
Multivariant Transcriptome Analysis Identifies Modules and Hub Genes Associated with Poor Outcomes in Newly Diagnosed Multiple Myeloma Patients

 , , , ,
, , , ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
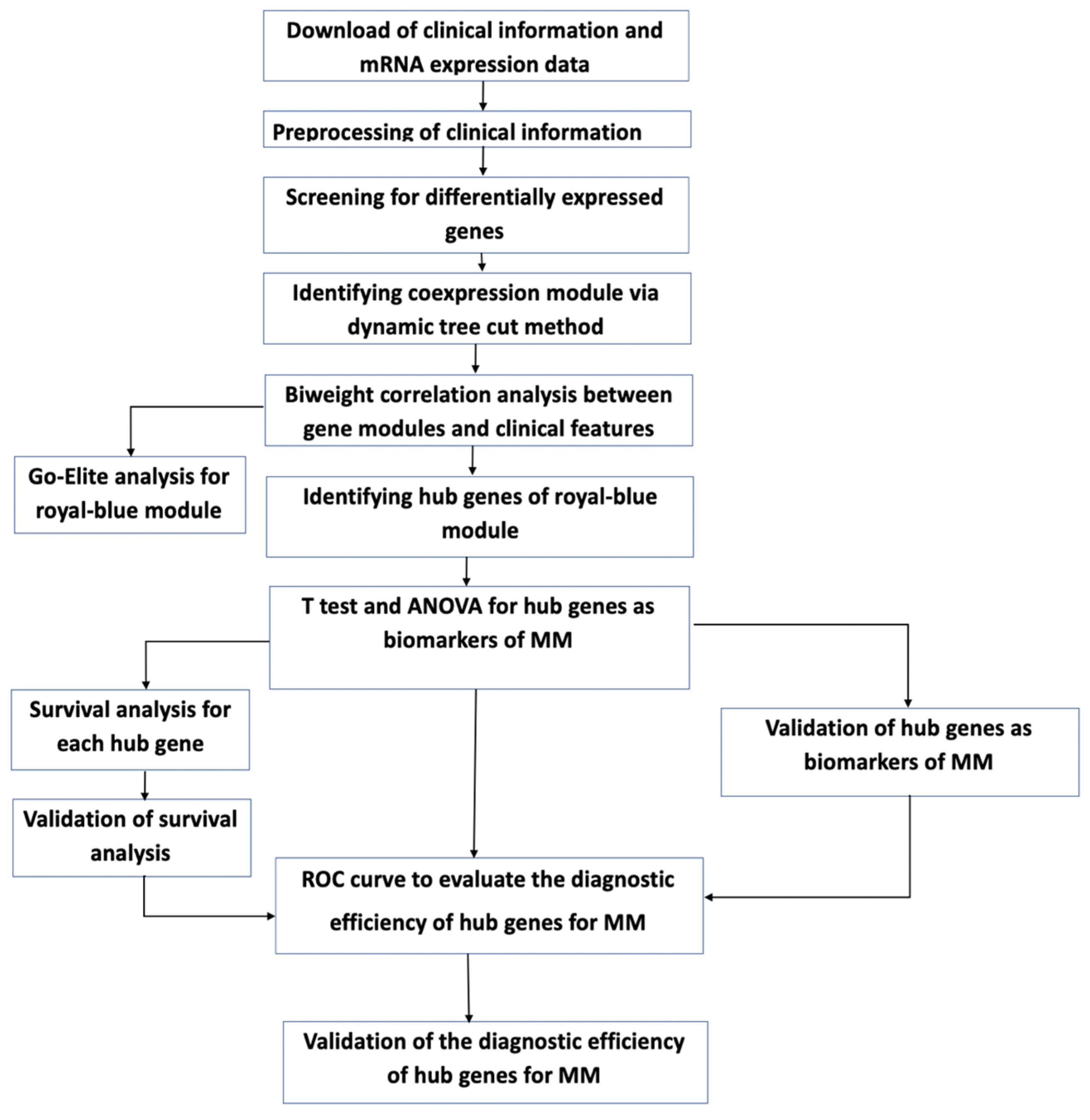
2. Materials and Methods
2.1. Data Curation and Normalization
2.2. Detecting Low Counts, Batch Effect Correction, and Removal of Outliers
2.3. Coding Clinical Metadata for Biological Network Analysis
2.4. Gene Clustering and Network Analysis
2.5. Gene Ontology (GO) Enrichment and Upstream Regulator Analysis
2.6. Differential Gene Expression
2.7. Protein-Protein Interaction within Modules of Interest
2.8. Geneset Enrichment Analysis
2.9. ROC Analyses
2.10. Survival Analyses
3. Results
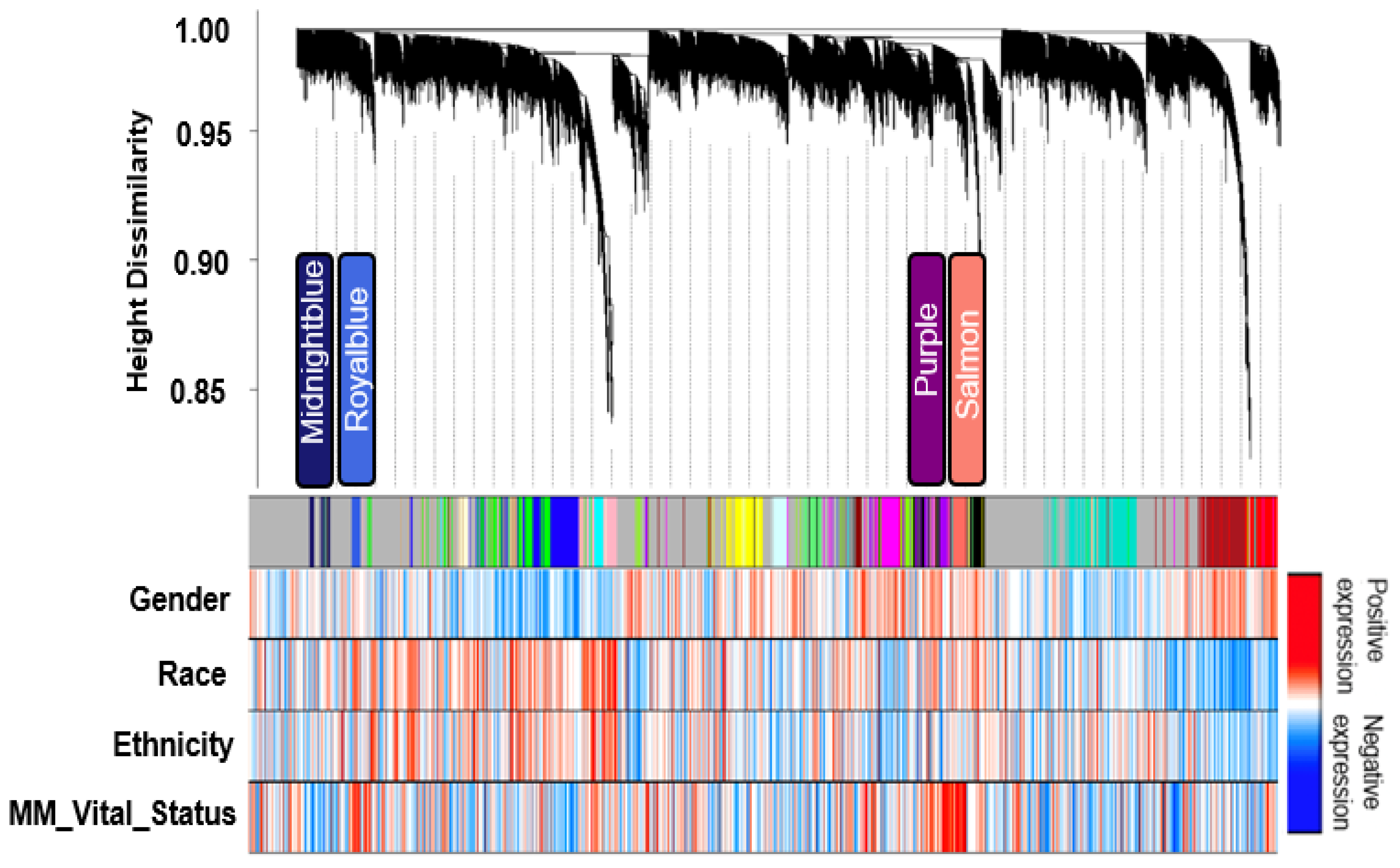
3.1. Transcriptomic Analysis Defines a Network of MM Co-Expression Modules
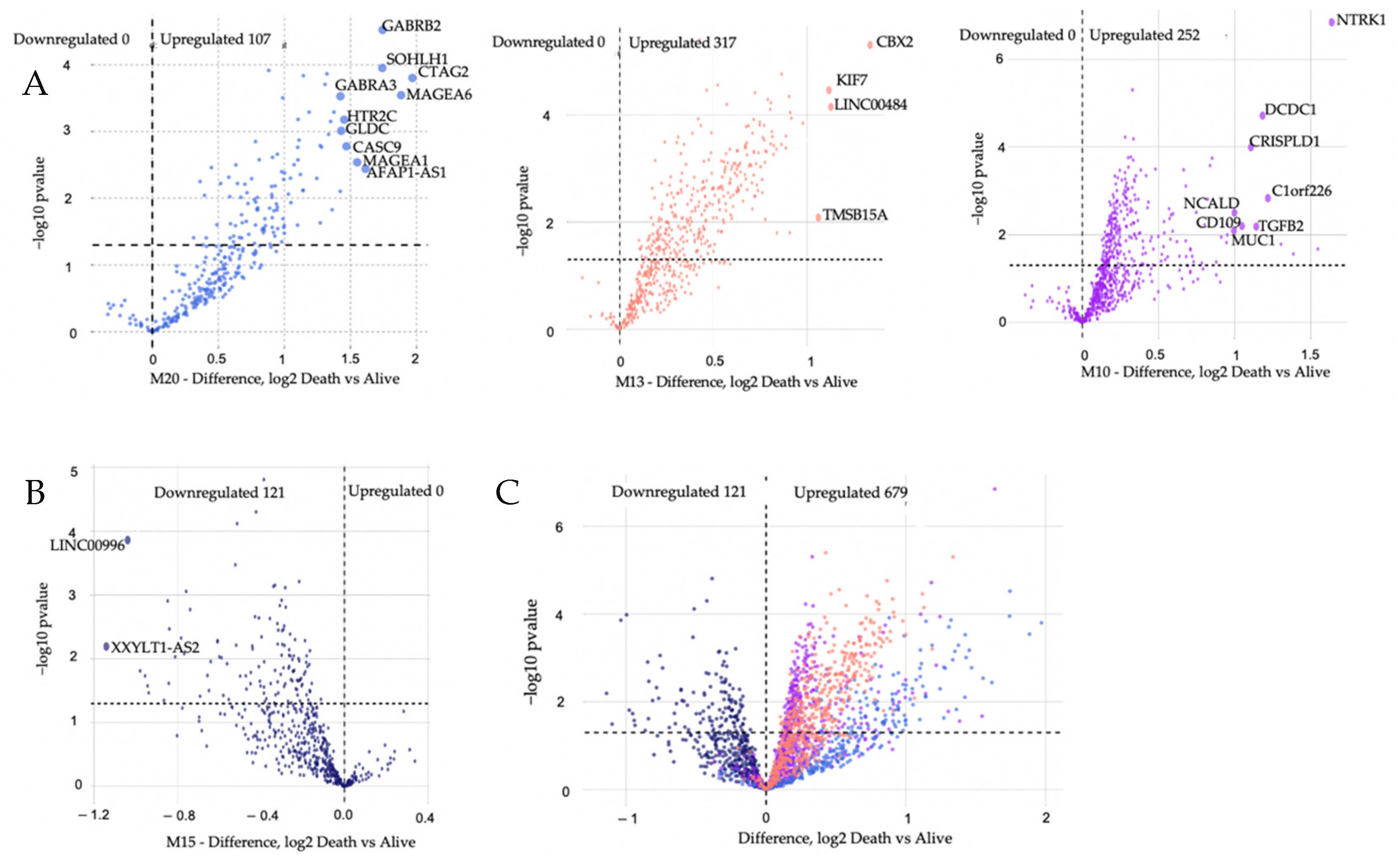
3.2. Identification of Transcript Significant Modules Associated with Mortality and Functional Annotation
3.3. Modules Differentiating between Alive and Dead Patients Represent Known Biological Groundworks of MM
3.4. Gene Set Enrichment, ROC Curve, and Kaplan–Meier Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cowan, A.J.; Allen, C.; Barac, A.; Basaleem, H.; Bensenor, I.; Curado, M.P.; Foreman, K.; Gupta, R.; Harvey, J.; Hosgood, H.D.; et al. Global Burden of multiple myeloma: A systematic analysis for the Global Burden of Disease study 2016. JAMA Oncol. 2018, 4, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.J.; Mink, P.J.; Devesa, S.S.; Anderson, W.F.; Weiss, B.M.; Kristinsson, S.Y.; McGlynn, K.A.; Landgren, O. Racial disparities in incidence and outcome in multiple myeloma: A population-based study. Blood 2010, 116, 5501–5506. [Google Scholar] [CrossRef] [Green Version]
- Lewis, T.; Corcoran, D.B.; Thurston, D.E.; Giles, P.J.; Ashelford, K.; Walsby, E.J.; Fegan, C.D.; Pepper, A.G.S.; Rahman, K.M. Novel pyrrolobenzodiazepine benzofused hybrid molecules inhibit NF-κB activity and synergise with bortezomib and ibrutinib in hematological cancers. Haematologica 2021, 106, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N.; Fend, F. A review on tumor heterogeneity and evolution in multiple myeloma: Pathological, radiological, molecular genetics, and clinical integration. Virchows Arch. 2020, 476, 337–351. [Google Scholar] [CrossRef]
- Pabst, C.; Zustin, J.; Jacobsen, F.; Luetkens, T.; Kröger, N.; Schilling, G.; Bokemeyer, C.; Sauter, G.; Atanackovic, D.; Marx, A. Expression and prognostic relevance of MAGE-C1/CT7 and MAGE-C2/CT10 in osteolytic lesions of patients with multiple myeloma. Exp. Mol. Pathol. 2010, 89, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Attal, M.; Harousseau, J.-L.; Stoppa, A.-M.; Sotto, J.-J.; Fuzibet, J.-G.; Rossi, J.-F.; Casassus, P.; Maisonneuve, H.; Facon, T.; Ifrah, N.; et al. A Prospective, Randomized Trial of Autologous Bone Marrow Transplantation and Chemotherapy in Multiple Myeloma. N. Engl. J. Med. 1996, 335, 91–97. [Google Scholar] [CrossRef]
- Child, J.A.; Morgan, G.J.; Davies, F.E.; Owen, R.G.; Bell, S.E.; Hawkins, K.; Brown, J.; Drayson, M.T.; Selby, P.J. High-dose chemotherapy with hematopoietic stem-cell rescue for multiple myeloma. N. Engl. J. Med. 2003, 348, 1875–1883. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.K.; Barlogie, B.; Van Rhee, F.; Zangari, M.; Walker, B.A.; Rosenthal, A.; Schinke, C.; Thanendrarajan, S.; Davies, F.E.; Hoering, A.; et al. Long-term outcomes after autologous stem cell transplantation for multiple myeloma. Blood Adv. 2020, 4, 422–431. [Google Scholar] [CrossRef]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Boise, L.H.; Kaufman, J.L.; Bahlis, N.J.; Lonial, S.; Lee, K.P. The Tao of myeloma. Blood 2014, 124, 1873–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringhen, S.; D’Agostino, M.; Paris, L.; Ballanti, S.; Pescosta, N.; Spada, S.; Pezzatti, S.; Grasso, M.; Rota-Scalabrini, D.; De Rosa, L.; et al. Lenalidomide-based induction and maintenance in elderly newly diagnosed multiple myeloma patients: Updated results of the EMN01 randomized trial. Haematologica 2020, 105, 1937–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonello, F.; Pulini, S.; Ballanti, S.; Gentile, M.; Spada, S.; Annibali, O.; Omedé, P.; Ronconi, S.; Cangialosi, C.; Podda, L.; et al. Lenalidomide Maintenance with or without Prednisone in Newly Diagnosed Myeloma Patients: A Pooled Analysis. Cancers 2019, 11, 1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barwick, B.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar] [CrossRef] [Green Version]
- Griffen, T.L.; Dammer, E.B.; Dill, C.D.; Carey, K.M.; Young, C.D.; Nunez, S.K.; Ohandjo, A.Q.; Kornblau, S.M.; Lillard, J.W. Multivariate transcriptome analysis identifies networks and key drivers of chronic lymphocytic leukemia relapse risk and patient survival. BMC Med. Genom. 2021, 14, 171. [Google Scholar] [CrossRef]
- Ohandjo, A.Q.; Liu, Z.; Dammer, E.B.; Dill, C.D.; Griffen, T.; Carey, K.M.; Hinton, D.E.; Meller, R.; Lillard, J.W. Transcriptome Network Analysis Identifies CXCL13-CXCR5 Signaling Modules in the Prostate Tumor Immune Microenvironment. Sci. Rep. 2019, 9, 14963. [Google Scholar] [CrossRef]
- Dill, C.D.; Dammer, E.B.; Griffen, T.L.; Seyfried, N.T.; Lillard, J.W. A network approach reveals driver genes associated with survival of patients with triple-negative breast cancer. iScience 2021, 24, 102451. [Google Scholar] [CrossRef]
- Johnson, E.C.B.; Dammer, E.B.; Duong, D.; Ping, L.; Zhou, M.; Yin, L.; Higginbotham, L.A.; Guajardo, A.; White, B.; Troncoso, J.C.; et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med. 2020, 26, 769–780. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Zambon, A.C.; Gaj, S.; Ho, I.; Hanspers, K.; Vranizan, K.; Evelo, C.; Conklin, B.R.; Pico, A.; Salomonis, N. GO-Elite: A flexible solution for pathway and ontology over-representation. Bioinformatics 2012, 28, 2209–2210. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Shi, M.; Li, J.; Zhang, H.; Chen, B.; Chen, L.; Gao, W.; Giuliani, N.; Zhao, R.C. Elevated tumor necrosis factor-alpha suppresses TAZ expression and impairs osteogenic potential of Flk-1+ mesen-chymal stem cells in patients with multiple myeloma. Stem Cells Dev. 2007, 16, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Yin, J.-J.; Wang, Z.-L.; Zhan, X.-R. Effect of G Protein-coupled Receptor Kinase 6 on Proliferation and Apoptosis of Multiple Myeloma Cells and Its Mechanisms. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2016, 24, 1801–1806. [Google Scholar]
- Mancino, M.; Ametller, E.; Gascón, P.; Almendro, V. The neuronal influence on tumor progression. Biochim. Biophys. Acta BBA Rev. Cancer 2011, 1816, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Klimienė, I.; Radzevičius, M.; Matuzevičienė, R.; Sinkevič-Belliot, K.; Kučinskienė, Z.A.; Pečeliūnas, V. Adhesion molecule immunophenotype of bone marrow multiple myeloma plasma cells impacts the presence of malignant circulating plasma cells in peripheral blood. Int. J. Lab. Hematol. 2021, 43, 403–408. [Google Scholar] [CrossRef]
- Abusamra, D.B.; Aleisa, F.A.; Al-Amoodi, A.S.; Ahmed, H.M.J.; Chin, C.J.; Abuelela, A.F.; Bergam, P.; Sougrat, R.; Merzaban, J.S. Not just a marker: CD34 on human hematopoietic stem/progenitor cells dominates vascular selectin binding along with CD44. Blood Adv. 2017, 1, 2799–2816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, I.; Subbarao, R.B.; Rho, G.J. Human mesenchymal stem cells-current trends and future prospective. Biosci. Rep. 2015, 35, e00191. [Google Scholar] [CrossRef]
- Kapoor, S.; Shenoy, S.P.; Bose, B. CD34 cells in somatic, regenerative and cancer stem cells: Developmental biology, cell therapy, and omics big data perspective. J. Cell. Biochem. 2020, 121, 3058–3069. [Google Scholar] [CrossRef]
- Gangenahalli, G.U.; Singh, V.K.; Verma, Y.K.; Gupta, P.; Sharma, R.K.; Chandra, R.; Luthra, P.M. Hematopoietic Stem Cell Antigen CD34: Role in Adhesion or Homing. Stem Cells Dev. 2006, 15, 305–313. [Google Scholar] [CrossRef]
- Kuranda, K.; Berthon, C.; Leprêtre, F.; Polakowska, R.; Jouy, N.; Quesnel, B. Expression of CD34 in hematopoietic cancer cell lines reflects tightly regulated stem/progenitor-like state. J. Cell. Biochem. 2011, 112, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, Y.; He, X.; Liu, X.; Zhang, Y.; Yang, Z.; Yang, P.; Wang, J.; Hu, K.; Liu, W.; et al. Prognosis value of RBBP8 expression in plasma cell myeloma. Cancer Gene Ther. 2020, 27, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoudi-Nejad, A.; Bidkhori, G.; Ashtiani, S.H.; Najafi, A.; Bozorgmehr, J.H.; Wang, E. Cancer systems biology and modeling: Microscopic scale and multiscale approaches. Semin. Cancer Biol. 2015, 30, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Gaiteri, C.; Ding, Y.; French, B.; Tseng, G.C.; Sibille, E. Beyond modules and hubs: The potential of gene coexpression networks for investigating molecular mechanisms of complex brain disorders. Genes Brain Behav. 2014, 13, 13–24. [Google Scholar] [CrossRef]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [Green Version]
- Mei, A.H.-C.; Tung, K.; Han, J.; Perumal, D.; Laganà, A.; Keats, J.; Auclair, D.; Chari, A.; Jagannath, S.; Parekh, S.; et al. MAGE-A inhibit apoptosis and promote proliferation in multiple myeloma through regulation of BIM and p21Cip1. Oncotarget 2020, 11, 727–739. [Google Scholar] [CrossRef]
- Kryukov, F.; Dementyeva, E.; Kubiczkova, L.; Jarkovsky, J.; Brozova, L.; Petrik, J.; Nemec, P.; Sevcikova, S.; Minarik, J.; Stefanikova, Z.; et al. Cell cycle genes co-expression in multiple myeloma and plasma cell leukemia. Genomics 2013, 102, 243–249. [Google Scholar] [CrossRef]
- Schooten, E.; Di Maggio, A.; en Henegouwen, P.M.v.B.; Kijanka, M.M. MAGE-A antigens as targets for cancer immunotherapy. Cancer Treat. Rev. 2018, 67, 54–62. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCRs and Signal Transducers: Interaction Stoichiometry. Trends Pharmacol. Sci. 2018, 39, 672–684. [Google Scholar] [CrossRef]
- Prud’Homme, G.J.; Glinka, Y.; Wang, Q. Immunological GABAergic interactions and therapeutic applications in autoimmune diseases. Autoimmun. Rev. 2015, 14, 1048–1056. [Google Scholar] [CrossRef]
- Agrawal, J.; Dwivedi, Y. GABAA Receptor Subunit Transcriptional Regulation, Expression Organization, and Mediated Calmodulin Signaling in Prefrontal Cortex of Rats Showing Testosterone-Mediated Impulsive Behavior. Front. Neurosci. 2020, 14, 1156. [Google Scholar] [CrossRef] [PubMed]
- Schwirtlich, M.; Emri, Z.; Antal, K.; Máté, Z.; Katarova, Z.; Szabó, G. GABA(A) and GABA(B) receptors of distinct properties affect oppositely the proliferation of mouse embryonic stem cells through synergistic elevation of intracellular Ca2+. FASEB J. 2010, 24, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.A.; Chiang, J.; Villanueva-Meyer, J.E.; Alexandrescu, S.; Eschbacher, J.M.; Wang, W.; Mafra, M.; Din, N.U.; Carr-Boyd, E.; Watson, M.; et al. Intracranial mesenchymal tumor with FET-CREB fusion—A unifying diagnosis for the spectrum of intracranial myxoid mesenchymal tumors and angiomatoid fibrous histiocytoma-like neoplasms. Brain Pathol. 2021, 31, e12918. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Gong, Y.; Shao, M.; Ruan, G.; Xie, H.; Liao, X.; Wang, X.; Han, Q.; Zhou, X.; Zhu, L.; et al. Distinct diagnostic and prognostic values of γ-aminobutyric acid type A receptor family genes in patients with colon adenocarcinoma. Oncol. Lett. 2020, 20, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.P. Mechanisms of action of gabapentin. Rev. Neurol. 1997, 153, S39–S45. [Google Scholar]
- Loprinzi, C.L.; Lacchetti, C.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Hertz, D.L.; Kelley, M.R.; Lavino, A.; Lustberg, M.B.; Paice, J.A.; et al. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3325–3348. [Google Scholar] [CrossRef]
- Bhandage, A.K.; Barragan, A. GABAergic signaling by cells of the immune system: More the rule than the exception. Cell. Mol. Life Sci. 2021, 78, 5667–5679. [Google Scholar] [CrossRef]
- Hemminger, J.A.; Toland, A.E.; Scharschmidt, T.J.; Mayerson, J.L.; Guttridge, D.C.; Iwenofu, O.H. Expression of cancer-testis antigens MAGEA1, MAGEA3, ACRBP, PRAME, SSX2, and CTAG2 in myxoid and round cell liposarcoma. Mod. Pathol. 2014, 27, 1238–1245. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Gao, Q.; Zhao, N.; Zhang, X.; Cui, W.; Sun, J.; Fu, J.; Hao, J. Sohlh1 suppresses glioblastoma cell proliferation, migration, and invasion by inhibition of Wnt/β-catenin signaling. Mol. Carcinog. 2018, 57, 494–502. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, Y.; Mao, F.; Lin, Y.; Shen, S.; Sun, Q. lncRNA AFAP1-AS1 promotes triple negative breast cancer cell proliferation and invasion via targeting miR-145 to regulate MTH1 expression. Sci. Rep. 2020, 10, 7662. [Google Scholar] [CrossRef]
- Brown, R.D.; Yuen, B.P.E.; Gibson, J.; Joshua, D.E. The Expression of T Cell Related Costimulatory Molecules in Multiple Myeloma. Leuk. Lymphoma 1998, 31, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Carpenter, K.; Mollard, A.; Vankayalapati, H.; Warner, S.L.; Sharma, S.; Tricot, G.; Zhan, F.; Bearss, D.J. Inhibition of Nek2 by Small Molecules Affects Proteasome Activity. BioMed Res. Int. 2014, 2014, 273180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Yang, Y.; Fu, X.; Wang, Z.; Liu, Y.; Li, M.; Zhang, Y.; Li, Y.; Li, P.-F.; Yu, T.; et al. Long noncoding RNA XXYLT1-AS2 regulates proliferation and adhesion by targeting the RNA binding protein FUS in HUVEC. Atherosclerosis 2020, 298, 58–69. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Traits | Classification | Number of Patients |
|---|---|---|
| Gender | Male | 156/270 |
| Female | 114/270 | |
| Race | European American | 184/270 |
| African American | 34/270 | |
| Others | 52/270 | |
| Tumor stages | I | 83/270 |
| II | 87/270 | |
| III | 93/270 | |
| NA | 7/270 | |
| MM vital status | Alive | 212/270 |
| Dead | 58/270 |
| Table of Modules | ||
|---|---|---|
| Module Name | Module Number | Number of Cluster Gene per Module |
| turquoise | M1 | 1861 |
| blue | M2 | 1295 |
| brown | M3 | 1274 |
| yellow | M4 | 1246 |
| green | M5 | 1216 |
| red | M6 | 1037 |
| black | M7 | 938 |
| pink | M8 | 931 |
| magenta | M9 | 840 |
| purple | M10 | 690 |
| greenyellow | M11 | 659 |
| tan | M12 | 635 |
| salmon | M13 | 568 |
| cyan | M14 | 560 |
| midnightblue | M15 | 534 |
| lightcyan | M16 | 531 |
| grey60 | M17 | 528 |
| lightgreen | M18 | 494 |
| lightyellow | M19 | 435 |
| royalblue | M20 | 307 |
| darkred | M21 | 216 |
| Module Name | Gene | AUC | z | p-Value |
|---|---|---|---|---|
| Royalblue | CTAG2 | 0.64854 | 3.5529 | 3.8 × 10−4 |
| MAGEA6 | 0.63382 | 3.0658 | 2.17 × 10−3 | |
| GABRB2 | 0.66859 | 4.19179 | 3.00 × 10−5 | |
| SOHLH1 | 0.64846 | 3.47324 | 5.4 × 10−4 | |
| AFAP1_AS1 | 0.62386 | 3.05481 | 2.30 × 10−3 | |
| MAGEA1 | 0.62911 | 3.14134 | 1.68 × 10−3 | |
| CASC9 | 0.61675 | 2.63932 | 8.31 × 10−3 | |
| HTR2C | 0.6261 | 2.87452 | 4.05 × 10−3 | |
| GLDC | 0.63476 | 3.31424 | 9.2 × 10−4 | |
| GABRA3 | 0.63484 | 3.15689 | 2.60 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adebayo, O.O.; Dammer, E.B.; Dill, C.D.; Adebayo, A.O.; Oseni, S.O.; Griffen, T.L.; Ohandjo, A.Q.; Yan, F.; Jain, S.; Barwick, B.G.; et al. Multivariant Transcriptome Analysis Identifies Modules and Hub Genes Associated with Poor Outcomes in Newly Diagnosed Multiple Myeloma Patients. Cancers 2022, 14, 2228. https://doi.org/10.3390/cancers14092228
Adebayo OO, Dammer EB, Dill CD, Adebayo AO, Oseni SO, Griffen TL, Ohandjo AQ, Yan F, Jain S, Barwick BG, et al. Multivariant Transcriptome Analysis Identifies Modules and Hub Genes Associated with Poor Outcomes in Newly Diagnosed Multiple Myeloma Patients. Cancers. 2022; 14(9):2228. https://doi.org/10.3390/cancers14092228
Chicago/Turabian StyleAdebayo, Olayinka O., Eric B. Dammer, Courtney D. Dill, Adeyinka O. Adebayo, Saheed O. Oseni, Ti’ara L. Griffen, Adaugo Q. Ohandjo, Fengxia Yan, Sanjay Jain, Benjamin G. Barwick, and et al. 2022. "Multivariant Transcriptome Analysis Identifies Modules and Hub Genes Associated with Poor Outcomes in Newly Diagnosed Multiple Myeloma Patients" Cancers 14, no. 9: 2228. https://doi.org/10.3390/cancers14092228
APA StyleAdebayo, O. O., Dammer, E. B., Dill, C. D., Adebayo, A. O., Oseni, S. O., Griffen, T. L., Ohandjo, A. Q., Yan, F., Jain, S., Barwick, B. G., Singh, R., Boise, L. H., & Lillard, Jr., J. W. (2022). Multivariant Transcriptome Analysis Identifies Modules and Hub Genes Associated with Poor Outcomes in Newly Diagnosed Multiple Myeloma Patients. Cancers, 14(9), 2228. https://doi.org/10.3390/cancers14092228