
Chronic Cancer Pain: Opioids within Tumor Microenvironment Affect Neuroinflammation, Tumor and Pain Evolution

{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Chronic Cancer and Non-Cancer Pain as Disease in Itself
2. Is Cancer Pain “Different”?
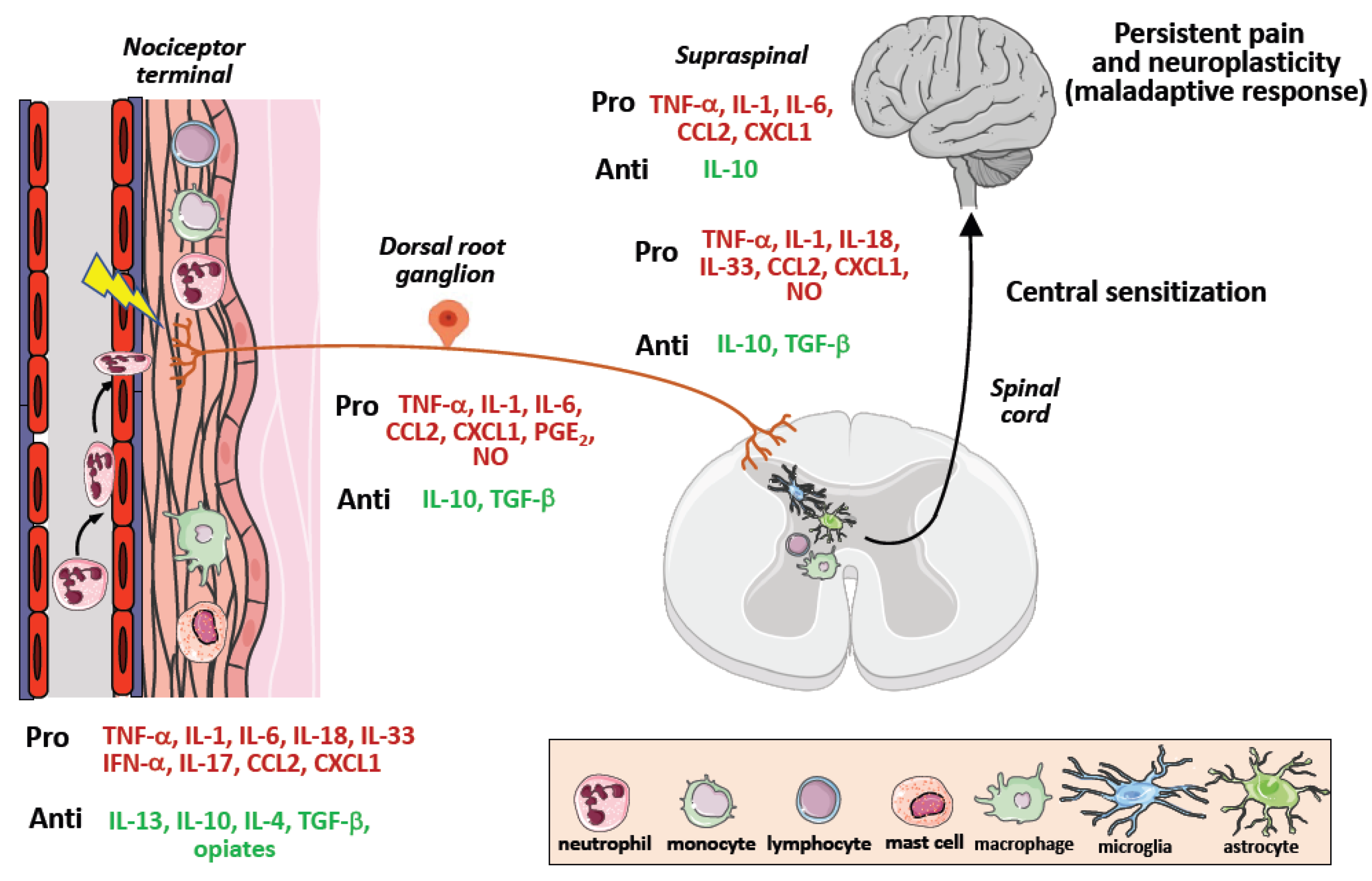
3. How Immunology Contributed to Modify the View of Chronic Pain
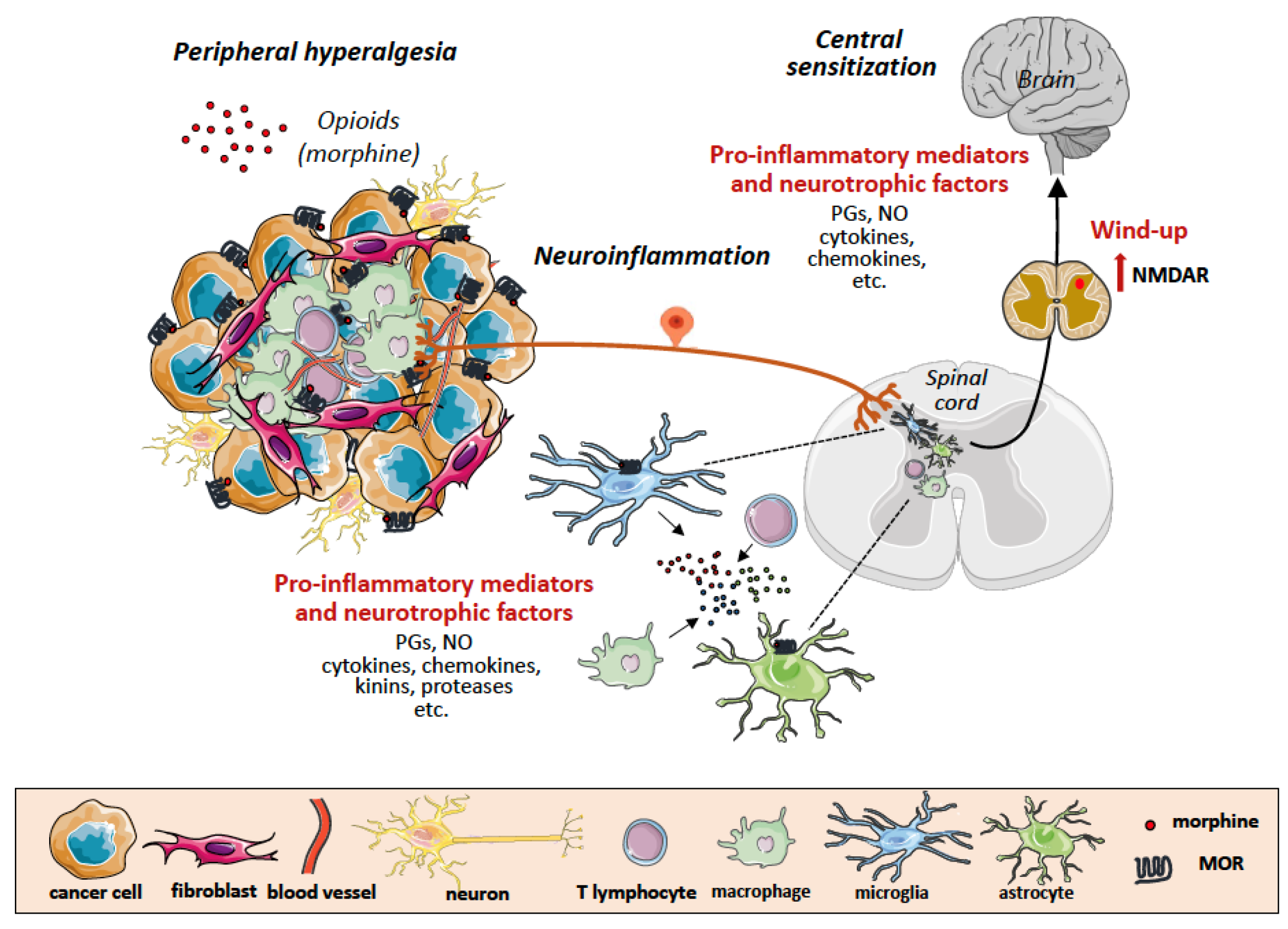
4. Neuroinflammation and Central Sensitization
5. The Complexity of Tumor Microenvironment
6. Tumor Microenvironment as Scenario of Neuroinflammation and Chronic Pain
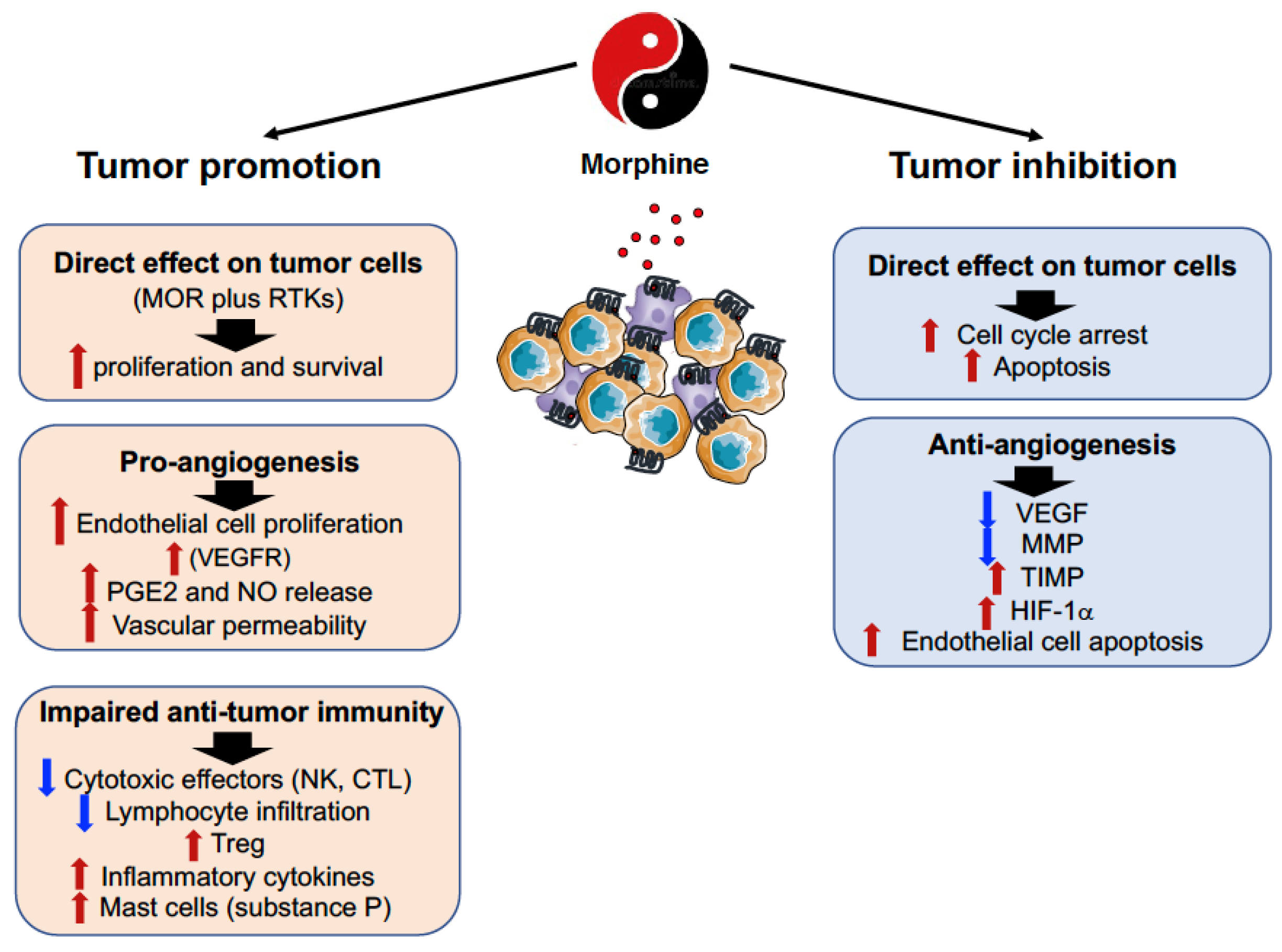
7. The Double-Edged Effects of Opioids and Their Receptors in the Tumor Microenvironment
8. Opioids (Morphine) and MOR Interference with the Neoplastic Process
9. When and How MOR Became a Relevant Bridge between Pain and Immunological Research
10. Cancer Immunotherapy and Pain
11. Immunotherapy of Pain
12. Conclusions
Funding
Conflicts of Interest
References
- Treede, R.D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. Chronic pain as a symptom or a disease: The IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 2019, 160, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, M.; Vlaeyen, J.W.S.; Rief, W.; Barke, A.; Aziz, Q.; Benoliel, R.; Cohen, M.; Evers, S.; Giamberardino, M.A.; Goebel, A.; et al. The IASP classification of chronic pain for ICD-11: Chronic primary pain. Pain 2019, 160, 28–37. [Google Scholar] [CrossRef]
- Bennett, M.I.; Kaasa, S.; Barkee, A.; Korwisie, B.; Riefe, W.; Treede, R.; The IASP Taskforce for the Classification of Chronic Pain. The IASP classification of chronic pain for ICD-11: Chronic cancer-related pain. Pain 2019, 160, 38–44. [Google Scholar] [CrossRef]
- Barke, A.; Korwisi, B.; Hans-Raimund, C.; Fors, E.A.; Geber, C.; Schug, S.A.; Stubhaug, A.; Ushida, T.; Wetterling, T.; Rief, W.; et al. Pilot field testing of the chronic pain classification for ICD-11: The results of ecological coding. BMC Public Health 2018, 18, 1239. [Google Scholar] [CrossRef]
- Breivik, H.; Cherny, N.; Collett, B.; de Conno, F.; Filbet, M.; Foubert, A.J.; Cohen, R.; Dow, L. Cancer-related pain: A pan-European survey of prevalence, treatment, and patient attitudes. Ann. Oncol. 2009, 20, 1420–1433. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Blalock, J.E.; Smith, E.M. Conceptual development of the immune system as a sixth sense. Brain Behav. Immun. 2007, 21, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. Pain 2011, 152, S2–S15. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.R.; Chen, O.; Ji, R.R. How do sensory neurons sense danger signals? Trends Neurosci. 2020, 43, 822–838. [Google Scholar] [CrossRef] [PubMed]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef] [PubMed]
- Kuner, R.; Flor, H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci. 2016, 18, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Berta, T.; Nedergaard, M. Glia and pain: Is chronic pain a gliopathy? Pain 2013, 154 (Suppl. S1), S10–S28. [Google Scholar] [CrossRef] [PubMed]
- Santoni, A.; Mercadante, S.; Arcuri, E. Chronic cancer and non-cancer pain and opioid-induced hyperalgesia share common mechanisms: Neuroinflammation and central sensitization. Minerva Anestesiol. 2021, 87, 210–222. [Google Scholar] [CrossRef]
- Kosek, E.; Cohen, M.; Baron, R.; Gebhart, G.F.; Mico, J.A.; Rice, A.S.; Rief, W.; Sluka, A.K. Do we need a third mechanistic descriptor for chronic pain states? Pain 2016, 157, 1382–1386. [Google Scholar] [CrossRef]
- Virchow, R.A. Die Cellular Pathologie in Ihrer Begründung auf Physiologische und Pathologische Gewebelehre; Hirschwald: Berlin, Germany, 1858; Volume XVI, 440p. [Google Scholar]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Maman, S.; Witz, I.P. A history of exploring cancer in context. Nat. Rev. Cancer 2018, 18, 359–376. [Google Scholar]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Young, H.H. On the presence of nerves in tumors and of other structures in them as revealed by a modification of Ehrlich’s method of “vital staining” with methylene blue. J. Exp. Med. 1897, 2, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boilly, B.; Faulkner, S.; Jobling, P.; Hondermarck, H. Nerve dependence: From regeneration to cancer. Cancer Cell 2017, 31, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, D.; Kuner, R. Molecular players of tumor-nerve interactions. Pain 2015, 156, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Jobling, P.; Pundavela, J.; Oliveira, M.R.S.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve–Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [Green Version]
- Amit, M.; Na’ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. Cancer 2016, 16, 399–408. [Google Scholar] [CrossRef]
- Marchesi, F.; Piemonti, L.; Mantovani, A.; Allavena, P. Molecular mechanisms of perineural invasion, a forgotten pathway of dissemination and metastasis. Cytokine Growth Factor Rev. 2010, 21, 77–82. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.P.; Firlej, V.; Allory, Y.; Roméo, P.H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef]
- Schwei, M.J.; Honore, P.; Rogers, S.D.; Salak-Johnson, J.L.; Finke, M.P.; Ramnaraine, M.L.; Clohisy, D.R.; Mantyh, P.W. Neurochemical and cellular reorganization of the spinal cord in a murine model of bone cancer pain. J. Neurosci. 1999, 19, 10886–10897. [Google Scholar] [CrossRef] [Green Version]
- Mantyh, P.W.; Clohisy, D.R.; Koltzenburg, M.; Hunt, S.P. Molecular mechanisms of cancer pain. Nat. Rev. Cancer 2002, 2, 201–209. [Google Scholar] [CrossRef]
- Sabino, M.A.; Luger, N.M.; Mach, D.B.; Rogers, S.D.; Schwei, M.J.; Mantyh, P.W. Different tumors in bone each give rise to a distinct pattern of skeletal destruction, bone cancer-related pain behaviors and neurochemical changes in the central nervous system. Int. J. Cancer 2003, 104, 550–558. [Google Scholar] [CrossRef]
- Mantyh, P.W. Cancer pain and its impact on diagnosis, survival and quality of life. Nat. Rev. Neurosci. 2006, 7, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Schäfer, M.; Machelska, H. Attacking pain at its source: New perspectives on opioids. Nat. Med. 2003, 9, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Luger, N.M.; Sabino, M.A.; Schwei, M.J.; Mach, D.B.; Pomonis, J.D.; Keyser, C.P.; Rathbun, M.; Clohisy, D.R.; Honore, P.; Yaksh, T.L.; et al. Efficacy of systemic morphine suggests a fundamental difference in the mechanisms that generate bone cancer vs inflammatory pain. Pain 2002, 99, 397–406. [Google Scholar] [CrossRef]
- Mercadante, S.; Arcuri, E.; Santoni, A. Opioid-induced tolerance and hyperalgesia. CNS Drugs 2019, 33, 943–955. [Google Scholar] [CrossRef]
- Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef]
- Scroope, C.A.; Singleton, Z.; Hollmann, M.W.; Parat, M.O. Opioid receptor-mediated and non-opioid receptor-mediated roles of opioids in tumour growth and metastasis. Front. Oncol. 2021, 11, 792290. [Google Scholar] [CrossRef]
- Amaram-Davila, J.; Davis, M.; Reddy, A. Opioids and cancer mortality. Curr. Treat. Options Oncol. 2020, 21, 22. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Liang, X.Y.; Yan, Y.; Dai, Z.; Chu, H.C. Morphine: Double-faced roles in the regulation of tumor development. Clin. Transl. Oncol. 2018, 20, 808–814. [Google Scholar] [CrossRef]
- Tuerxun, H.; Cui, J. The dual effect of morphine on tumor development. Clin. Transl. Oncol. 2019, 21, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, G.; Tao, T.; Kang, X.; Liu, C.; Zhang, X.; Wang, C.; Li, C.; Guo, X. The μ-opioid receptor (MOR) promotes tumor initiation in hepatocellular carcinoma. Cancer Lett. 2019, 453, 1–9. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, H.; Weng, M.L.; Zhang, J.; Jiang, N.; Cata, J.P.; Ma, D.; Chen, W.K.; Miao, C.H. Morphine promotes tumorigenesis and cetuximab resistance via EGFR signaling activation in human colorectal cancer. J. Cell. Physiol. 2021, 236, 4445–4454. [Google Scholar] [CrossRef] [PubMed]
- Tagirasa, R.; Yoo, E. Role of Serine Proteases at the Tumor-Stroma Interface. Front. Immunol. 2022, 13, 832418. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Nunez, V.; Noriega-Prieto, J.A.; Rodriguez, R.E. Morphine modulates cell proliferation through mir133b & mir128 in the neuroblastoma SH-SY5Y cell line. Biochim. Biophys. Acta 2014, 1842, 566–572. [Google Scholar] [PubMed] [Green Version]
- Lennon, F.E.; Mirzapoiazova, T.; Mambetsariev, B.; Poroyko, V.A.; Salgia, R.; Moss, J.; Singleton, P.A. The Mu Opioid Receptor Promotes Opioid and Growth Factor-Induced Proliferation, Migration and Epithelial Mesenchymal Transition (EMT) in Human Lung Cancer. PLoS ONE 2014, 9, e91577. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Luk, K.; Vang, D.; Soto, W.; Vincent, L.; Robiner, S.; Saavedra, R.; Li, Y.; Gupta, P.; Gupta, K. Morphine stimulates cancer progression and mast cell activation and impairs survival in transgenic mice with breast cancer. Br. J. Anaesth. 2014, 113, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Lennon, F.E.; Mirzapoiazova, T.; Mambetsariev, B.; Salgia, R.; Moss, J.; Singleton, P.A. Overexpression of the m-Opioid Receptor in Human Non-Small Cell Lung Cancer Promotes Akt and mTOR Activation, Tumor Growth, and Metastasis. Anesthesiology 2012, 116, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Gach, K.; Szemraj, J. The influence of opioids on matrix metalloproteinase-2 and -9 secretion and mRNA levels in MCF-7 breast cancer cell line. Mol. Biol. Rep. 2011, 38, 1231–1236. [Google Scholar] [CrossRef]
- Xie, N.; Khabbazi, S.; Nassar, Z.D.; Gregory, K.; Vithanage, T.; Anand-Apte, B.; Cabot, P.J.; Sturgess, D.; Shaw, P.N.; and Parat, M. Morphine alters the circulating proteolytic profile in mice: Functional consequences on cellular migration and invasion. FASEB J. 2017, 31, 5208–5216. [Google Scholar] [CrossRef] [Green Version]
- Singleton, P.A.; Lingen, M.W.; Fekete, M.J.; Garcia, J.G.; Moss, J. Methylnaltrexone inhibits opiate and VEGF-induced angiogenesis: Role of receptor transactivation. Microvasc. Res. 2006, 72, 3–11. [Google Scholar] [CrossRef]
- Gupta, K.; Kshirsagar, S.; Chang, L.; Schwartz, R.; Law, P.Y.; Yee, D.; Hebbel, R.P. Morphine stimulates angiogenesis by activating proangiogenic and survival-promoting signaling and promotes breast tumor growth. Cancer Res. 2002, 62, 4491–4498. [Google Scholar]
- Farooqui, M.; Li, Y.; Rogers, T.; Poonawala, T.; Griffin, R.J.; Song, C.W.; Gupta, K. COX-2 inhibitor celecoxib prevents chronic morphine-induced promotion of angiogenesis, tumour growth, metastasis and mortality, without compromising analgesia. Br. J. Cancer 2007, 97, 1523–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Liu, C.H.; Conway, R.; Han, D.K.; Nithipatikom, K.; Trifan, O.C.; Lane, T.F.; Hla, T. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc. Natl. Acad. Sci. USA 2004, 101, 591–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, M.; Zhu, W.; Tønnesen, J.; Cadet, P.; Tønnesen, E.; Stefano, G.B. Effects of morphine on tumour growth. Neuroendocrinol. Lett. 2002, 23, 193–198. [Google Scholar] [PubMed]
- Koodie, L.; Ramakrishnan, S.; Roy, S. Morphine suppresses tumor angiogenesis through a HIF-1alpha/p38MAPK pathway. Am. J. Pathol. 2010, 177, 984–997. [Google Scholar] [CrossRef]
- Hsiaoa, P.; Changa, M.; Cheng, W.; Chen, C.; Lin, H.; Hsieh, C.; Sun, W. Morphine induces apoptosis of human endothelial cells through nitric oxide and reactive oxygen species pathways. Toxicology 2009, 256, 83–91. [Google Scholar] [CrossRef]
- Eisenstein, T.K. The Role of Opioid Receptors in Immune System Function. Front. Immunol. 2019, 10, 2904. [Google Scholar] [CrossRef] [Green Version]
- Wybran, J.; Appelboom, T.; Famaey, J.P.; Govaerts, A. Suggestive evidence for receptors for morphine and methionine-enkephalin on normal human blood T lymphocytes. J. Immunol. 1979, 123, 1068–1070. [Google Scholar]
- Plein, L.M.; Rittner, H.L. Opioids and the immune system—Friend or foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef]
- Gong, L.; Dong, C.; Ouyang, W.; Quin, Q. Regulatory T cells: A possible promising approach to cancer recurrence induced by morphine. Med. Hypotheses 2013, 80, 308–310. [Google Scholar] [CrossRef]
- Koodie, L.; Yuan, H.; Pumper, J.A.; Yu, H.; Charboneau, R.; Ramkrishnan, S. Morphine inhibits migration of tumor-infiltrating leukocytes and suppresses angiogenesis associated with tumor growth in mice. Am. J. Pathol. 2014, 184, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- Kawase, M.; Sakagami, H.; Furuya, K.; Kikuchi, H.; Nishikawa, H.; Motohashi, N.; Morimoto, Y.; Varga, A.; Molnar, J. Cell death-inducing activity of opiates in human oral tumor cell lines. Anticancer Res. 2002, 22, 211–214. [Google Scholar] [PubMed]
- Sueoka, E.; Sueoka, N.; Kai, Y.; Okabe, S.; Suganuma, M.; Kanematsu, K.; Yamamoto, T.; Fujiki, H. Anticancer activity of morphine and its synthetic derivative, KT-90, mediated through apoptosis and Inhibition of NF-kB activation. Biochem. Biophys. Res. Commun. 1998, 252, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qin, Y.; Li, L.; Chen, J.; Zhang, X.; Xie, Y. Morphine can inhibit the growth of breast cancer MCF-7 cells by arresting the cell cycle and inducing apoptosis. Biol. Pharm. Bull. 2017, 40, 1686–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gach, K.; Wyrębska, A.; Fichna, J.; Janecka, A. The role of morphine in regulation of cancer cell growth. Arch. Pharmacol. 2011, 384, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Weingaertner, I.R.; Koutnik, S.; Ammer, H. Chronic morphine treatment attenuates cell growth of human BT474 breast cancer cells by rearrangement of the ErbB signalling network. PLoS ONE 2013, 8, e53510. [Google Scholar] [CrossRef] [Green Version]
- Zagon, I.S.; Verderameb, M.F.; McLaughlin, P.J. The biology of the opioid growth factor receptor (OGFr). Brain Res. Rev. 2002, 38, 351–376. [Google Scholar] [CrossRef]
- Kim, J.Y.; Ahn, H.J.; Kim, J.K.; Kim, J.; Lee, S.H.; Chae, H.B. Morphine suppresses lung cancer cell proliferation through the interaction with opioid growth factor receptor: An in vitro and human lung tissue study. Anesth. Analg. 2016, 123, 1429–1436. [Google Scholar] [CrossRef]
- Sasamura, T.; Nakamura, S.; Iida, Y.; Fujii, H.; Murata, J.; Saiki, I.; Nojima, H.; Kuraishi, Y. Morphine analgesia suppresses tumor growth and metastasis in a mouse model of cancer pain produced by orthotopic tumor inoculation. Eur. J. Pharmacol. 2002, 441, 185–191. [Google Scholar] [CrossRef]
- Harimaya, Y.; Koizumi, K.; Andoh, T.; Nojima, H.; Kuraishi, Y.; Saiki, I. Potential ability of morphine to inhibit the adhesion, invasion and metastasis of metastatic colon 26-L5 carcinoma cells. Cancer Lett. 2002, 187, 121–127. [Google Scholar] [CrossRef]
- Tegeder, I.; Grosch, S.; Schmidtko, A.; Haussler, A.; Schmidt, H.; Niederberger, E.; Scholich, K.; Geisslinger, G. G protein-independent G1 cell cycle block and apoptosis with morphine in adenocarcinoma cells: Involvement of p53 phosphorylation. Cancer Res. 2003, 63, 1846–1852. [Google Scholar]
- Li, C.; Li, L.; Qin, Y.; Jiang, Y.; Wei, Y.; Chen, J.; Yubo, X. Exogenous morphine inhibits the growth of human gastric tumor in vivo. Ann. Transl. Med. 2020, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Loram, L.C.; Ramos, K.; de Jesus, A.J.; Thomas, J.; Cheng, K.; Reddy, A.; Somogyi, A.A.; Hutchinson, M.R.; Watkins, L.R.; et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc. Natl. Acad. Sci. USA 2012, 109, 6325–6330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, L.R.; Hutchinson, M.R.; Rice, K.C.; Maier, S.F. The “toll” of opioid-induced glial activation: Improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol. Sci. 2009, 30, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabr, M.M.; Saeed, I.; Miles, J.A.; Ross, B.P.; Shaw, P.N.; Hollmann, M.W.; Parat, M.O. Interaction of opioids with TLR4-mechanisms and ramifications. Cancers 2021, 13, 5274. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, J.; Liu, T.; Yu, W.; Dong, N.; Zhang, C.; Xia, W.; Wei, F.; Yang, L.; Ren, X. Morphine-3-glucuronide upregulates PD-L1 expression via TLR4 and promotes the immune escape of non-small cell lung cancer. Cancer Biol. Med. 2021, 18, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, M.; Zhou, D.; Gorur, A.; Sun, Z.; Zeng, W.; Cata, J.P.; Chen, W.; Miao, C. Increased mu-opioid receptor expression is associated with reduced disease-free and overall survival in laryngeal squamous cell carcinoma. Br. J. Anaesth. 2020, 125, 722–729. [Google Scholar] [CrossRef]
- Chen, D.T.; Pan, J.H.; Chen, Y.H.; Xing, W.; Yan, Y.; Yuan, Y.F.; Zeng, W.A. The mu-opioid receptor is a molecular marker for poor prognosis in hepatocellular carcinoma and represents a potential therapeutic target. Br. J. Anaesth. 2019, 122, e157–e167. [Google Scholar] [CrossRef]
- Yao, Y.S.; Yao, R.Y.; Zhuang, L.K.; Qi, W.W.; Lv, J.; Zhou, F.; Qiu, W.S.; Yue, L. MOR1 expression in gastric cancer: A biomarker associated with poor outcome. Clin. Transl. Sci. 2015, 8, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Zylla, D.; Gourley, B.L.; Vang, D.; Jackson, S.; Boatman, S.; Lindgren, B.; Kuskowski, M.A.; Le, C.; Gupta, K.; Gupta, P. Opioid requirement, opioid receptor expression, and clinical outcomes in patients with advanced prostate cancer. Cancer 2013, 119, 4103–4110. [Google Scholar] [CrossRef]
- Cata, J.P. Outcomes of regional anesthesia in cancer patients. Curr. Opin. Anaesthesiol. 2018, 31, 593–600. [Google Scholar] [CrossRef]
- Forget, P.; Aguirre, J.A.; Bencic, I.; Borgeat, A.; Cama, A.; Condron, C.; Eintrei, C.; Eroles, P.; Gupta, A.; Hales, T.G.; et al. How anesthetic, analgesic and other non-surgical techniques during cancer surgery might affect postoperative oncologic outcomes: A summary of current state of evidence. Cancers 2019, 11, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novy, D.M.; Nelson, D.V.; Koyyalagunta, D.; Cata, J.P.; Gupta, P.; Gupta, K. Pain, opioid therapy, and survival: A needed discussion. Pain 2020, 161, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Johnson, L.K.; Karp, D.D.; Atkins, J.T.; Singleton, P.A.; Moss, J. Treatment with methylnaltrexone is associated with increased survival in patients with advanced cancer. Ann Oncol. 2016, 27, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Missair, A.; Cata, J.P.; Votta-Velis, G.; Johnson, M.; Borgeat, A.; Tiouririne, M.; Gottumukkala, V.; Buggy, D.; Vallejo, R.; Marrero, E.B.; et al. Impact of perioperative pain management on cancer recurrence: An ASRA/ESRA special article. Reg. Anesth. Pain Med. 2019, 44, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Peterson, P.K.; Molitor, T.W.; Chao, C.C. The opioid–cytokine connection. J. Neuroimmunol. 1998, 83, 63–69. [Google Scholar] [CrossRef]
- Collin, E.; Poulain, P.; Gauvain-Piquard, A.; Petit, G.; Pichard-Leandri, E. Is disease progression the major factor in morphine “tolerance” in cancer pain treatment? Pain 1993, 55, 319–326. [Google Scholar] [CrossRef]
- Arcuri, E.; Ginobbi, P.; Tirelli, W.; Froldi, R.; Citro, G.; Santoni, A. Preliminary in vivo experimental evidence on intratumoral morphine uptake. Possible clinical implications in cancer pain and opioid responsiveness. J. Pain Symptom Manag. 2002, 24, 1–3. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Kolesnikov, Y.A.; Babey, A.M. Perspectives on the N-methyl-D-aspartate/nitric oxide cascade and opioid tolerance. Neuropsychopharmacology 1995, 13, 309–313. [Google Scholar] [CrossRef]
- Kolesnikov, Y.A.; Pick, C.G.; Ciszewska, G.; Pasternak, G.W. Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc. Natl. Acad. Sci. USA 1993, 90, 5162–5166. [Google Scholar] [CrossRef] [Green Version]
- Fimiani, C.; Arcuri, E.; Santoni, A.; Rialas, C.M.; Bilfinger, T.V.; Peter, D.; Salzet, B.; Stefano, G.B. Mu3 opiate receptor expression in lung and lung carcinoma: Ligand binding and coupling to nitric oxide release. Cancer Lett. 1999, 146, 45–51. [Google Scholar] [CrossRef]
- DeLeo, J.A.; Yezierski, R.P. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 2001, 90, 1–6. [Google Scholar] [CrossRef]
- Arcuri, E. Can tumors act as opioid traps, mimicking opioid tolerance? J. Pain Symptom Manag. 1998, 16, 78–79. [Google Scholar]
- Sjøgren, P.; Jensen, N.H.; Jensen, T.S. Disappearance of morphine-induced hyperalgesia after discontinuing or substituting morphine with other opioid agonists. Pain 1994, 59, 313–316. [Google Scholar] [CrossRef]
- Mercadante, S.; Ferrera, P.; Villari, P.; Arcuri, E. Hyperalgesia: An emerging iatrogenic syndrome. J. Pain Symptom Manag. 2003, 26, 769–775. [Google Scholar] [CrossRef]
- Mercadante, S.; Arcuri, E. Breakthrough pain in cancer patients: Pathophysiology and treatment. Cancer Treat. Rev. 1998, 24, 425–432. [Google Scholar] [CrossRef]
- Arcuri, E.; Ginobbi, P.; Tirelli, W.; Ayers, G.; Milana, R. Breakthrough pain: A single mask for varying painful situations: Therapeutic reflexes in cancer pain. Transl. Med. 2012, 1, 40–42. [Google Scholar]
- Mastronicola, D.; Arcuri, E.; Arese, M.; Bacchi, A.; Mercadante, S.; Cardelli, P.; Citro, G.; Sarti, P. Morphine but not fentanyl and methadone affects mitochondrial membrane potential by inducing nitric oxide release in glioma cells. Cell. Mol. Life Sci. 2004, 61, 2991–2997. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Chen, G.; Kim, Y.H.; Li, H.; Luo, H.; Liu, D.L.; Zhang, Z.J.; Lay, M.; Chang, W.; Zhang, Y.Q.; Ji, R.R. PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat. Neurosci. 2017, 20, 917–926. [Google Scholar] [CrossRef]
- Liu, B.L.; Cao, Q.L.; Zhao, X.; Liu, H.Z.; Zhang, Y.Q. Inhibition of TRPV1 by SHP-1 in nociceptive primary sensory neurons is critical in PD-L1 analgesia. JCI Insight 2020, 5, e137386. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gu, Y.; Liao, Y.; Bang, S.; Donnelly, C.R.; Chen, O.; Tao, X.; Mirando, A.J.; Hilton, M.J.; Ji, R.R. PD-1 blockade inhibits osteoclast formation and murine bone cancer pain. J. Clin. Investig. 2020, 130, 3603–3620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Jiang, C.; He, Q.; Matsuda, M.; Han, Q.; Wang, K.; Bang, S.; Ding, H.; Ko, M.C.; Ji, R.R. Anti-PD-1 treatment impairs opioid antinociception in rodents and nonhuman primates. Sci. Transl. Med. 2020, 12, eaaw6471. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Abdelhakim, S.; Klapholz, J.D.; Roy, B.; Weiss, S.A.; McGuone, D.; Corbin, Z.A. Mononeuritis multiplex as a rare and severe neurological complication of immune checkpoint inhibitors: A case report. J. Med. Case Rep. 2022, 16, 81. [Google Scholar] [CrossRef]
- Patel, A.S.; Snook, R.J.; Amikar, S. Chronic inflammatory demyelinating polyradiculoneuropathy secondary to immune checkpoint inhibitors in melanoma patients. Discov. Med. 2019, 28, 107–111. [Google Scholar]
- Koldenhof, J.J.; van der Baan, F.H.; Verberne, E.G.; Kamphuis, A.M.; Verheijden, R.J.; Tonk, E.H.; van Lindert, A.S.; van der Stap, J.; Teunissen, S.C.; Witteveen, P.O.; et al. Patient-reported outcomes during checkpoint inhibition: Insight into symptom burden in daily clinical practice. J. Pain Symptom Manag. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Kwatra, S.G.; Ständer, S.; Kang, H. PD-1 Blockade-Induced Pruritus Treated with a Mu-Opioid Receptor Antagonist. N. Engl. J. Med. 2018, 379, 1578–1579. [Google Scholar] [CrossRef]
- Ikoma, A.; Steinhoff, M.S.; Yosipovitch, G.; Schmelz, M. The neurobiology of itch. Nat. Rev. Neurosci. 2006, 7, 535–547. [Google Scholar] [CrossRef]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Ji, R.R.; Xu, Z.Z.; Gao, Y.J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonçalves Dos Santos, G.; Delay, L.; Yaksh, T.L.; Corr, M. Neuraxial Cytokines in Pain States. Front. Immunol. 2020, 10, 3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderwall, A.G.; Milligan, E.D. Cytokines in Pain: Harnessing Endogenous Anti-Inflammatory Signaling for Improved Pain Management. Front. Immunol. 2019, 10, 3009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.R.; Xu, Z.Z.; Strichartz, G.; Serhan, C.N. Emerging roles of resolvins in the resolution of inflammation and pain. Trends Neurosci. 2011, 34, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Pannell, M.; Labuz, D.; Celik, M.Ö.; Keye, J.; Batra, A.; Siegmund, B.; Machelska, H. Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J. Neuroinflamm. 2016, 13, 262. [Google Scholar] [CrossRef]
- Celik, M.Ö.; Labuz, D.; Keye, J.; Glauben, R.; Machelska, H. IL-4 induces M2 macrophages to produce sustained analgesia via opioids. JCI Insight 2020, 5, e133093. [Google Scholar] [CrossRef] [Green Version]
- Laumet, G.; Ma, J.; Robison, A.J.; Kumari, S.; Heijnen, C.J.; Kavelaars, A. T Cells as an Emerging Target for Chronic Pain Therapy. Front. Mol. Neurosci. 2019, 12, 216. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Qiao, G.; Ren, J.; Wang, X.; Wang, S.; Zhu, S.; Yuan, Y.; Morse, M.A.; Hobeika, A.; Lyerly, H.K. Adoptive immunotherapy with autologous T-cell infusions reduces opioid requirements in advanced cancer patients. Pain 2020, 161, 127–134. [Google Scholar] [CrossRef]
- Clauw, D. Hijacking the endogenous opioid system to treat pain: Who thought it would be so complicated? Pain 2017, 158, 2283–2284. [Google Scholar] [CrossRef]
- Blendon, R.J.; Benson, J.M. The Public and the Opioid-Abuse Epidemic. N. Engl. J. Med. 2018, 378, 407–411. [Google Scholar] [CrossRef]
- Boland, J.W.; Pockley, A.G. Clinically relevant concentrations of opioids for in vitro studies. J. Opioid Manag. 2016, 12, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Rivat, C.; Ballantyne, J. The dark side of opioids in pain management: Basic science explains clinical observation. Pain Rep. 2016, 1, e570. [Google Scholar] [CrossRef] [PubMed]
- Santoni, A.; Arcuri, E. The ambiguity of opioids revealed by immunology is changing the knowledge and the therapeutic approach in cancer and non-cancer pain: A narrative review. Immunol. Lett. 2020, 226, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, E.; Mercadante, S.; Santoni, A. Immunity and pain: Is it time for the birth of Immunoalgology? Minerva Anestesiol. 2021, 87, 845–847. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoni, A.; Santoni, M.; Arcuri, E. Chronic Cancer Pain: Opioids within Tumor Microenvironment Affect Neuroinflammation, Tumor and Pain Evolution. Cancers 2022, 14, 2253. https://doi.org/10.3390/cancers14092253
Santoni A, Santoni M, Arcuri E. Chronic Cancer Pain: Opioids within Tumor Microenvironment Affect Neuroinflammation, Tumor and Pain Evolution. Cancers. 2022; 14(9):2253. https://doi.org/10.3390/cancers14092253
Chicago/Turabian StyleSantoni, Angela, Matteo Santoni, and Edoardo Arcuri. 2022. "Chronic Cancer Pain: Opioids within Tumor Microenvironment Affect Neuroinflammation, Tumor and Pain Evolution" Cancers 14, no. 9: 2253. https://doi.org/10.3390/cancers14092253
APA StyleSantoni, A., Santoni, M., & Arcuri, E. (2022). Chronic Cancer Pain: Opioids within Tumor Microenvironment Affect Neuroinflammation, Tumor and Pain Evolution. Cancers, 14(9), 2253. https://doi.org/10.3390/cancers14092253