
The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1—A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting

 ,
,  , ,
, ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
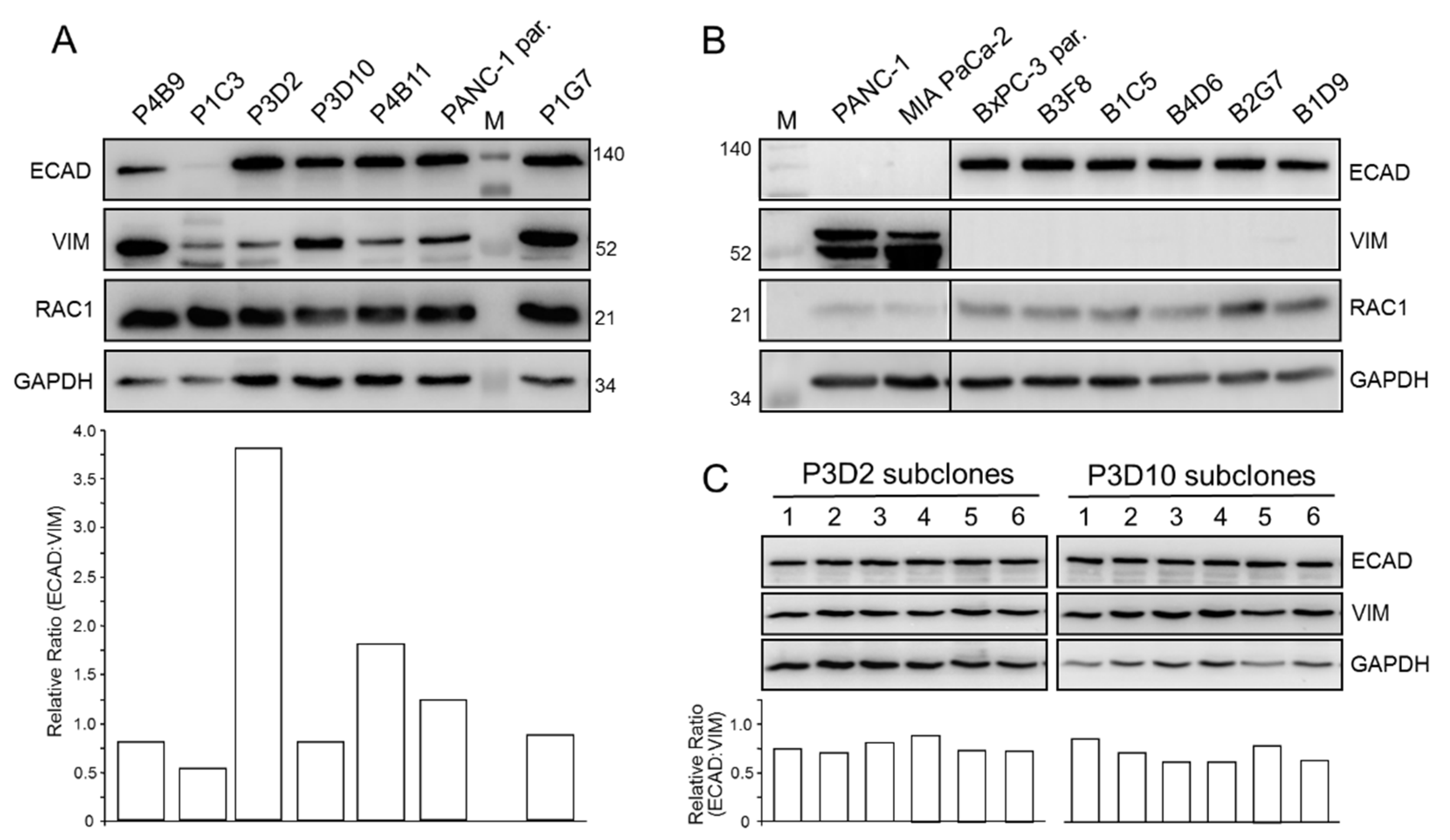
2.1. Single Cell-Derived Clones of PANC-1 Reveal Heterogeneity in Morphology and EMT Marker Expression
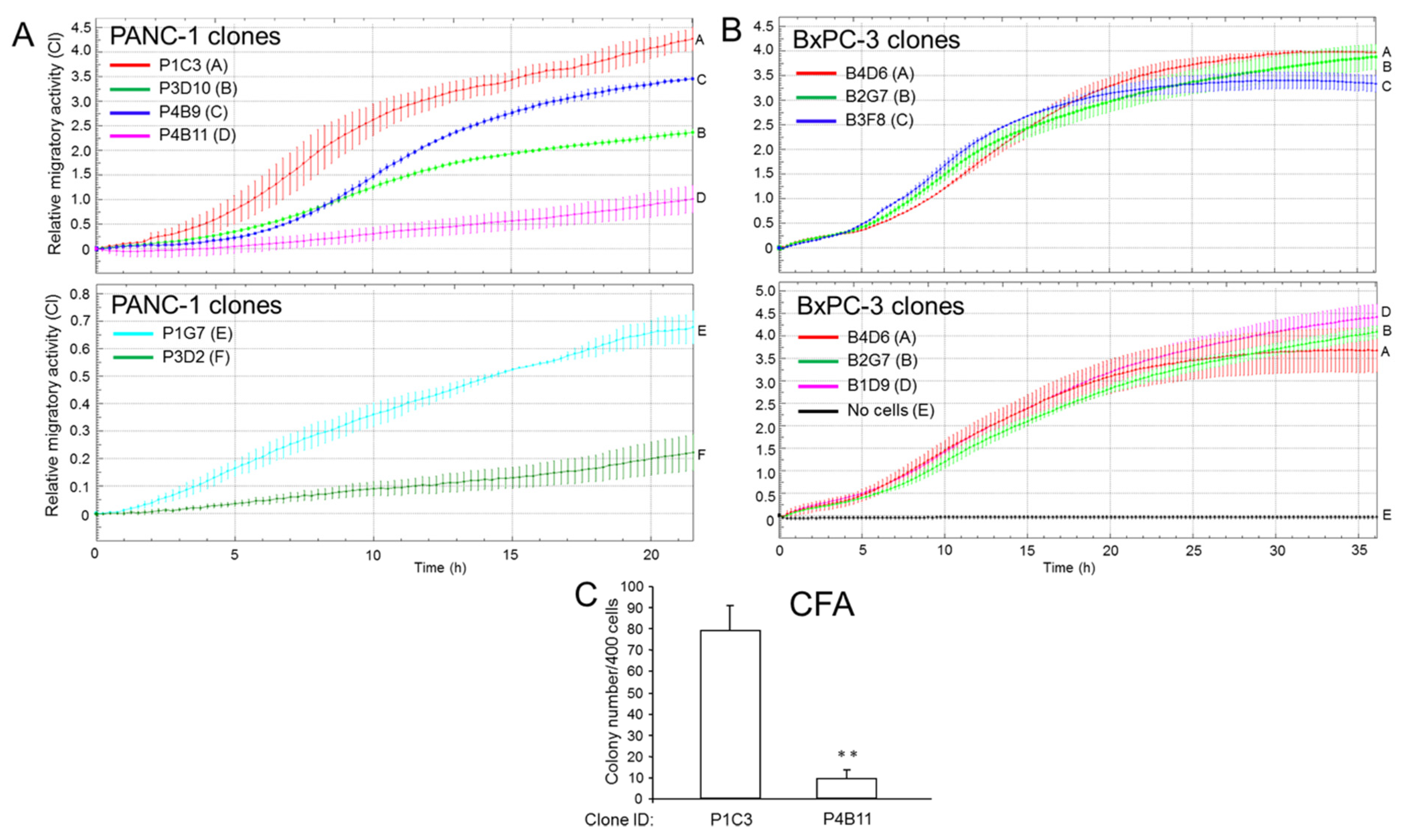
2.2. Single Cell-Derived Clones of PANC-1 Reveal Heterogeneity in Tumorigenic Potential with Respect to Stemness and Migratory Activity
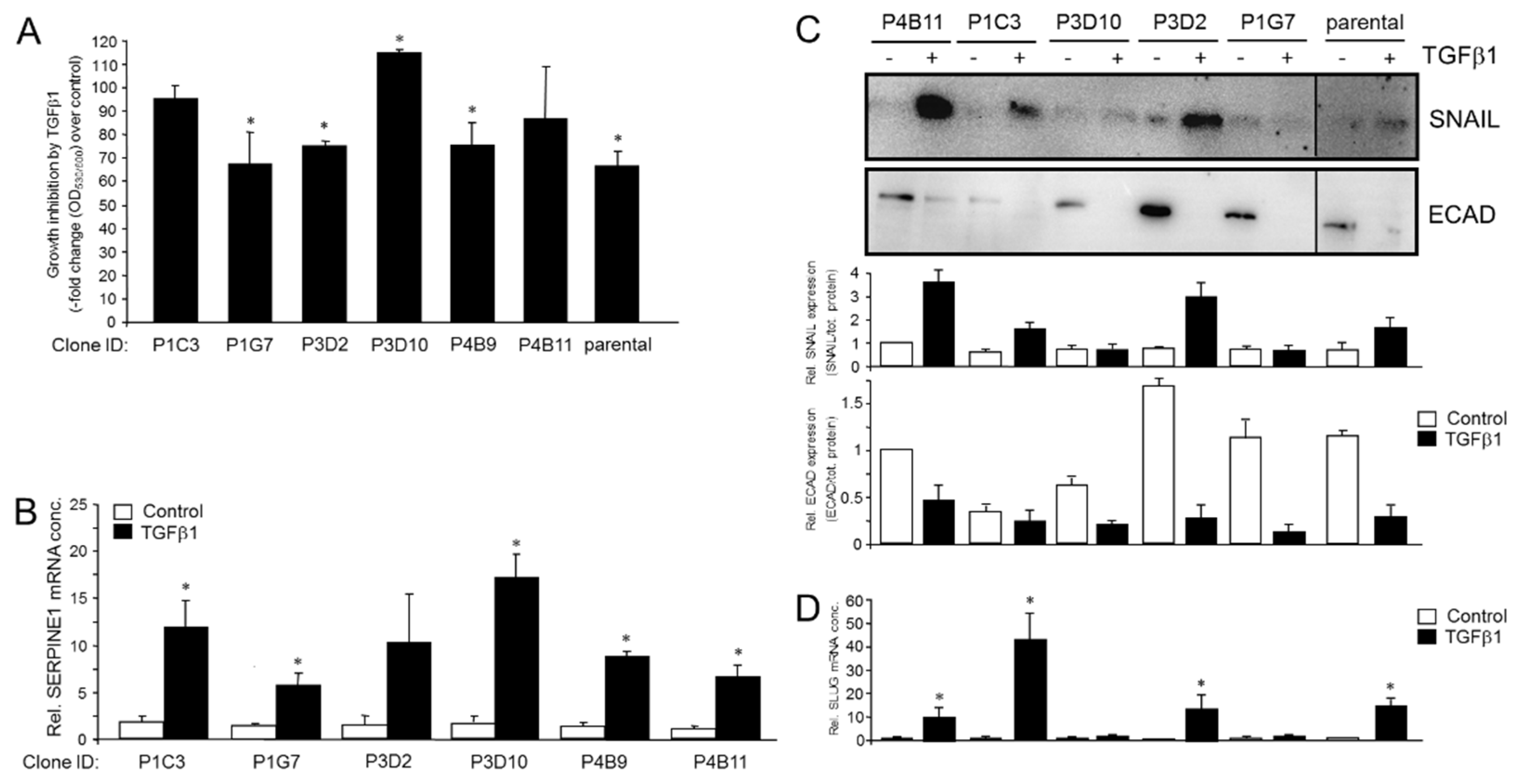
2.3. Single Cell-Derived Clones of PANC-1 Reveal Heterogeneity in TGFβ1-Mediated Regulation of Growth and Individual TGFβ Target Genes
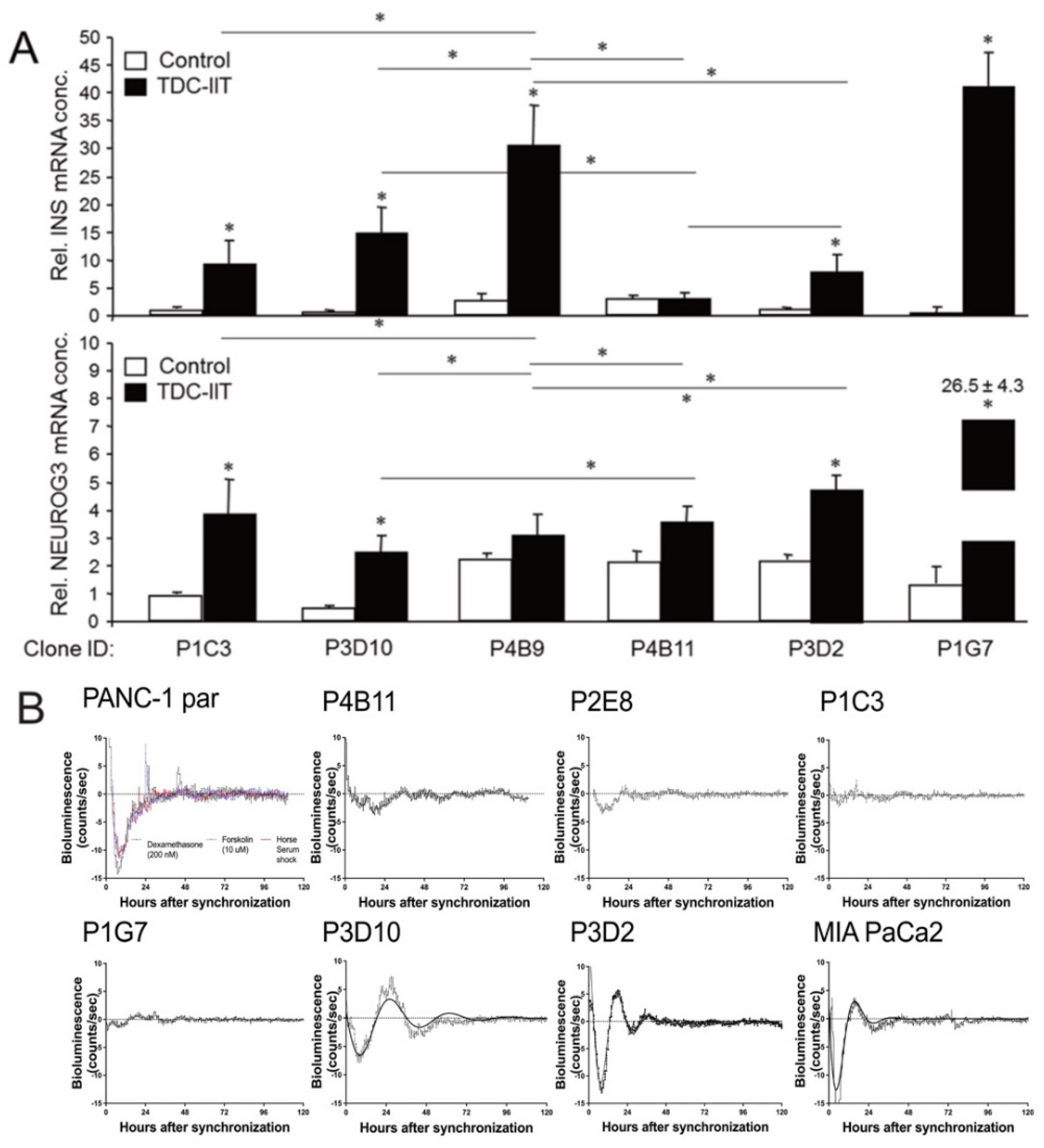
2.4. Single Cell-Derived Clones of PANC-1 Reveal Heterogeneity in Ductal-to-Endocrine Transdifferentiation Potential and Circadian Clock Function
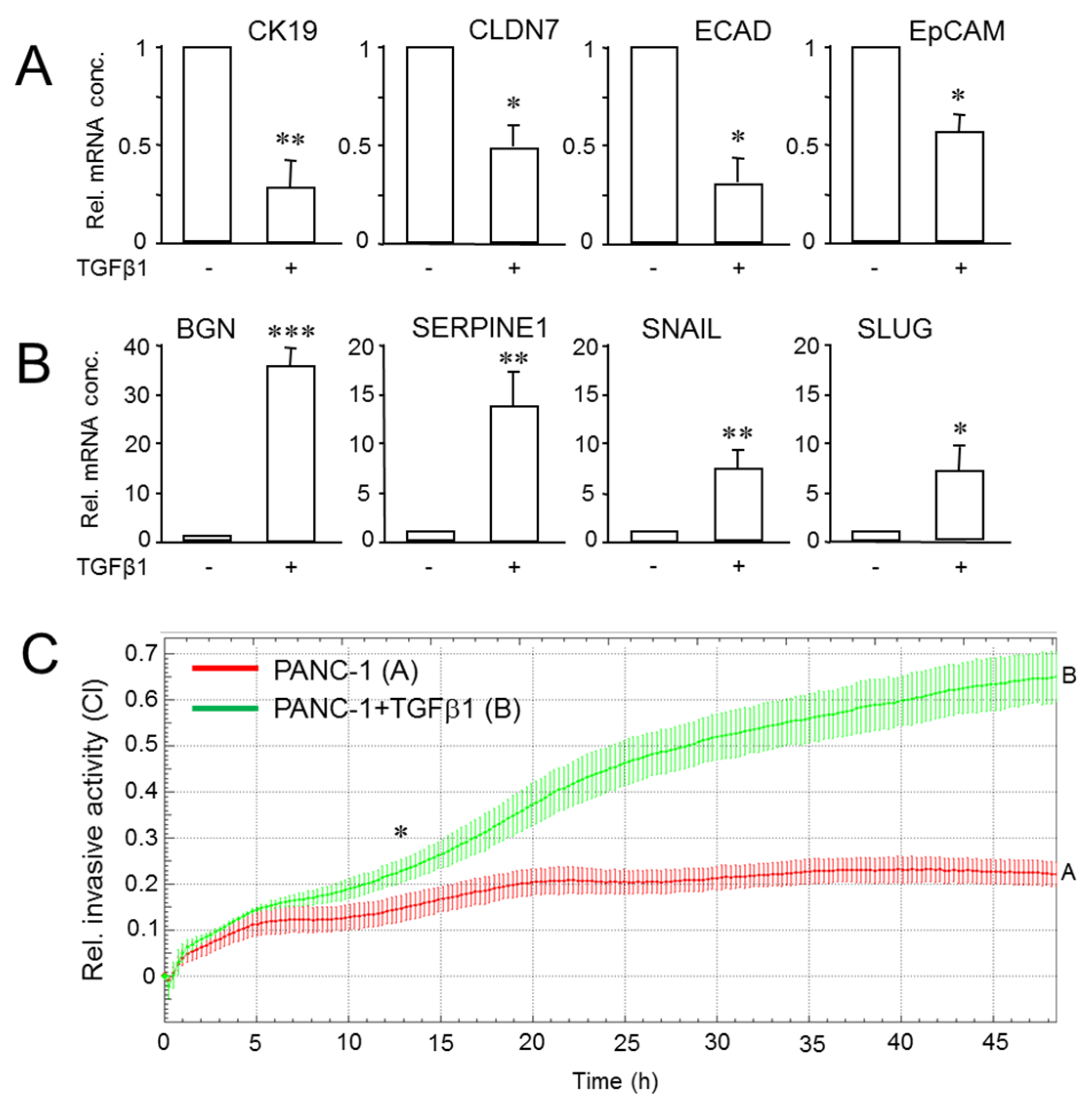
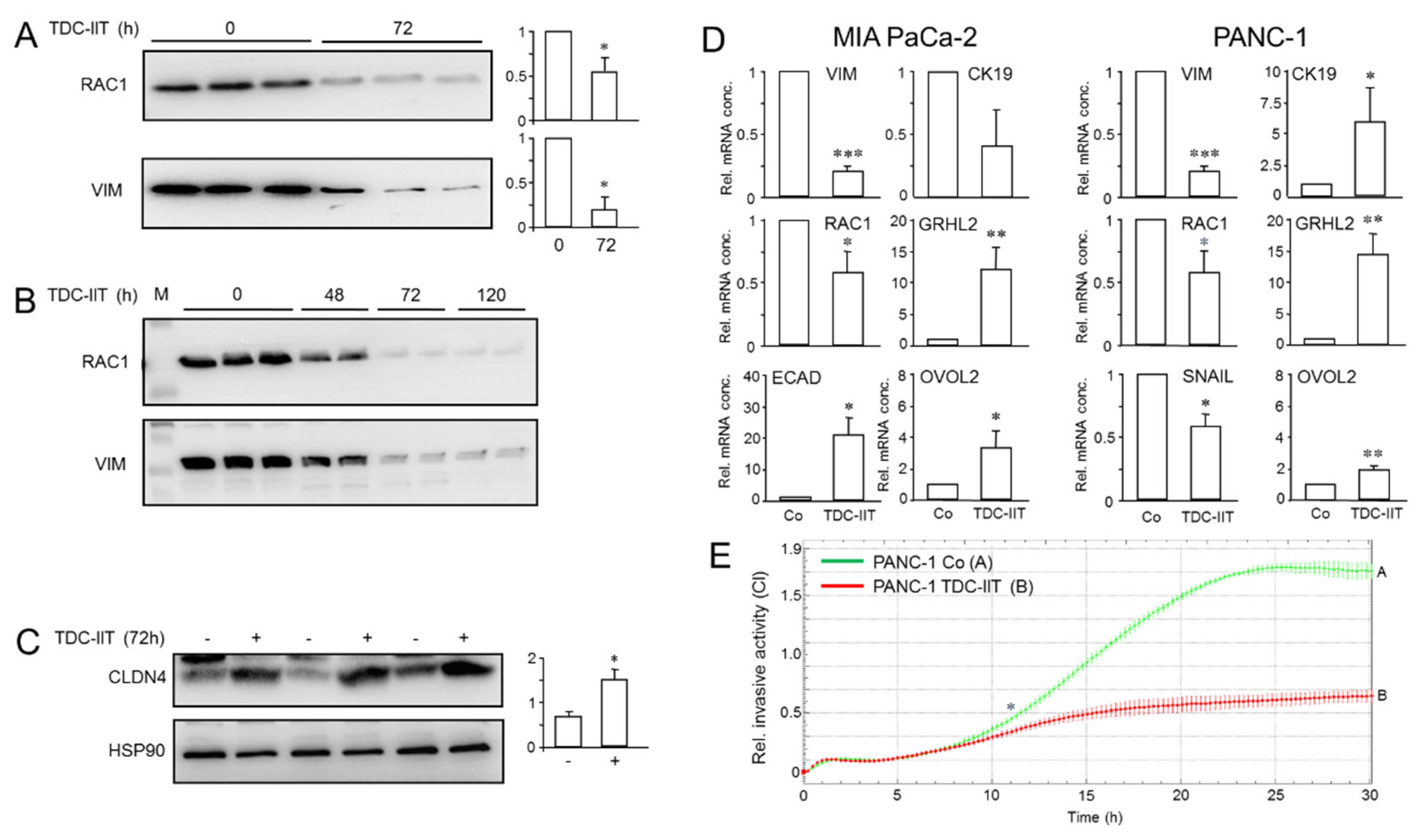
2.5. Exogenous TGFβ1 Induces a More Mesenchymal and Invasive Phenotype in Parental Cultures of PANC-1 Cells
2.6. A Combination of IFNγ, IL1β and TNFα Induces a More Epithelial and Less Invasive Phenotype
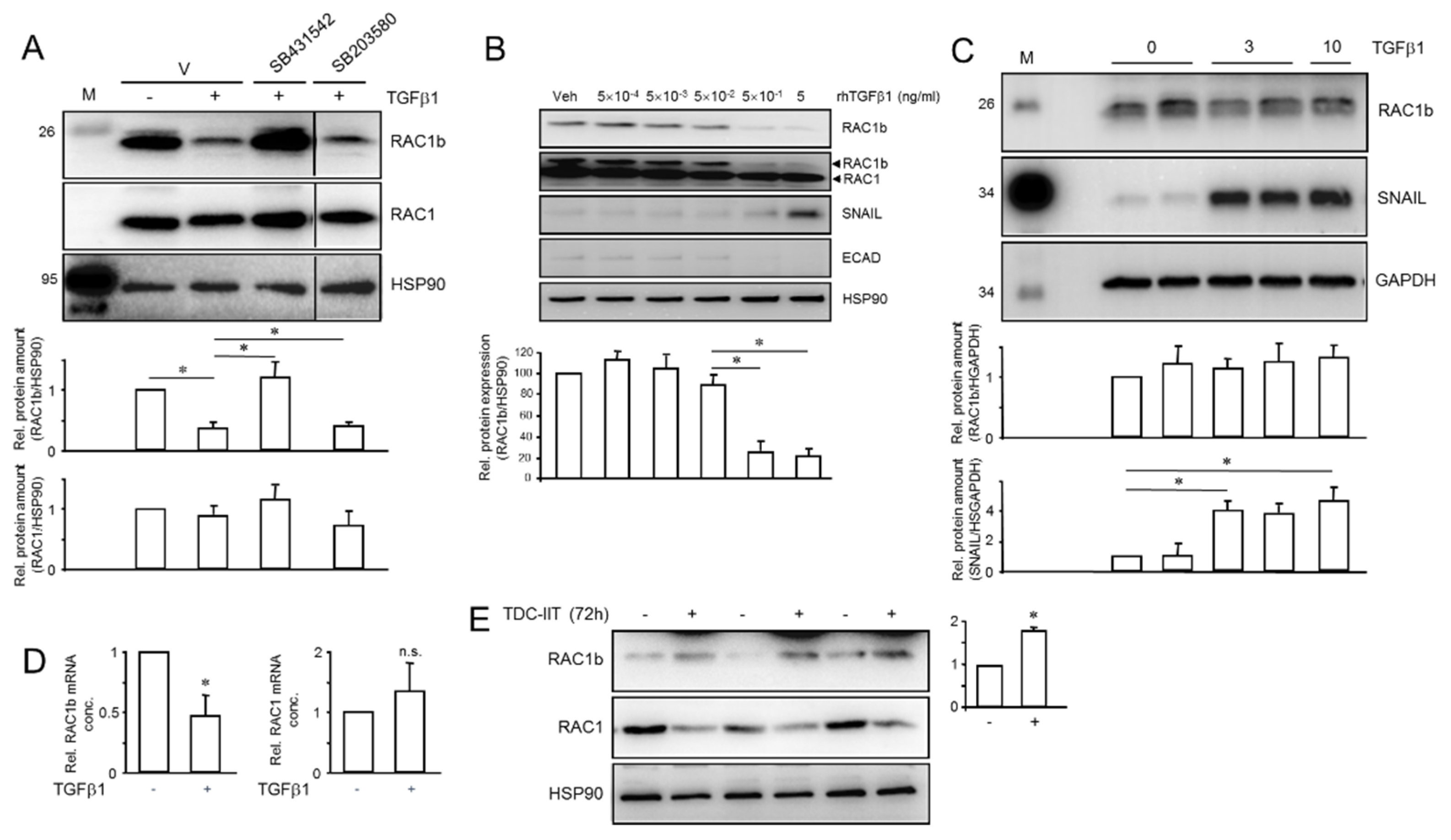
2.7. TGFβ1 and IIT Differentially Regulate RAC1b—TGFβ1 and TII Converge on Shifting the Ratio of RAC1 to RAC1b
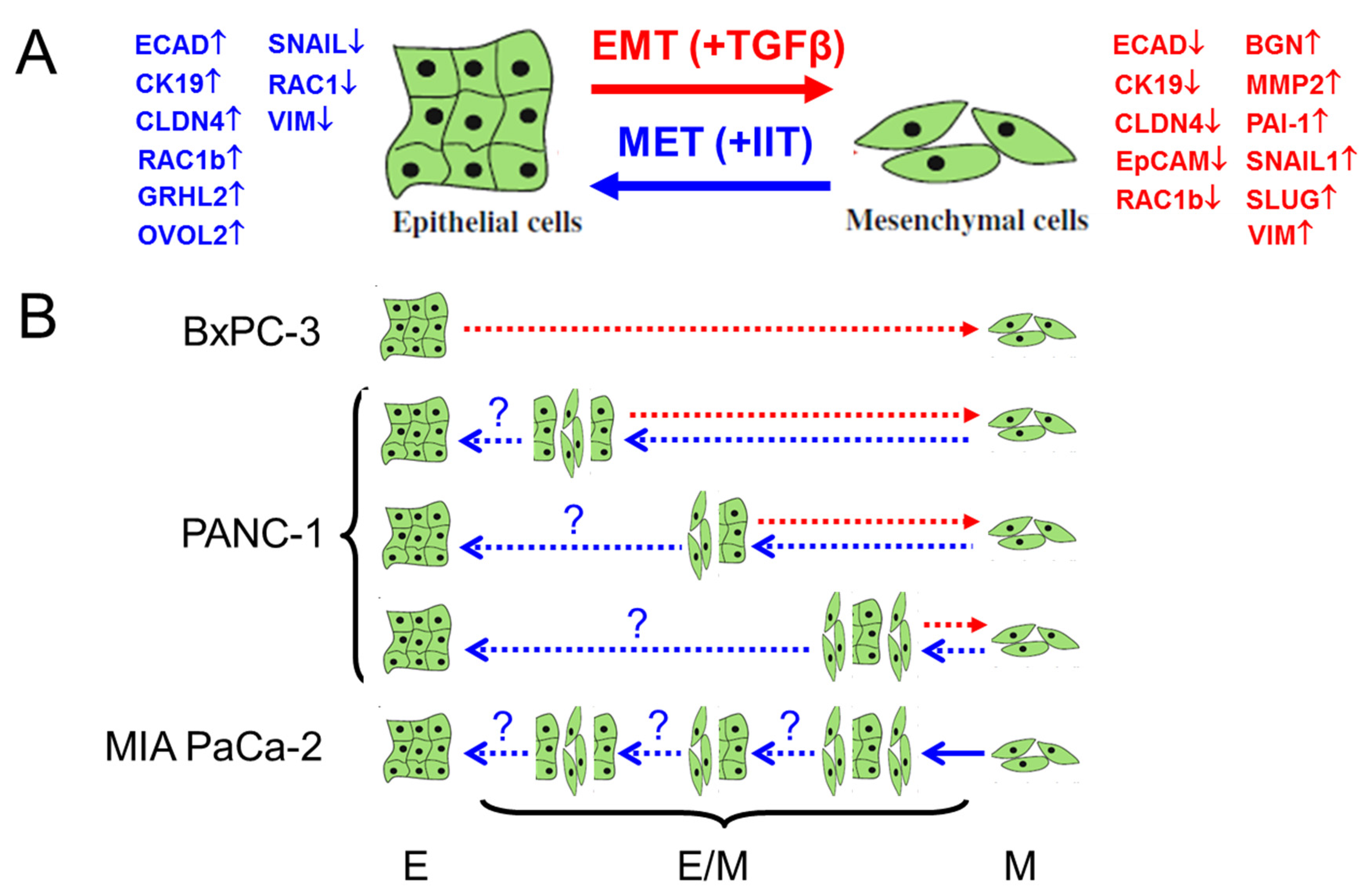
3. Discussion
4. Material and Methods
4.1. Cell Culture and Generation of Single Cell-Derived Clones of PANC-1 and BxPC-3
4.2. Quantitative RT-PCR Analysis
4.3. Immunoblotting
4.4. Cell Proliferation and Viability Assays
4.5. Colony Formation Assay
4.6. Measurement of Circadian Rhythmicity
4.7. In Vitro Transdifferentiation to Endocrine Progenitors and Insulin-Expressing Cells and Induction of MET
4.8. Migration and Invasion Assays
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brabletz, S.; Schuhwerk, H.; Brabletz, T.; Stemmler, M.P. Dynamic EMT: A multi-tool for tumor progression. EMBO J. 2021, 40, e108647. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; von der Ohe, J.; Ungefroren, H. The Intimate Relationship Among EMT, MET and TME: A T(ransdifferentiation) E(nhancing) M(ix) to Be Exploited for Therapeutic Purposes. Cancers 2020, 12, 3674. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Dernyck, R. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Monkman, J.; Toh, A.K.L.; Nagaraj, S.H.; Thompson, E.W. Targeting epithelial-mesenchymal plasticity in cancer: Clinical and preclinical advances in therapy and monitoring. Biochem. J. 2017, 474, 3269–3306. [Google Scholar] [CrossRef]
- Ye, X.; Weinberg, R.A. Epithelial-Mesenchymal Plasticity: A Central Regulator of Cancer Progression. Trends Cell Biol. 2015, 25, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, R.S.; Dumont, N.; Ritter, C.A.; Dugger, T.C.; Brantley, D.M.; Chen, J.; Easterly, E.; Roebuck, L.R.; Ryan, S.; Gotwals, P.J.; et al. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J. Clin. Investig. 2002, 109, 1551–1559. [Google Scholar] [CrossRef]
- Garg, M. Epithelial plasticity and cancer stem cells: Major mechanisms of cancer pathogenesis and therapy resistance. World J. Stem Cells 2017, 9, 118–126. [Google Scholar] [CrossRef]
- Brabletz, T. EMT and MET in metastasis: Where are the cancer stem cells? Cancer Cell 2012, 22, 699–701. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aiello, N.M.; Kang, Y. Context-dependent EMT programs in cancer metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Monkman, J.; Blick, T.; Pinto, C.; Waltham, M.; Nagaraj, S.H.; Thompson, E.W. Interrogation of Phenotypic Plasticity between Epithelial and Mesenchymal States in Breast Cancer. J. Clin. Med. 2019, 8, 893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi-Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial-mesenchymal plasticity and suppresses metastatic, castration-resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Weinberg, R.A. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat. Rev. Cancer 2009, 9, 265–273. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [Green Version]
- Schmidtlein, P.M.; Volz, C.; Braun, R.; Thürling, I.; Lapshyna, O.; Wellner, U.F.; Konukiewitz, B.; Lehnert, H.; Marquardt, J.U.; Ungefroren, H. A Comparative Endocrine Trans-Differentiation Approach to Pancreatic Ductal Adenocarcinoma Cells with Different EMT Phenotypes Identifies Quasi-Mesenchymal Tumor Cells as Those with Highest Plasticity. Cancers 2021, 13, 4663. [Google Scholar] [CrossRef]
- Schmidtlein, P.M.; Volz, C.; Hackel, A.; Thürling, I.; Castven, D.; Braun, R.; Wellner, U.F.; Konukiewitz, B.; Riemekasten, G.; Lehnert, H.; et al. Activation of a Ductal-to-Endocrine Transdifferentiation Transcriptional Program in the Pancreatic Cancer Cell Line PANC-1 Is Controlled by RAC1 and RAC1b through Antagonistic Regulation of Stemness Factors. Cancers 2021, 13, 5541. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell. 2018, 45, 681–695.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A guardian of the epithelial phenotype and protector against epithelial-mesenchymal transition. Cells 2019, 8, 1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1—Pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6, 21648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frescas, D.; Roux, C.M.; Aygun-Sunar, S.; Gleiberman, A.S.; Krasnov, P.; Kurnasov, O.V.; Strom, E.; Virtuoso, L.P.; Wrobel, M.; Osterman, A.L.; et al. Senescent cells expose and secrete an oxidized form of membrane-bound vimentin as revealed by a natural polyreactive antibody. Proc. Natl. Acad. Sci. USA 2017, 114, E1668–E1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Wang, J.; Dapeng, C.; Wu, D.; Cai, G.; Chen, X. Bioinformatics analysis of proteomics profiles in senescent human primary proximal tubule epithelial cells. BMC Nephrol. 2016, 17, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiden, C.; Ungefroren, H. The Ratio of RAC1B to RAC1 Expression in Breast Cancer Cell Lines as a Determinant of Epithelial/Mesenchymal Differentiation and Migratory Potential. Cells 2021, 10, 351. [Google Scholar] [CrossRef]
- Soundararajan, R.; Paranjape, A.N.; Maity, S.; Aparicio, A.; Mani, S.A. EMT, stemness and tumor plasticity in aggressive variant neuroendocrine prostate cancers. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 229–238. [Google Scholar] [CrossRef]
- Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers 2019, 11, 691. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Groth, S.; Sebens, S.; Lehnert, H.; Gieseler, F.; Fändrich, F. Differential roles of Smad2 and Smad3 in the regulation of TGF-β1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: Control by Rac1. Mol. Cancer 2011, 10, 67. [Google Scholar] [CrossRef] [Green Version]
- Valdez, I.A.; Dirice, E.; Gupta, M.K.; Shirakawa, J.; Teo, A.K.K.; Kulkarni, R.N. Proinflammatory Cytokines Induce Endocrine Differentiation in Pancreatic Ductal Cells via STAT3-Dependent NGN3 Activation. Cell Rep. 2016, 15, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Basti, A.; Yalçin, M.; Relógio, A. Circadian Dysregulation of the TGFbeta/SMAD4 Pathway Modulates Metastatic Properties and Cell Fate Decisions in Pancreatic Cancer Cells. iScience 2020, 23, 101551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Devocelle, A.; Desterke, C.; de Souza, L.E.B.; Hadadi, É.; Acloque, H.; Foudi, A.; Xiang, Y.; Ballesta, A.; Chang, Y.; et al. BMAL1 Knockdown Leans Epithelial-Mesenchymal Balance toward Epithelial Properties and Decreases the Chemoresistance of Colon Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 5247. [Google Scholar] [CrossRef] [PubMed]
- de Jesus, D.S.; Mak, T.C.S.; Wang, Y.F.; von Ohlen, Y.; Bai, Y.; Kane, E.; Chabosseau, P.; Chahrour, C.M.; Distaso, W.; Salem, V.; et al. Dysregulation of the Pdx1/Ovol2/Zeb2 axis in dedifferentiated β-cells triggers the induction of genes associated with epithelial-mesenchymal transition in diabetes. Mol. Metab. 2021, 53, 101248. [Google Scholar] [CrossRef] [PubMed]
- Michl, P.; Barth, C.; Buchholz, M.; Lerch, M.M.; Rolke, M.; Holzmann, K.H.; Menke, A.; Fensterer, H.; Giehl, K.; Löhr, M.; et al. Claudin-4 expression decreases invasiveness and metastatic potential of pancreatic cancer. Cancer Res. 2003, 63, 6265–6271. [Google Scholar]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [Green Version]
- Roy Burman, D.; Das, S.; Das, C.; Bhattacharya, R. Alternative splicing modulates cancer aggressiveness: Role in EMT/metastasis and chemoresistance. Mol. Biol. Rep. 2021, 48, 897–914. [Google Scholar] [CrossRef]
- Ishii, H.; Saitoh, M.; Sakamoto, K.; Kondo, T.; Katoh, R.; Tanaka, S.; Motizuki, M.; Masuyama, K.; Miyazawa, K. Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J. Biol. Chem. 2014, 289, 27386–27399. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Witte, D.; Otterbein, H.; Förster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fandrich, F.; Rocken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.W.; Nagaraj, S.H. Transition states that allow cancer to spread. Nature 2018, 556, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jolly, M.K.; Celià-Terrassa, T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J. Clin. Med. 2019, 8, 1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinouchi, K.; Sassone-Corsi, P. Metabolic rivalry: Circadian homeostasis and tumorigenesis. Nat. Rev. Cancer 2020, 20, 645–661. [Google Scholar] [CrossRef]
- Hadadi, E.; Acloque, H. Role of circadian rhythm disorders on EMT and tumour-immune interactions in endocrine-related cancers. Endocr. Relat. Cancer 2021, 28, R67–R80. [Google Scholar] [CrossRef]
- Kuo, T.T.; Ladurner, A.G. Exploiting the Circadian Clock for Improved Cancer Therapy: Perspective from a Cell Biologist. Front. Genet. 2019, 10, 1210. [Google Scholar] [CrossRef]
- Masri, S.; Sassone-Corsi, P. The emerging link between cancer, metabolism, and circadian rhythms. Nat. Med. 2018, 24, 1795–1803. [Google Scholar] [CrossRef]
- Relógio, A.; Thomas, P.; Medina-Pérez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schäfer, R.; et al. Ras-mediated deregulation of the circadian clock in cancer. PLoS Genet. 2014, 10, e1004338. [Google Scholar] [CrossRef] [PubMed]
- Mathis, R.A.; Sokol, E.S.; Gupta, P.B. Cancer cells exhibit clonal diversity in phenotypic plasticity. Open Biol. 2017, 7, 160283. [Google Scholar] [CrossRef] [PubMed]
- Rios, A.C.; Capaldo, B.D.; Vaillant, F.; Pal, B.; van Ineveld, R.; Dawson, C.A.; Chen, Y.; Nolan, E.; Fu, N.Y.; 3DTCLSM Group; et al. Intraclonal Plasticity in Mammary Tumors Revealed through Large-Scale Single-Cell Resolution 3D Imaging. Cancer Cell 2019, 35, 618–632.e6. [Google Scholar] [CrossRef] [Green Version]
- Ischenko, I.; Liu, J.; Petrenko, O.; Hayman, M.J. Transforming growth factor-beta signaling network regulates plasticity and lineage commitment of lung cancer cells. Cell Death Differ. 2014, 21, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tian, X.-J.; Zhang, H.; Teng, Y.; Li, R.; Bai, F.; Elankumaran, S.; Xing, J. TGF-β–induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal. 2014, 7, ra91. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bracken, C.P.; Smith, E.; Bert, A.G.; Wright, J.A.; Roslan, S.; Morris, M.; Wyatt, L.; Farshid, G.; Lim, Y.Y.; et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell. 2011, 22, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.D.; Parvani, J.G.; Schiemann, W.P. The relevance of the TGF-β Paradox to EMT-MET programs. Cancer Lett. 2013, 341, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef] [Green Version]
- Bonyadi Rad, E.; Hammerlindl, H.; Wels, C.; Popper, U.; Ravindran Menon, D.; Breiteneder, H.; Kitzwoegerer, M.; Hafner, C.; Herlyn, M.; Bergler, H.; et al. Notch4 Signaling Induces a Mesenchymal-Epithelial-like Transition in Melanoma Cells to Suppress Malignant Behaviors. Cancer Res. 2016, 76, 1690–1697. [Google Scholar] [CrossRef] [Green Version]
- Nishino, H.; Takano, S.; Yoshitomi, H.; Suzuki, K.; Kagawa, S.; Shimazaki, R.; Shimizu, H.; Furukawa, K.; Miyazaki, M.; Ohtsuka, M. Grainyhead-like 2 (GRHL2) regulates epithelial plasticity in pancreatic cancer progression. Cancer Med. 2017, 6, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Somarelli, J.A.; Shetler, S.; Jolly, M.K.; Wang, X.; Dewitt, S.B.; Hish, A.J.; Gilja, S.; Eward, W.C.; Ware, K.E.; Levine, H.; et al. Mesenchymal-Epithelial Transition in Sarcomas Is Controlled by the Combinatorial Expression of MicroRNA 200s and GRHL2. Mol. Cell. Biol. 2016, 36, 2503–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieply, B.; Riley, P.; Pifer, P.M.; Widmeyer, J.; Addison, J.B.; Ivanov, A.V.; Denvir, J.; Frisch, S.M. Suppression of the epithelial-mesenchymal transition by grainyhead-like-2. Cancer Res. 2012, 72, 2440–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.H.; Chang, Y.W.; Hong, M.X.; Hsu, T.C.; Lee, K.C.; Lin, C.; Lee, J.L. STAT3 phosphorylation at Ser727 and Tyr705 differentially regulates the EMT-MET switch and cancer metastasis. Oncogene 2021, 40, 791–805. [Google Scholar] [CrossRef]
- Kamohara, H.; Ogawa, M.; Ishiko, T.; Sakamoto, K.; Baba, H. Leukemia inhibitory factor functions as a growth factor in pancreas carcinoma cells: Involvement of regulation of LIF and its receptor expression. Int. J. Oncol. 2007, 30, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Kamohara, H.; Sakamoto, K.; Ishiko, T.; Masuda, Y.; Abe, T.; Ogawa, M. Leukemia inhibitory factor induces apoptosis and proliferation of human carcinoma cells through different oncogene pathways. Int. J. Cancer 1997, 72, 687–695. [Google Scholar] [CrossRef]
- Takano, S.; Reichert, M.; Bakir, B.; Das, K.K.; Nishida, T.; Miyazaki, M.; Heeg, S.; Collins, M.A.; Marchand, B.; Hicks, P.D.; et al. Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes Dev. 2016, 30, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Ishay-Ronen, D.; Diepenbruck, M.; Kalathur, R.K.R.; Sugiyama, N.; Tiede, S.; Ivanek, R.; Bantug, G.; Morini, M.F.; Wang, J.; Hess, C.; et al. Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell 2019, 35, 17–32.e6. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Esquer, H.; Zhou, Q.; Nemkov, T.; Abraham, A.D.; Rinaldetti, S.; Chen, Y.C.; Zhang, X.; Orman, M.V.; D’Alessandro, A.; Ferrer, M.; et al. Isolating and targeting the real-time plasticity and malignant properties of epithelial-mesenchymal transition in cancer. Oncogene 2021, 40, 2884–2897. [Google Scholar] [CrossRef]
- Parsons, R.; Parsons, R.; Garner, N.; Oster, H.; Rawashdeh, O. CircaCompare: A method to estimate and statistically support differences in mesor, amplitude and phase, between circadian rhythms. Bioinformatics 2020, 36, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Mandel, K.; Seidl, D.; Rades, D.; Lehnert, H.; Gieseler, F.; Hass, R.; Ungefroren, H. Characterization of spontaneous and TGF-β-induced cell motility of primary human normal and neoplastic mammary cells in vitro using novel real-time technology. PLoS ONE 2013, 8, e56591. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungefroren, H.; Thürling, I.; Färber, B.; Kowalke, T.; Fischer, T.; De Assis, L.V.M.; Braun, R.; Castven, D.; Oster, H.; Konukiewitz, B.; et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1—A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers 2022, 14, 2057. https://doi.org/10.3390/cancers14092057
Ungefroren H, Thürling I, Färber B, Kowalke T, Fischer T, De Assis LVM, Braun R, Castven D, Oster H, Konukiewitz B, et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1—A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers. 2022; 14(9):2057. https://doi.org/10.3390/cancers14092057
Chicago/Turabian StyleUngefroren, Hendrik, Isabel Thürling, Benedikt Färber, Tanja Kowalke, Tanja Fischer, Leonardo Vinícius Monteiro De Assis, Rüdiger Braun, Darko Castven, Henrik Oster, Björn Konukiewitz, and et al. 2022. "The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1—A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting" Cancers 14, no. 9: 2057. https://doi.org/10.3390/cancers14092057
APA StyleUngefroren, H., Thürling, I., Färber, B., Kowalke, T., Fischer, T., De Assis, L. V. M., Braun, R., Castven, D., Oster, H., Konukiewitz, B., Wellner, U. F., Lehnert, H., & Marquardt, J. -U. (2022). The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1—A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers, 14(9), 2057. https://doi.org/10.3390/cancers14092057