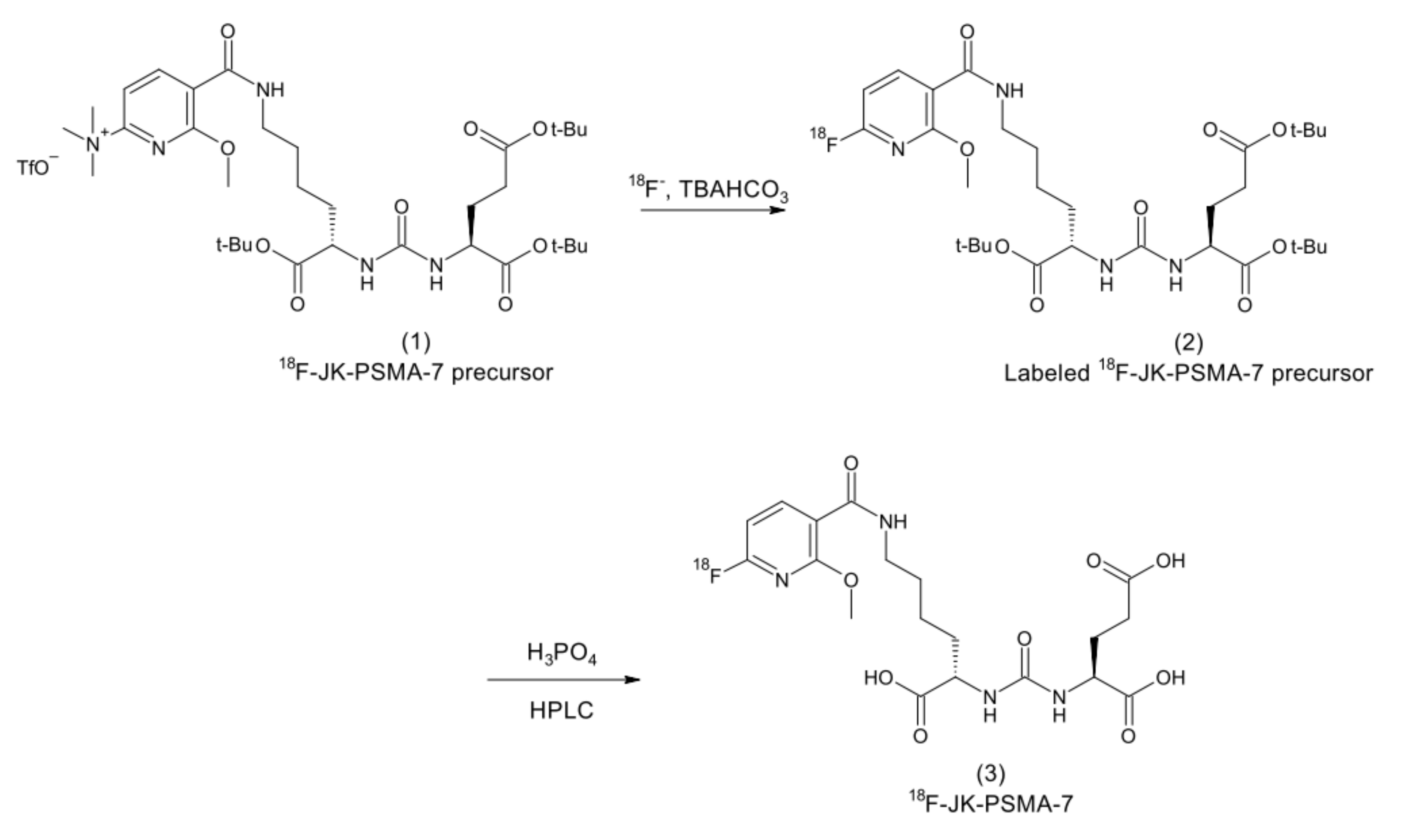
2.10. 18F-JK-PSMA-7 Synthesis
2.11. Animals, Feeding, Husbandry, Preparation, and Animal Experiments
2.12. 64Cu-TM Positron Emission Tomography
2.13. 18F-JK-PSMA-7 Positron Emission Tomography
2.14. X-ray CT Imaging
2.15. Statistical Analysis
3. Results
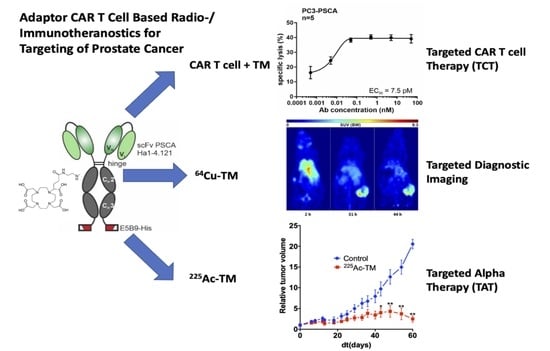
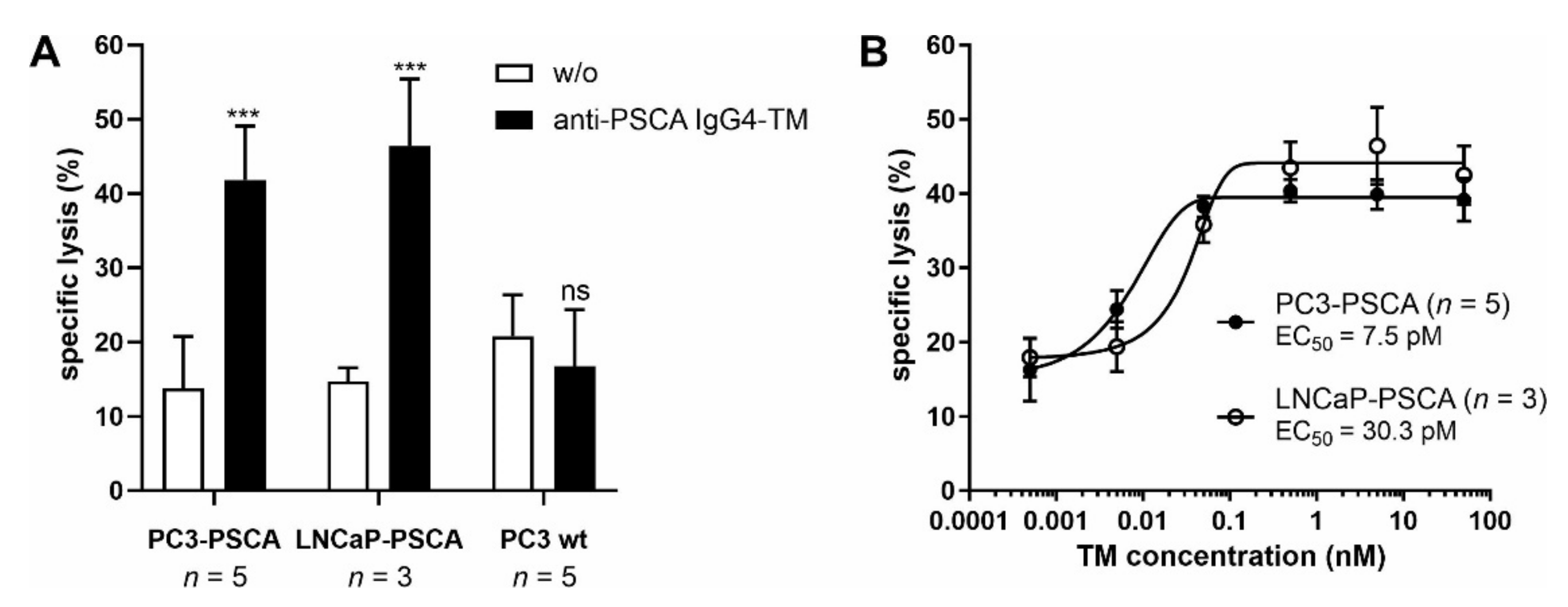
3.1. Antibody Preparation and Characterization
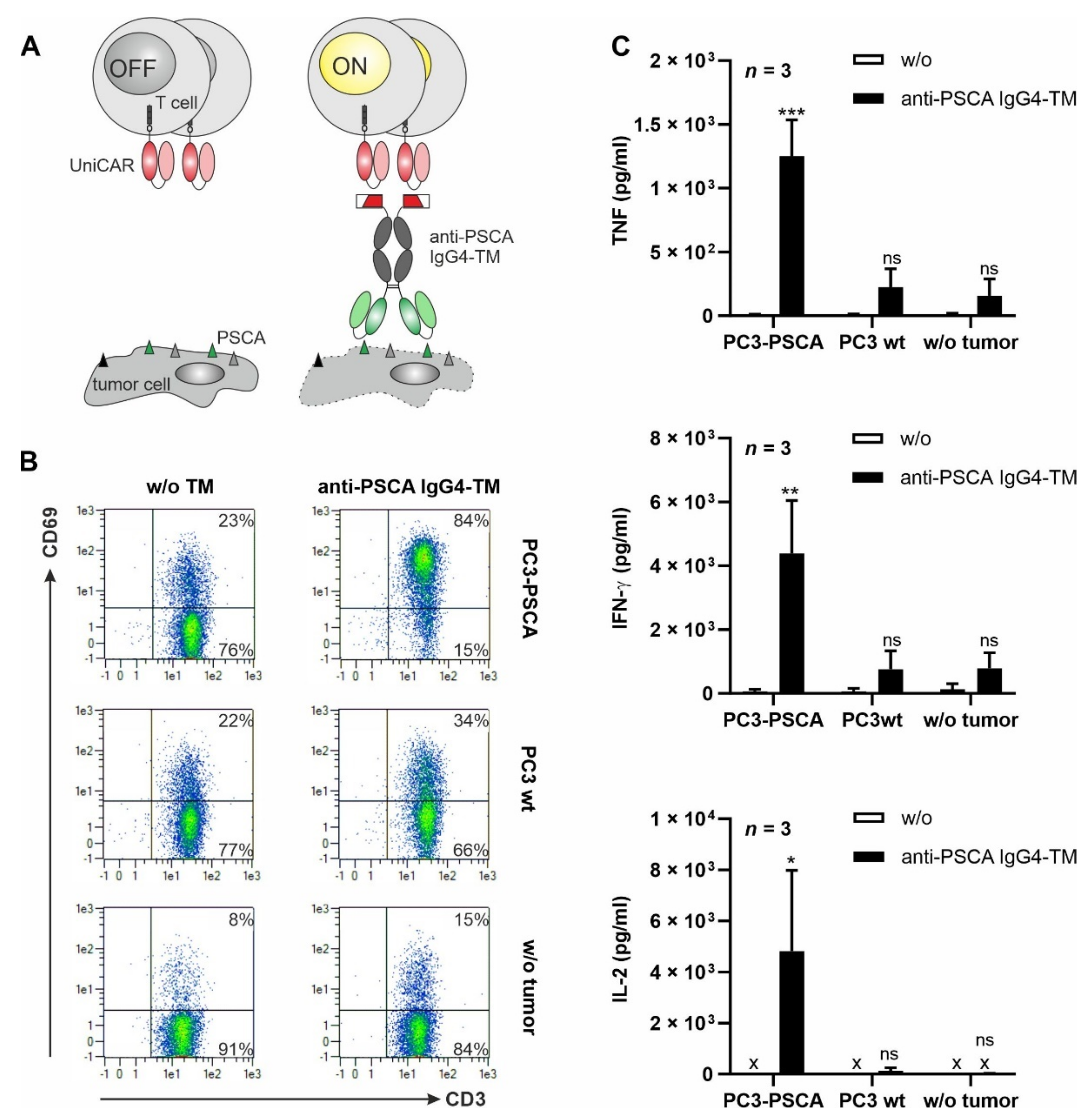
3.2. UniCAR T Cell Immunotherapy with the Anti-PSCA IgG4-TM
3.3. Radiolabeling of the Anti-PSCA IgG4-TM with Cu-64 and Ac-225
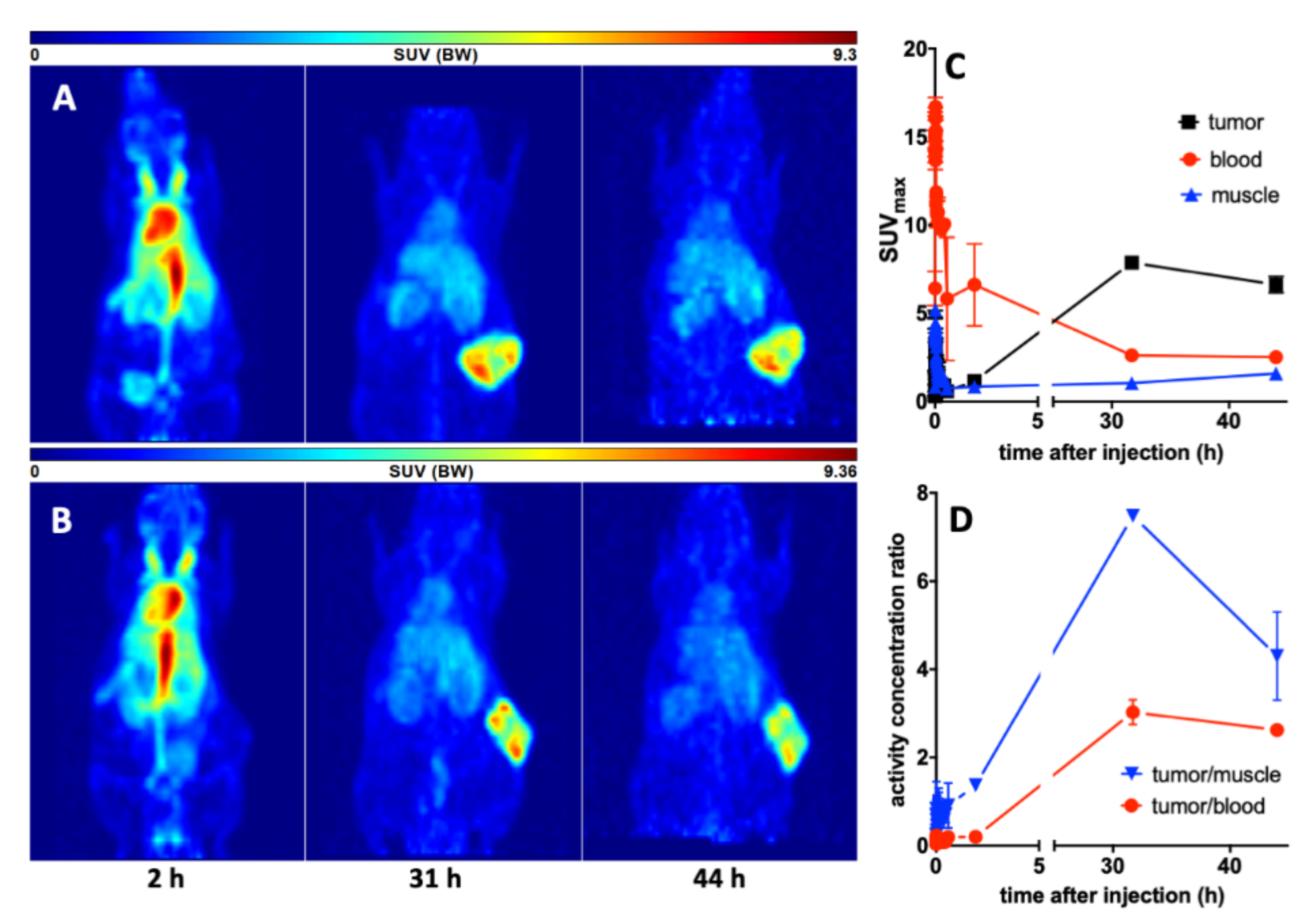
3.4. Imaging with 64Cu-TM in Xenotransplanted Mice
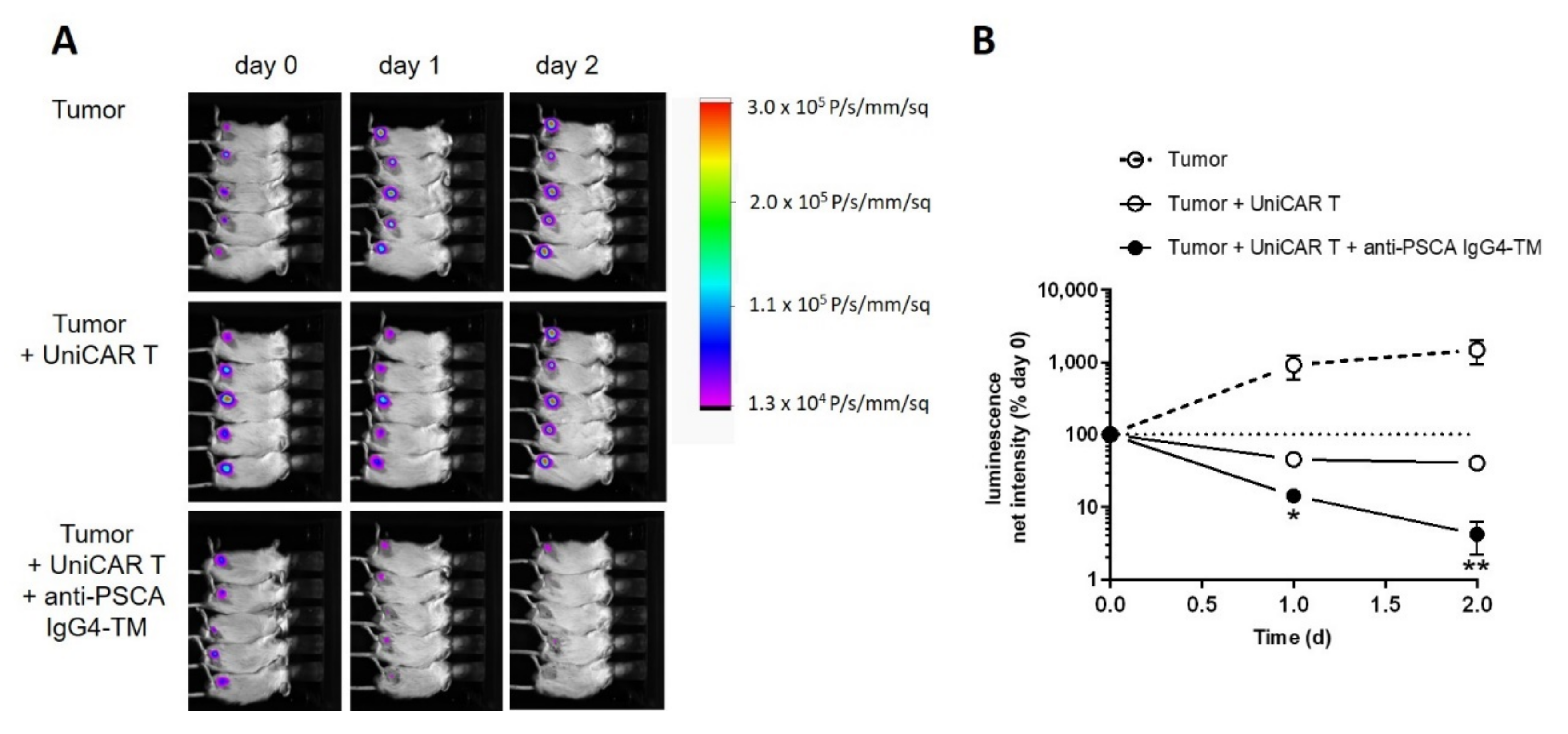
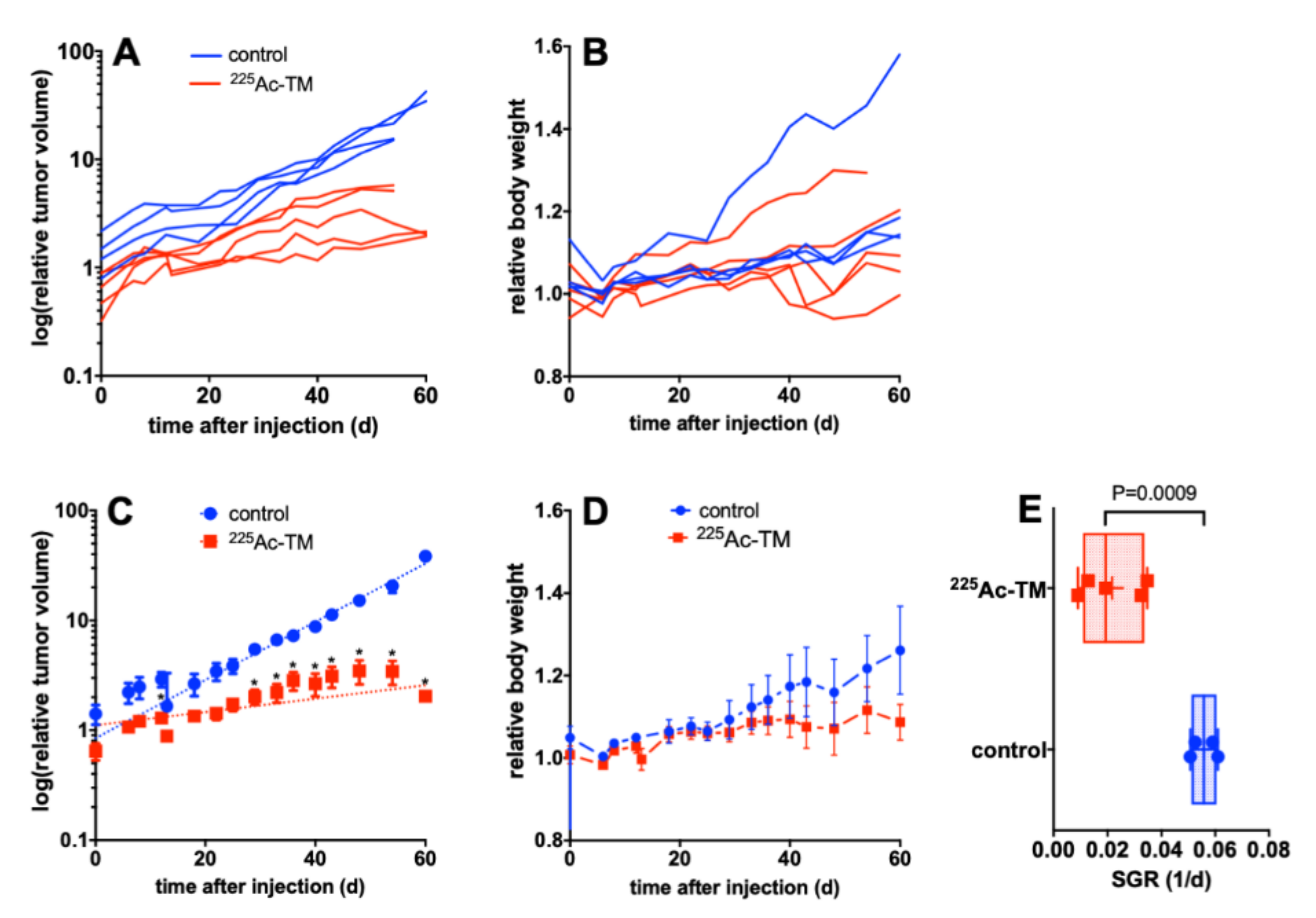
3.5. Targeted Alpha Therapy with the 225Ac-TM in a Prostate Cancer Mouse Model
3.5.1. Tumor Growth Delay after 225Ac-TM Treatment
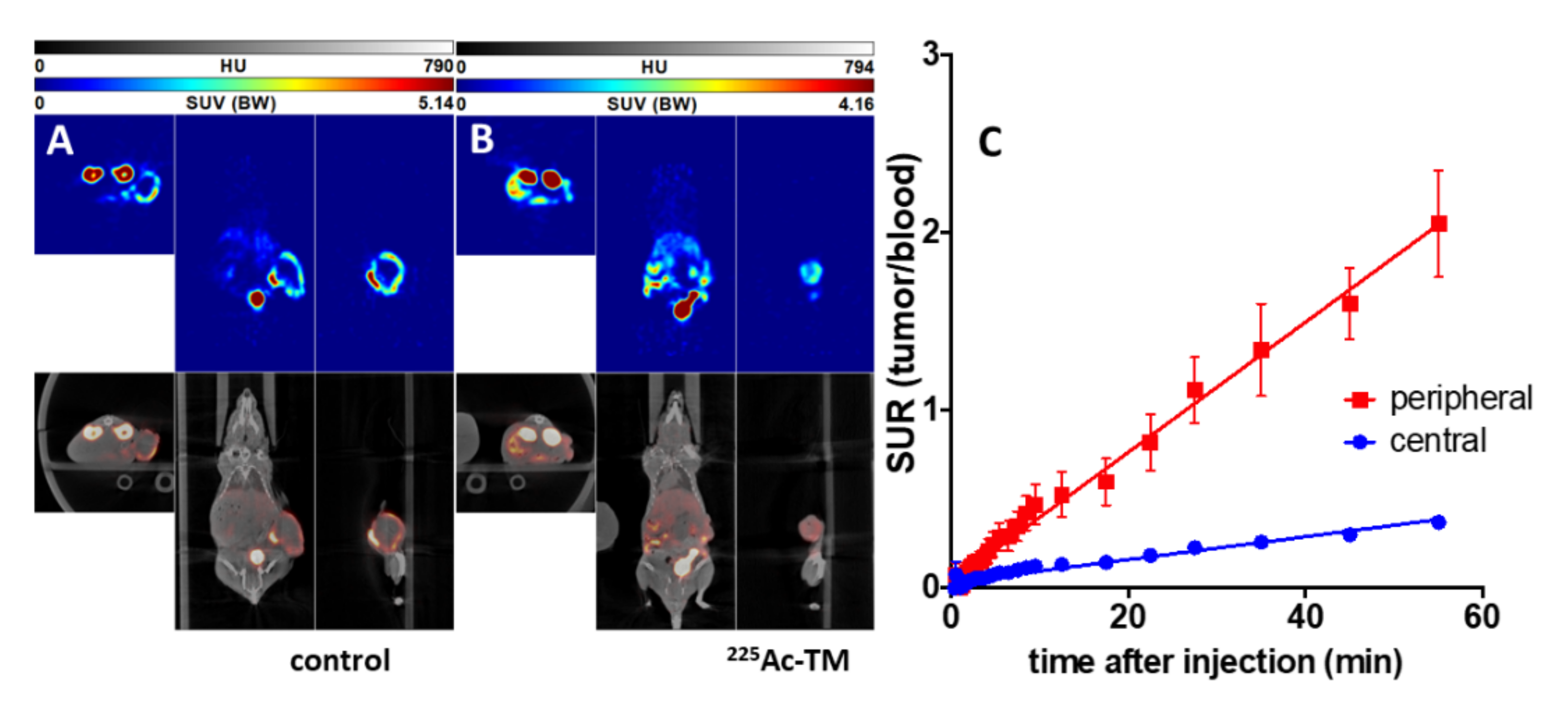
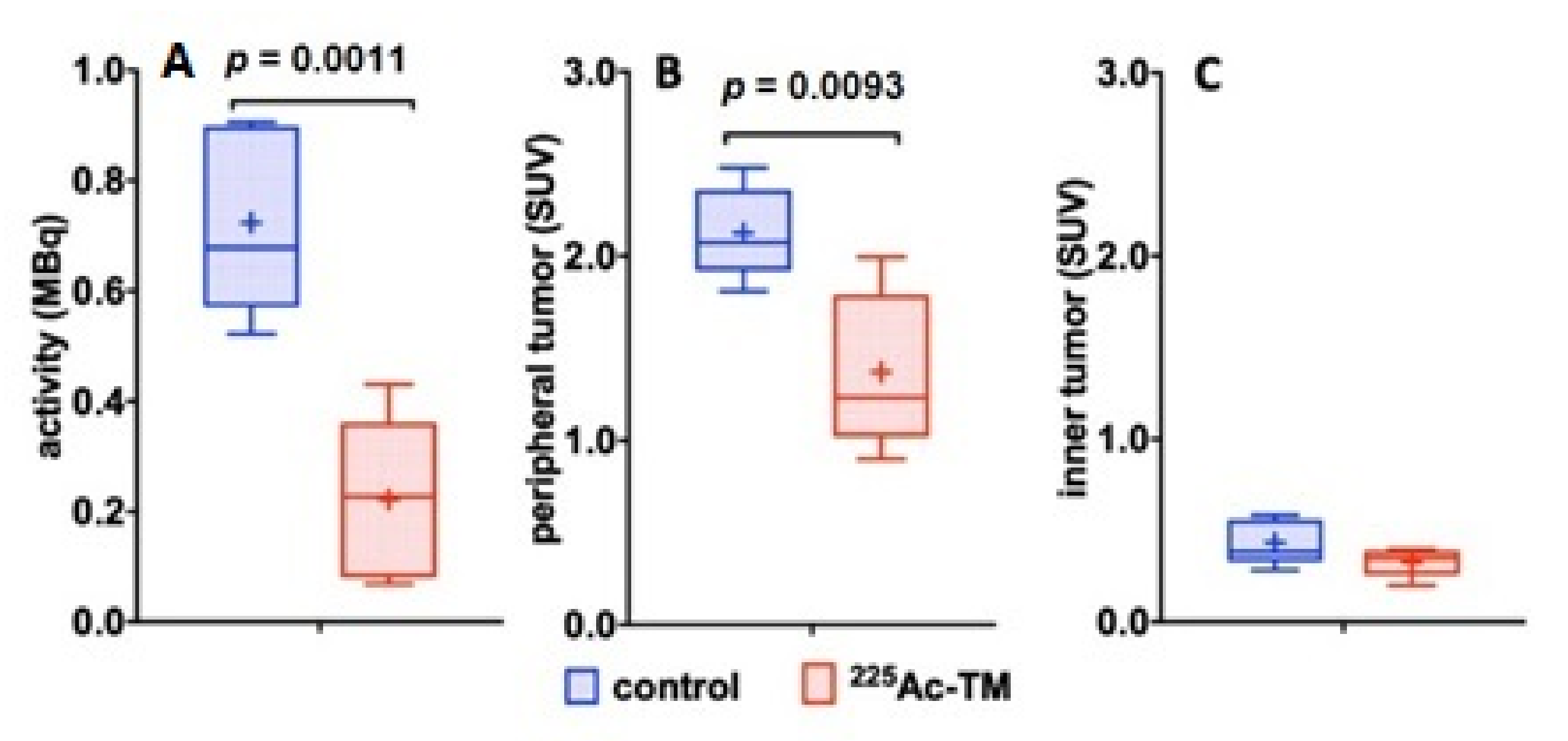
3.5.2. Imaging of the Tumors with 18F-JK-PSMA-7
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Farolfi, A.; Hadaschik, B.; Hamdy, F.C.; Herrmann, K.; Hofman, M.S.; Murphy, D.G.; Ost, P.; Padhani, A.R.; Fanti, S. Positron Emission Tomography and Whole-body Magnetic Resonance Imaging for Metastasis-directed Therapy in Hormone-sensitive Oligometastatic Prostate Cancer After Primary Radical Treatment: A Systematic Review. Eur. Urol. Oncol. 2021, 4, 714–730. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Zhou, N.; McLaughlin, M.; Rice, S.; Pillai, A.K.; Hao, G.; Sun, X. PSMA-Targeting Imaging and Theranostic Agents-Current Status and Future Perspective. Int. J. Mol. Sci. 2022, 23, 1158. [Google Scholar] [CrossRef] [PubMed]
- Farolfi, A.; Mei, R.; Ali, S.; Castellucci, P. Theragnostics in prostate cancer. Q. J. Nucl. Med. Mol. Imaging 2021, 65, 333–341. [Google Scholar] [CrossRef]
- FDA Approves First PSMA-Targeted PET Drug. J. Nucl. Med. 2021, 62, 11N. Available online: https://pubmed.ncbi.nlm.nih.gov/33468545/ (accessed on 5 March 2022).
- FDA Approves (18)F-DCFPyL PET Agent in Prostate Cancer. J. Nucl. Med. 2021, 62, 11N. Available online: https://pubmed.ncbi.nlm.nih.gov/34330741/ (accessed on 5 March 2022).
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Perera, M.P.J.; Thomas, P.B.; Risbridger, G.P.; Taylor, R.; Azad, A.; Hofman, M.S.; Williams, E.D.; Vela, I. Chimeric Antigen Receptor T-Cell Therapy in Metastatic Castrate-Resistant Prostate Cancer. Cancers 2022, 14, 503. [Google Scholar] [CrossRef]
- Siegel, D.A.; O’Neil, M.E.; Richards, T.B.; Dowling, N.F.; Weir, H.K. Prostate Cancer Incidence and Survival, by Stage and Race/Ethnicity—United States, 2001–2017. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1473–1480. [Google Scholar] [CrossRef]
- Bouchkouj, N.; Kasamon, Y.L.; de Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, M.C.; Lu, X.; Huang, Y.; Lin, X.; Mahmood, I.; Przepiorka, D.; Gavin, D.; Lee, S.; Liu, K.; George, B.; et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2019, 25, 1142–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Kanapuru, B.; George, B.; Lin, X.; Xu, Z.; Bryan, W.W.; Pazdur, R.; Theoret, M.R. FDA Approval Summary: Idecabtagene Vicleucel for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2022, OF1–OF6. [Google Scholar] [CrossRef] [PubMed]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Berish, R.B.; Ali, A.N.; Telmer, P.G.; Ronald, J.A.; Leong, H.S. Translational models of prostate cancer bone metastasis. Nat. Rev. Urol. 2018, 15, 403–421. [Google Scholar] [CrossRef]
- Arndt, C.; Fasslrinner, F.; Loureiro, L.R.; Koristka, S.; Feldmann, A.; Bachmann, M. Adaptor CAR Platforms-Next Generation of T Cell-Based Cancer Immunotherapy. Cancers 2020, 12, 1302. [Google Scholar] [CrossRef]
- Koristka, S.; Cartellieri, M.; Feldmann, A.; Arndt, C.; Loff, S.; Michalk, I.; Aliperta, R.; von Bonin, M.; Bornhäuser, M.; Ehninger, G.; et al. Flexible Antigen-Specific Redirection of Human Regulatory T Cells Via a Novel Universal Chimeric Antigen Receptor System. Blood 2014, 4, 3494. [Google Scholar] [CrossRef]
- Cartellieri, M.; Loff, S.; von Bonin, M.; Bejestani, E.P.; Ehninger, A.; Feldmann, A.; Koristka, S.; Arndt, C.; Ehninger, G.; Bachmann, M. Unicar: A novel modular retargeting platform technology for CAR T cells. Blood 2015, 126, 5549. [Google Scholar] [CrossRef]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef] [Green Version]
- Arndt, C.; Bachmann, M.; Bergmann, R.; Berndt, N.; Feldmann, A.; Koristka, S. Theranostic CAR T cell targeting: A brief review. J. Label. Comp. Radiopharm. 2019, 62, 533–540. [Google Scholar] [CrossRef]
- Koristka, S.; Cartellieri, M.; Theil, A.; Feldmann, A.; Arndt, C.; Stamova, S.; Michalk, I.; Topfer, K.; Temme, A.; Kretschmer, K.; et al. Retargeting of human regulatory T cells by single-chain bispecific antibodies. J. Immunol. 2012, 188, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Mitwasi, N.; Feldmann, A.; Bergmann, R.; Berndt, N.; Arndt, C.; Koristka, S.; Kegler, A.; Jureczek, J.; Hoffmann, A.; Ehninger, A.; et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget 2017, 8, 108584–108603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, M. Anti-La Antibodies and Their Use for Immunotargeting. U.S. Patent 9,540,446 B2, 10 January 2017. [Google Scholar]
- Kremerskothen, J.; Nettermann, M.; op de Bekke, A.; Bachmann, M.; Brosius, J. Identification of human autoantigen La/SS-B as BC1/BC200 RNA-binding protein. DNA Cell Biol. 1998, 17, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Carmo-Fonseca, M.; Pfeifer, K.; Schroder, H.C.; Vaz, M.F.; Fonseca, J.E.; Muller, W.E.; Bachmann, M. Identification of La ribonucleoproteins as a component of interchromatin granules. Exp. Cell Res. 1989, 185, 73–85. [Google Scholar] [CrossRef]
- Yiannaki, E.E.; Tzioufas, A.G.; Bachmann, M.; Hantoumi, J.; Tsikaris, V.; Sakarellos-Daitsiotis, M.; Sakarellos, C.; Moutsopoulos, H.M. The value of synthetic linear epitope analogues of La/SSB for the detection of autoantibodies to La/SSB; specificity, sensitivity and comparison of methods. Clin. Exp. Immunol. 1998, 112, 152–158. [Google Scholar] [CrossRef]
- Koristka, S.; Cartellieri, M.; Arndt, C.; Bippes, C.C.; Feldmann, A.; Michalk, I.; Wiefel, K.; Stamova, S.; Schmitz, M.; Ehninger, G.; et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J. Autoimmun. 2013, 42, 105–116. [Google Scholar] [CrossRef]
- Albert, S.; Arndt, C.; Koristka, S.; Berndt, N.; Bergmann, R.; Feldmann, A.; Schmitz, M.; Pietzsch, J.; Steinbach, J.; Bachmann, M. From mono- to bivalent: Improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget 2018, 9, 25597–25616. [Google Scholar] [CrossRef] [Green Version]
- Koristka, S.; Kegler, A.; Bergmann, R.; Arndt, C.; Feldmann, A.; Albert, S.; Cartellieri, M.; Ehninger, A.; Ehninger, G.; Middeke, J.M.; et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J. Autoimmun. 2018, 90, 116–131. [Google Scholar] [CrossRef]
- Loureiro, L.R.; Feldmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Pietzsch, J.; Novo, C.; Videira, P.; Bachmann, M. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018, 8, 81. [Google Scholar] [CrossRef]
- Wermke, M.; Kraus, S.; Ehninger, A.; Bargou, R.C.; Goebeler, M.E.; Middeke, J.M.; Kreissig, C.; von Bonin, M.; Koedam, J.; Pehl, M.; et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood 2021, 137, 3145–3148. [Google Scholar] [CrossRef]
- Link, T.; Kuithan, F.; Ehninger, A.; Kuhlmann, J.D.; Kramer, M.; Werner, A.; Gatzweiler, A.; Richter, B.; Ehninger, G.; Baretton, G.; et al. Exploratory investigation of PSCA-protein expression in primary breast cancer patients reveals a link to HER2/neu overexpression. Oncotarget 2017, 8, 54592–54603. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Feldmann, A.; Topfer, K.; Koristka, S.; Cartellieri, M.; Temme, A.; Ehninger, A.; Ehninger, G.; Bachmann, M. Redirection of CD4+ and CD8+ T lymphocytes via a novel antibody-based modular targeting system triggers efficient killing of PSCA+ prostate tumor cells. Prostate 2014, 74, 1347–1358. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, A.; Arndt, C.; Bergmann, R.; Loff, S.; Cartellieri, M.; Bachmann, D.; Aliperta, R.; Hetzenecker, M.; Ludwig, F.; Albert, S.; et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget 2017, 8, 31368–31385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgenroth, A.; Cartellieri, M.; Schmitz, M.; Gunes, S.; Weigle, B.; Bachmann, M.; Abken, H.; Rieber, E.P.; Temme, A. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate 2007, 67, 1121–1131. [Google Scholar] [CrossRef]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Mathe, D.; Hegedus, N.; et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [Green Version]
- Stamova, S.; Cartellieri, M.; Feldmann, A.; Arndt, C.; Koristka, S.; Bartsch, H.; Bippes, C.C.; Wehner, R.; Schmitz, M.; von Bonin, M.; et al. Unexpected recombinations in single chain bispecific anti-CD3-anti-CD33 antibodies can be avoided by a novel linker module. Mol. Immunol. 2011, 49, 474–482. [Google Scholar] [CrossRef]
- Feldmann, A.; Stamova, S.; Bippes, C.C.; Bartsch, H.; Wehner, R.; Schmitz, M.; Temme, A.; Cartellieri, M.; Bachmann, M. Retargeting of T cells to prostate stem cell antigen expressing tumor cells: Comparison of different antibody formats. Prostate 2011, 71, 998–1011. [Google Scholar] [CrossRef]
- Feldmann, A.; Hoffmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Rodrigues Loureiro, L.; Kittel-Boselli, E.; Mitwasi, N.; Kegler, A.; et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. Oncoimmunology 2020, 9, 1785608. [Google Scholar] [CrossRef]
- Cartellieri, M.; Koristka, S.; Arndt, C.; Feldmann, A.; Stamova, S.; von Bonin, M.; Topfer, K.; Kruger, T.; Geib, M.; Michalk, I.; et al. A novel ex vivo isolation and expansion procedure for chimeric antigen receptor engrafted human T cells. PLoS ONE 2014, 9, e93745. [Google Scholar] [CrossRef]
- Loureiro, L.R.; Feldmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Mathe, D.; Hegedus, N.; Szigeti, K.; Videira, P.A.; Bachmann, M.; et al. Extended half-life target module for sustainable UniCAR T-cell treatment of STn-expressing cancers. J. Exp. Clin. Cancer Res. 2020, 39, 77. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Topfer, K.; Stamova, S.; Krone, F.; Cartellieri, M.; Koristka, S.; Michalk, I.; Lindemann, D.; Schmitz, M.; et al. Novel humanized and highly efficient bispecific antibodies mediate killing of prostate stem cell antigen-expressing tumor cells by CD8+ and CD4+ T cells. J. Immunol. 2012, 189, 3249–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, C.; Koristka, S.; Feldmann, A.; Bergmann, R.; Bachmann, M. Coomassie Brilliant Blue Staining of Polyacrylamide Gels. Methods Mol. Biol. 2018, 1853, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Arndt, C.; Feldmann, A.; Bergmann, R.; Bachmann, D.; Koristka, S.; Ludwig, F.; Ziller-Walter, P.; Kegler, A.; Gartner, S.; et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology 2017, 6, e1287246. [Google Scholar] [CrossRef]
- Arndt, C.; Loureiro, L.R.; Feldmann, A.; Jureczek, J.; Bergmann, R.; Mathe, D.; Hegedus, N.; Berndt, N.; Koristka, S.; Mitwasi, N.; et al. UniCAR T cell immunotherapy enables efficient elimination of radioresistant cancer cells. Oncoimmunology 2020, 9, 1743036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, M.P.; Bartsch, T.; Bippes, C.C.; Bachmann, D.; Puentes-Cala, E.; Bachmann, J.; Bartsch, H.; Arndt, C.; Koristka, S.; Loureiro, L.R.; et al. T Cell Mediated Conversion of a Non-Anti-La Reactive B Cell to an Autoreactive Anti-La B Cell by Somatic Hypermutation. Int. J. Mol. Sci. 2021, 22, 1198. [Google Scholar] [CrossRef] [PubMed]
- Thieme, S.; Walther, M.; Pietzsch, H.J.; Henniger, J.; Preusche, S.; Mading, P.; Steinbach, J. Module-assisted preparation of 64Cu with high specific activity. Appl. Radiat. Isot. 2012, 70, 602–608. [Google Scholar] [CrossRef]
- David, T.; Hlinova, V.; Kubicek, V.; Bergmann, R.; Striese, F.; Berndt, N.; Szollosi, D.; Kovacs, T.; Mathe, D.; Bachmann, M.; et al. Improved Conjugation, 64-Cu Radiolabeling, in Vivo Stability, and Imaging Using Nonprotected Bifunctional Macrocyclic Ligands: Bis(Phosphinate) Cyclam (BPC) Chelators. J. Med. Chem. 2018, 61, 8774–8796. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Endepols, H.; Krapf, P.; Guliyev, M.; Urusova, E.A.; Richarz, R.; Hohberg, M.; Dietlein, M.; Drzezga, A.; Neumaier, B. Discovery of (18)F-JK-PSMA-7, a PET Probe for the Detection of Small PSMA-Positive Lesions. J. Nucl. Med. 2019, 60, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Mehrara, E.; Forssell-Aronsson, E.; Ahlman, H.; Bernhardt, P. Specific growth rate versus doubling time for quantitative characterization of tumor growth rate. Cancer Res. 2007, 67, 3970–3975. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef]
- Miyahira, A.K.; Pienta, K.J.; Morris, M.J.; Bander, N.H.; Baum, R.P.; Fendler, W.P.; Goeckeler, W.; Gorin, M.A.; Hennekes, H.; Pomper, M.G.; et al. Meeting report from the Prostate Cancer Foundation PSMA-directed radionuclide scientific working group. Prostate 2018, 78, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Pandit-Taskar, N.; O’Donoghue, J.A.; Durack, J.C.; Lyashchenko, S.K.; Cheal, S.M.; Beylergil, V.; Lefkowitz, R.A.; Carrasquillo, J.A.; Martinez, D.F.; Fung, A.M.; et al. A Phase I/II Study for Analytic Validation of 89Zr-J591 ImmunoPET as a Molecular Imaging Agent for Metastatic Prostate Cancer. Clin. Cancer Res. 2015, 21, 5277–5285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffran, D.C.; Raitano, A.B.; Hubert, R.S.; Witte, O.N.; Reiter, R.E.; Jakobovits, A. Anti-PSCA mAbs inhibit tumor growth and metastasis formation and prolong the survival of mice bearing human prostate cancer xenografts. Proc. Natl. Acad. Sci. USA 2001, 98, 2658–2663. [Google Scholar] [CrossRef] [Green Version]
- Olafsen, T.; Gu, Z.; Sherman, M.A.; Leyton, J.V.; Witkosky, M.E.; Shively, J.E.; Raubitschek, A.A.; Morrison, S.L.; Wu, A.M.; Reiter, R.E. Targeting, imaging, and therapy using a humanized antiprostate stem cell antigen (PSCA) antibody. J. Immunother. 2007, 30, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.M.; Tavare, R.; Zettlitz, K.A.; Rochefort, M.M.; Salazar, F.B.; Jiang, Z.K.; Reiter, R.E.; Wu, A.M. Applications of immunoPET: Using 124I-anti-PSCA A11 minibody for imaging disease progression and response to therapy in mouse xenograft models of prostate cancer. Clin. Cancer Res. 2014, 20, 6367–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.K.; Zettlitz, K.A.; Tavare, R.; Kobayashi, N.; Reiter, R.E.; Wu, A.M. Dual-Modality ImmunoPET/Fluorescence Imaging of Prostate Cancer with an Anti-PSCA Cys-Minibody. Theranostics 2018, 8, 5903–5914. [Google Scholar] [CrossRef]
- Kiessling, A.; Wehner, R.; Fussel, S.; Bachmann, M.; Wirth, M.P.; Schmitz, M. Tumor-associated antigens for specific immunotherapy of prostate cancer. Cancers 2012, 4, 193–217. [Google Scholar] [CrossRef] [Green Version]
- Cunha, A.C.; Weigle, B.; Kiessling, A.; Bachmann, M.; Rieber, E.P. Tissue-specificity of prostate specific antigens: Comparative analysis of transcript levels in prostate and non-prostatic tissues. Cancer Lett. 2006, 236, 229–238. [Google Scholar] [CrossRef]
- Arndt, C.; Feldmann, A.; Koristka, S.; Cartellieri, M.; Dimmel, M.; Ehninger, A.; Ehninger, G.; Bachmann, M. Simultaneous targeting of prostate stem cell antigen and prostate-specific membrane antigen improves the killing of prostate cancer cells using a novel modular T cell-retargeting system. Prostate 2014, 74, 1335–1346. [Google Scholar] [CrossRef]
- Bloem, K.; Hernandez-Breijo, B.; Martinez-Feito, A.; Rispens, T. Immunogenicity of Therapeutic Antibodies: Monitoring Antidrug Antibodies in a Clinical Context. Ther. Drug Monit. 2017, 39, 327–332. [Google Scholar] [CrossRef]
- Houen, G. Therapeutic Antibodies: An Overview. Methods Mol. Biol. 2022, 2313, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Simmons, E.; Abdeen, S.; Kinne, A.; Parmer, E.; Rinker, S.; Thystrup, J.; Ramaswamy, S.; Bowsher, R.R. Sensitive assay design for detection of anti-drug antibodies to biotherapeutics that lack an immunoglobulin Fc domain. Sci. Rep. 2021, 11, 15467. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E. Biotherapeutics: Challenges and Opportunities for Predictive Toxicology of Monoclonal Antibodies. Int. J. Mol. Sci. 2018, 19, 3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumet, C.; Pottier, J.; Gouilleux-Gruart, V.; Watier, H. Insights into the IgG heavy chain engineering patent landscape as applied to IgG4 antibody development. MAbs 2019, 11, 1341–1350. [Google Scholar] [CrossRef] [Green Version]
- Neuber, T.; Frese, K.; Jaehrling, J.; Jager, S.; Daubert, D.; Felderer, K.; Linnemann, M.; Hohne, A.; Kaden, S.; Kolln, J.; et al. Characterization and screening of IgG binding to the neonatal Fc receptor. MAbs 2014, 6, 928–942. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Arndt, C.; Feldmann, A.; Koristka, S.; Schafer, M.; Bergmann, R.; Mitwasi, N.; Berndt, N.; Bachmann, D.; Kegler, A.; Schmitz, M.; et al. A theranostic PSMA ligand for PET imaging and retargeting of T cells expressing the universal chimeric antigen receptor UniCAR. Oncoimmunology 2019, 8, 1659095. [Google Scholar] [CrossRef] [Green Version]
- Zechmann, C.M.; Afshar-Oromieh, A.; Armor, T.; Stubbs, J.B.; Mier, W.; Hadaschik, B.; Joyal, J.; Kopka, K.; Debus, J.; Babich, J.W.; et al. Radiation dosimetry and first therapy results with a (124)I/(131)I-labeled small molecule (MIP-1095) targeting PSMA for prostate cancer therapy. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Weineisen, M.; Schottelius, M.; Simecek, J.; Baum, R.P.; Yildiz, A.; Beykan, S.; Kulkarni, H.R.; Lassmann, M.; Klette, I.; Eiber, M.; et al. 68Ga- and 177Lu-Labeled PSMA I&T: Optimization of a PSMA-Targeted Theranostic Concept and First Proof-of-Concept Human Studies. J. Nucl. Med. 2015, 56, 1169–1176. [Google Scholar] [CrossRef] [Green Version]
- Benesova, M.; Bauder-Wust, U.; Schafer, M.; Klika, K.D.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Linker Modification Strategies To Control the Prostate-Specific Membrane Antigen (PSMA)-Targeting and Pharmacokinetic Properties of DOTA-Conjugated PSMA Inhibitors. J. Med. Chem. 2016, 59, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Asti, M.; Iori, M.; Capponi, P.C.; Atti, G.; Rubagotti, S.; Martin, R.; Brennauer, A.; Muller, M.; Bergmann, R.; Erba, P.A.; et al. Influence of different chelators on the radiochemical properties of a 68-Gallium labelled bombesin analogue. Nucl. Med. Biol. 2014, 41, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Anderson, C.J. Chelators for copper radionuclides in positron emission tomography radiopharmaceuticals. J. Label. Comp. Radiopharm. 2014, 57, 224–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, G.; Zarschler, K.; Hunoldt, S.; Martinez, I.I.S.; Ruehl, C.L.; Matterna, M.; Bergmann, R.; Mathe, D.; Hegedus, N.; Bachmann, M.; et al. Versatile Bispidine-Based Bifunctional Chelators for (64) Cu(II)-Labelling of Biomolecules. Chemistry 2020, 26, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Guo, Y.; White, A.G.; Cai, Z.; Modi, J.; Ferdani, R.; Anderson, C.J. Comparison of conjugation strategies of cross-bridged macrocyclic chelators with cetuximab for copper-64 radiolabeling and PET imaging of EGFR in colorectal tumor-bearing mice. Mol. Pharm. 2014, 11, 3980–3987. [Google Scholar] [CrossRef]
- Kennel, S.J.; Chappell, L.L.; Dadachova, K.; Brechbiel, M.W.; Lankford, T.K.; Davis, I.A.; Stabin, M.; Mirzadeh, S. Evaluation of 225Ac for vascular targeted radioimmunotherapy of lung tumors. Cancer Biother. Radiopharm. 2000, 15, 235–244. [Google Scholar] [CrossRef]
- Stein, B.W.; Morgenstern, A.; Batista, E.R.; Birnbaum, E.R.; Bone, S.E.; Cary, S.K.; Ferrier, M.G.; John, K.D.; Pacheco, J.L.; Kozimor, S.A.; et al. Advancing Chelation Chemistry for Actinium and Other +3 f-Elements, Am, Cm, and La. J. Am. Chem. Soc. 2019, 141, 19404–19414. [Google Scholar] [CrossRef]
- Morgenstern, A.; Lilley, L.M.; Stein, B.W.; Kozimor, S.A.; Batista, E.R.; Yang, P. Computer-Assisted Design of Macrocyclic Chelators for Actinium-225 Radiotherapeutics. Inorg. Chem. 2021, 60, 623–632. [Google Scholar] [CrossRef]
- Hatcher-Lamarre, J.L.; Sanders, V.A.; Rahman, M.; Cutler, C.S.; Francesconi, L.C. Alpha emitting nuclides for targeted therapy. Nucl. Med. Biol. 2021, 92, 228–240. [Google Scholar] [CrossRef]
- Sun, M.; Niaz, M.J.; Niaz, M.O.; Tagawa, S.T. Prostate-Specific Membrane Antigen (PSMA)-Targeted Radionuclide Therapies for Prostate Cancer. Curr. Oncol. Rep. 2021, 23, 59. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-Targeted alpha-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgqvist, J.; Frost, S.; Pouget, J.P.; Albertsson, P. The potential and hurdles of targeted alpha therapy—Clinical trials and beyond. Front. Oncol. 2014, 3, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, R.; Allen, K.J.H.; Dawicki, W.; Geoghegan, E.M.; Ludwig, D.L.; Dadachova, E. 225Ac-labeled CD33-targeting antibody reverses resistance to Bcl-2 inhibitor venetoclax in acute myeloid leukemia models. Cancer Med. 2021, 10, 1128–1140. [Google Scholar] [CrossRef]
- Gasser, G.; Tjioe, L.; Graham, B.; Belousoff, M.J.; Juran, S.; Walther, M.; Kunstler, J.U.; Bergmann, R.; Stephan, H.; Spiccia, L. Synthesis, Copper(II) Complexation, (64)Cu-Labeling, and Bioconjugation of a New Bis(2-pyridylmethyl) Derivative of 1,4,7-Triazacyclononane. Bioconjug. Chem. 2008, 19, 719–730. [Google Scholar] [CrossRef]
- Cutler, C.S.; Wuest, M.; Anderson, C.J.; Reichert, D.E.; Sun, Y.; Martell, A.E.; Welch, M.J. Labeling and in vivo evaluation of novel copper(II) dioxotetraazamacrocyclic complexes. Nucl. Med. Biol. 2000, 27, 375–380. [Google Scholar] [CrossRef]
- Zettlitz, K.A.; Tsai, W.K.; Knowles, S.M.; Kobayashi, N.; Donahue, T.R.; Reiter, R.E.; Wu, A.M. Dual-Modality Immuno-PET and Near-Infrared Fluorescence Imaging of Pancreatic Cancer Using an Anti-Prostate Stem Cell Antigen Cys-Diabody. J. Nucl. Med. 2018, 59, 1398–1405. [Google Scholar] [CrossRef]
- Zang, J.; Fan, X.; Wang, H.; Liu, Q.; Wang, J.; Li, H.; Li, F.; Jacobson, O.; Niu, G.; Zhu, Z.; et al. First-in-human study of (177)Lu-EB-PSMA-617 in patients with metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 148–158. [Google Scholar] [CrossRef]
- Van Simaeys, G.; Doumont, G.; De Maeseneire, C.; Passon, N.; Lacroix, S.; Lentz, C.; Horion, A.; Warnier, C.; Torres, D.; Martens, C.; et al. [(18)F]-JK-PSMA-7 and [(18)F]-FDG tumour PET uptake in treated xenograft human prostate cancer model in mice. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1773–1784. [Google Scholar] [CrossRef]
- Lunger, L.; Tauber, R.; Feuerecker, B.; Gschwend, J.E.; Eiber, M.; Heck, M.M. Narrative review: Prostate-specific membrane antigen-radioligand therapy in metastatic castration-resistant prostate cancer. Transl. Androl. Urol. 2021, 10, 3963–3971. [Google Scholar] [CrossRef]
- Lee, D.Y.; Kim, Y.I. Effects of 225Ac-labeled prostate-specific membrane antigen radioligand therapy in metastatic castration-resistant prostate cancer: A meta-analysis. J. Nucl. Med. 2021, jnumed.121.262017. [Google Scholar] [CrossRef]
- Rathke, H.; Kratochwil, C.; Hohenberger, R.; Giesel, F.L.; Bruchertseifer, F.; Flechsig, P.; Morgenstern, A.; Hein, M.; Plinkert, P.; Haberkorn, U.; et al. Initial clinical experience performing sialendoscopy for salivary gland protection in patients undergoing (225)Ac-PSMA-617 RLT. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Bander, N.H.; Milowsky, M.I.; Nanus, D.M.; Kostakoglu, L.; Vallabhajosula, S.; Goldsmith, S.J. Phase I trial of 177lutetium-labeled J591, a monoclonal antibody to prostate-specific membrane antigen, in patients with androgen-independent prostate cancer. J. Clin. Oncol. 2005, 23, 4591–4601. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Vallabhajosula, S.; Christos, P.J.; Jhanwar, Y.S.; Batra, J.S.; Lam, L.; Osborne, J.; Beltran, H.; Molina, A.M.; Goldsmith, S.J.; et al. Phase 1/2 study of fractionated dose lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 ((177) Lu-J591) for metastatic castration-resistant prostate cancer. Cancer 2019, 125, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.; Vallabhajosula, S.; Christos, P.; Akhtar, N.H.; Osborne, J.; Goldsmith, S.J.; Larson, S.; Taskar, N.P.; et al. Phase II study of Lutetium-177-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 5182–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niaz, M.J.; Batra, J.S.; Walsh, R.D.; Ramirez-Fort, M.K.; Vallabhajosula, S.; Jhanwar, Y.S.; Molina, A.M.; Nanus, D.M.; Osborne, J.R.; Bander, N.H.; et al. Pilot Study of Hyperfractionated Dosing of Lutetium-177-Labeled Antiprostate-Specific Membrane Antigen Monoclonal Antibody J591 ((177) Lu-J591) for Metastatic Castration-Resistant Prostate Cancer. Oncologist 2020, 25, 477-e895. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, E.; Chargari, C.; Galluzzi, L.; Kroemer, G. Optimising efficacy and reducing toxicity of anticancer radioimmunotherapy. Lancet Oncol. 2019, 20, e452–e463. [Google Scholar] [CrossRef]
- Flynn, J.P.; O’Hara, M.H.; Gandhi, S.J. Preclinical rationale for combining radiation therapy and immunotherapy beyond checkpoint inhibitors (i.e., CART). Transl. Lung Cancer Res. 2017, 6, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Eckert, F.; Jelas, I.; Oehme, M.; Huber, S.M.; Sonntag, K.; Welker, C.; Gillies, S.D.; Strittmatter, W.; Zips, D.; Handgretinger, R.; et al. Tumor-targeted IL-12 combined with local irradiation leads to systemic tumor control via abscopal effects in vivo. Oncoimmunology 2017, 6, e1323161. [Google Scholar] [CrossRef] [Green Version]
- Weiss, T.; Weller, M.; Guckenberger, M.; Sentman, C.L.; Roth, P. NKG2D-Based CAR T Cells and Radiotherapy Exert Synergistic Efficacy in Glioblastoma. Cancer Res. 2018, 78, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Minn, I.; Rowe, S.P.; Pomper, M.G. Enhancing CAR T-cell therapy through cellular imaging and radiotherapy. Lancet Oncol. 2019, 20, e443–e451. [Google Scholar] [CrossRef]
- DeSelm, C.; Palomba, M.L.; Yahalom, J.; Hamieh, M.; Eyquem, J.; Rajasekhar, V.K.; Sadelain, M. Low-Dose Radiation Conditioning Enables CAR T Cells to Mitigate Antigen Escape. Mol. Ther. 2018, 26, 2542–2552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menager, J.; Gorin, J.B.; Maurel, C.; Drujont, L.; Gouard, S.; Louvet, C.; Cherel, M.; Faivre-Chauvet, A.; Morgenstern, A.; Bruchertseifer, F.; et al. Combining alpha-Radioimmunotherapy and Adoptive T Cell Therapy to Potentiate Tumor Destruction. PLoS ONE 2015, 10, e0130249. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arndt, C.; Bergmann, R.; Striese, F.; Merkel, K.; Máthé, D.; Loureiro, L.R.; Mitwasi, N.; Kegler, A.; Fasslrinner, F.; González Soto, K.E.; et al. Development and Functional Characterization of a Versatile Radio-/Immunotheranostic Tool for Prostate Cancer Management. Cancers 2022, 14, 1996. https://doi.org/10.3390/cancers14081996
Arndt C, Bergmann R, Striese F, Merkel K, Máthé D, Loureiro LR, Mitwasi N, Kegler A, Fasslrinner F, González Soto KE, et al. Development and Functional Characterization of a Versatile Radio-/Immunotheranostic Tool for Prostate Cancer Management. Cancers. 2022; 14(8):1996. https://doi.org/10.3390/cancers14081996
Chicago/Turabian StyleArndt, Claudia, Ralf Bergmann, Franziska Striese, Keresztély Merkel, Domokos Máthé, Liliana R. Loureiro, Nicola Mitwasi, Alexandra Kegler, Frederick Fasslrinner, Karla Elizabeth González Soto, and et al. 2022. "Development and Functional Characterization of a Versatile Radio-/Immunotheranostic Tool for Prostate Cancer Management" Cancers 14, no. 8: 1996. https://doi.org/10.3390/cancers14081996
APA StyleArndt, C., Bergmann, R., Striese, F., Merkel, K., Máthé, D., Loureiro, L. R., Mitwasi, N., Kegler, A., Fasslrinner, F., González Soto, K. E., Neuber, C., Berndt, N., Kovács, N., Szöllősi, D., Hegedűs, N., Tóth, G., Emmermann, J. -P., Harikumar, K. B., Kovacs, T., ... Feldmann, A. (2022). Development and Functional Characterization of a Versatile Radio-/Immunotheranostic Tool for Prostate Cancer Management. Cancers, 14(8), 1996. https://doi.org/10.3390/cancers14081996