
CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
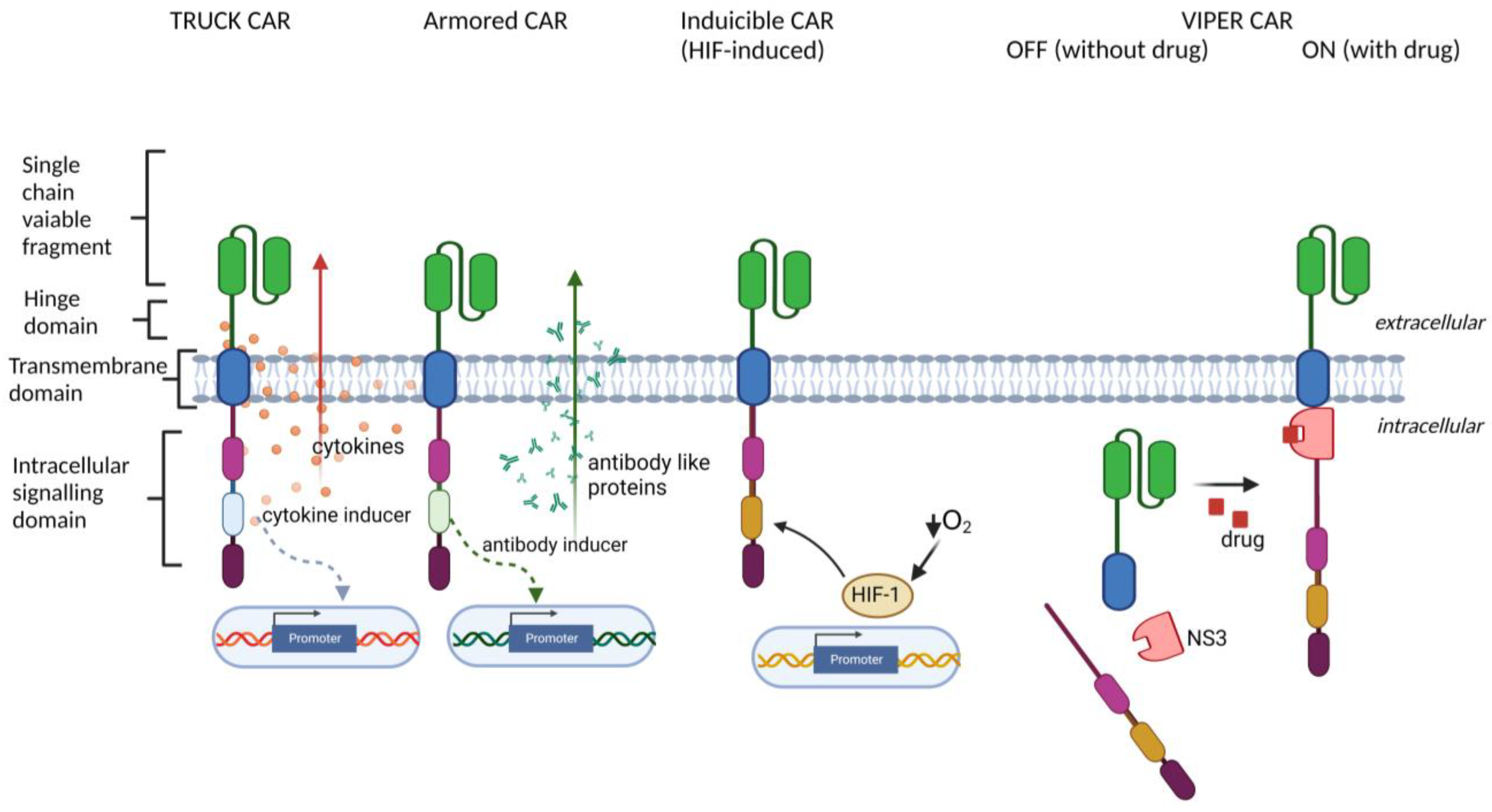
2. CAR Engineering
- Dual or multi-targeting, leading to the recognition of two different epitopes on the same target antigen (to intensify target antigen binding by CAR) or recognition of two or more different antigens (bispecific or multi-specific CARs recognizing antigenic pattern) on the surface of tumor cells (to prevent antigen escape by tumor cells; reviewed in [22]);
- Shorter extracellular fragment, such as single domain variable heavy-chain (VH), derived from camelid antibody (called nanobody; reviewed in [23]) or fully-human heavy-chain-only variable domain (FHVH) instead of conventional scFv fragment (for better expression of smaller CAR constructs on T cells and less immunogenicity induced in the patient’s organism toward foreign human protein [24]);
- switchable CAR-T cells (sCAR-T) with CAR molecules that do not directly recognize tumor antigens, but instead recognize the molecule that targets the antigen, such as the Fab fragment of an antigen-specific recombinant antibody [25,26,27,28] or the adaptor protein zipFv [29], consisting of scFv and a fragment of the leucine zipper, functioning in this case as a “switch” (for more precise control of CAR-T specificity and activity).
- TRUCK (T cells Redirected for antigen-Unrestricted Cytokine-initiated Killing) approach, based on engineering CAR-T cells to release particular transgenic cytokine upon CAR engagement, including IL-7 [32,33], IL-12 [34,35], IL-15 [36], IL-18 [37,38], IL-23 [39], and IL-33 [40]. TRUCK CARs stimulate the release of cytokines specifically at the tumor site to provide either an auto-stimulatory effect for CAR-bearing cells or activation of other immune cell types in the TME;
- Armored CAR-T cells engineered to express various proteins alongside the CAR (reviewed in [30], such as antibodies or their fragments, which are able to inhibit immune checkpoints [41,42], or dominant-negative TGF-β receptors [43,44], which are able to overcome TGF-β-induced T cell repression in the TME;
- Inducible CAR expression, regulated by specific cellular signaling and transcription factors, including synthetic Notch signaling [45,46], STAT5, AP-1, NFκB [44] (to improve the control over timing and magnitude of CAR expression), or HIF-1α [47] (to restrict the CAR expression to hypoxic areas of the solid tumors);
- ON- and OFF-controllable CAR signaling, regulated by clinically-approved drugs, including CAR regulated by lenalidomide-induced degradation [48], by dasatinib-induced downregulation [49] and by proteolytical cleavage [50,51] (to avoid CAR-T exhaustion and to obtain complete control over CAR activity by drug dosing);
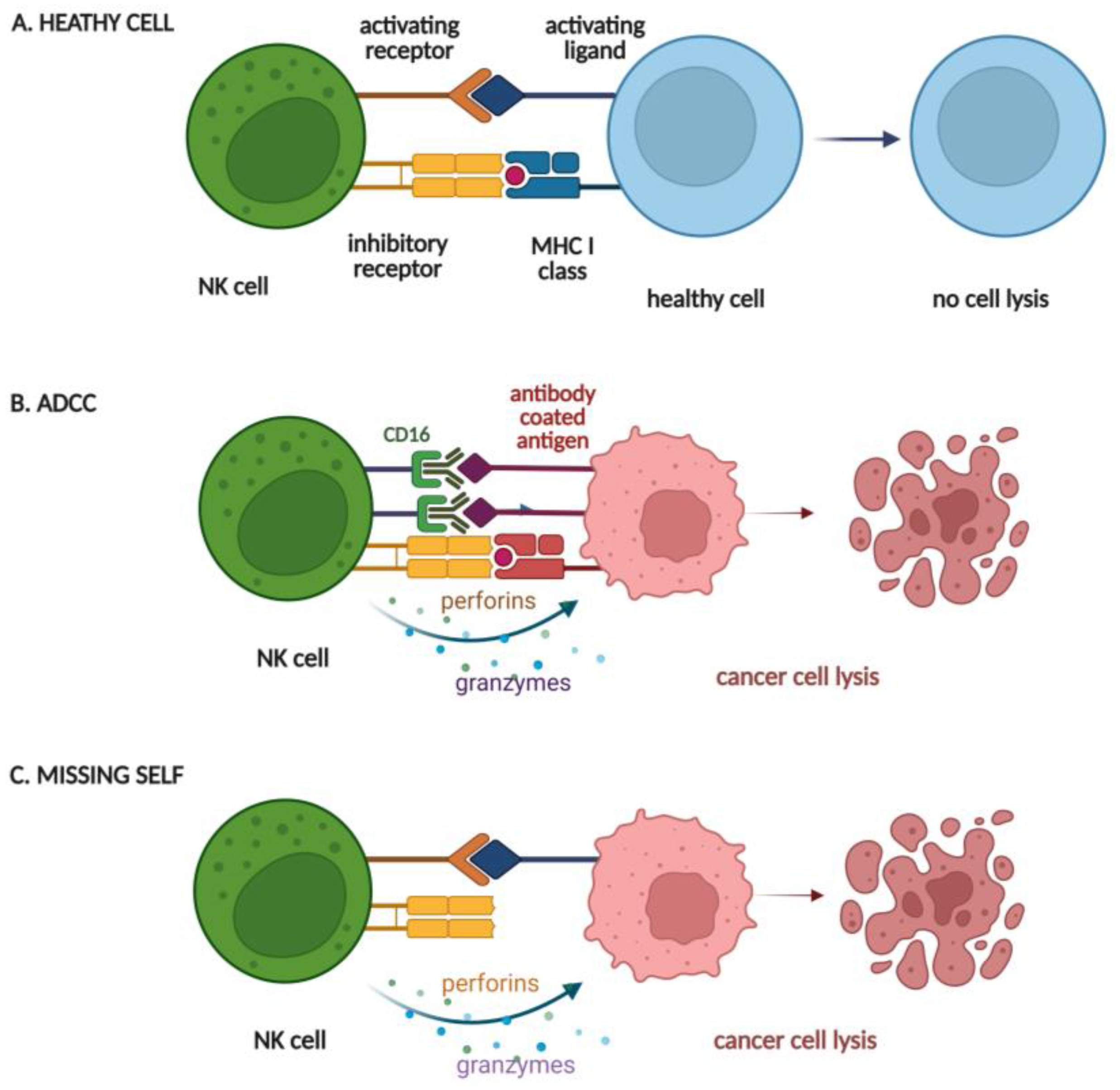
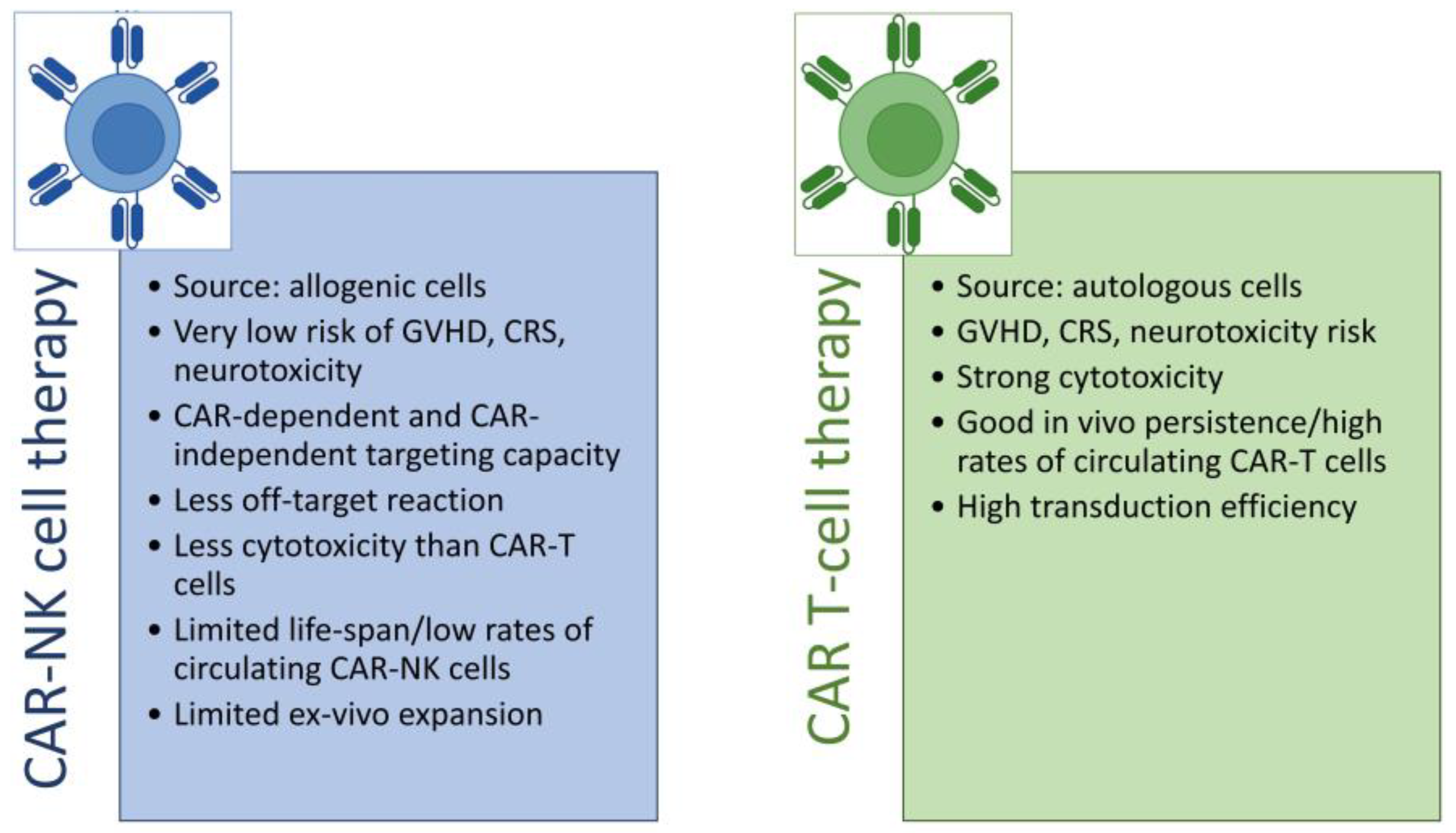
3. Advantages and Limitations of NK Cells as a CAR Platform
4. CAR-Engineered NK-92 Cell Line
5. CAR-Engineered Primary NK Cells
6. iPSCs-Derived CAR-NK Cells
7. CAR-NK as a Therapy for Solid Tumors
- Higher heterogeneity of tumor cells;
- Weak intratumoral penetration and trafficking of CAR-modified immune cells;
- Inhibition of immune cell activation by checkpoint molecules and immunosuppressive TME.
8. Clinical Trials Summary
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Prager, I.; Liesche, C.; van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandstrom, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verron, Q.; Forslund, E.; Brandt, L.; Leino, M.; Frisk, T.W.; Olofsson, P.E.; Onfelt, B. NK cells integrate signals over large areas when building immune synapses but require local stimuli for degranulation. Sci. Signal. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Grimm, E.A.; Mazumder, A.; Zhang, H.Z.; Rosenberg, S.A. Lymphokine-activated killer cell phenomenon: Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J. Exp. Med. 1982, 155, 1823–1841. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer. Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Terunuma, H.; Deng, X.; Nishino, N.; Watanabe, K. NK cell-based autologous immune enhancement therapy (AIET) for cancer. J. Stem. Cells. Regen. Med. 2013, 9, 9–13. [Google Scholar]
- Gross, G.; Gorochov, G.; Waks, T.; Eshhar, Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant. Proc. 1989, 21, 127–130. [Google Scholar]
- Decker, W.K.; da Silva, R.F.; Sanabria, M.H.; Angelo, L.S.; Guimaraes, F.; Burt, B.M.; Kheradmand, F.; Paust, S. Cancer Immunotherapy: Historical Perspective of a Clinical Revolution and Emerging Preclinical Animal Models. Front. Immunol. 2017, 8, 829. [Google Scholar] [CrossRef] [Green Version]
- Graham, C.; Hewitson, R.; Pagliuca, A.; Benjamin, R. Cancer immunotherapy with CAR-T cells—Behold the future. Clin. Med. 2018, 18, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Dirar, Q.; Russell, T.; Liu, L.; Ahn, S.; Dotti, G.; Aravamudhan, S.; Conforti, L.; Yun, Y. Activation and degranulation of CAR-T cells using engineered antigen-presenting cell surfaces. PLoS One 2020, 15, e0238819. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Wang, X.; Riviere, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncolytics 2016, 3, 16015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- First-Ever CAR T-cell Therapy Approved in U.S. Cancer Discov. 2017, 7, OF1. [CrossRef] [Green Version]
- Heymach, J.; Krilov, L.; Alberg, A.; Baxter, N.; Chang, S.M.; Corcoran, R.B.; Dale, W.; DeMichele, A.; Magid Diefenbach, C.S.; Dreicer, R.; et al. Clinical Cancer Advances 2018: Annual Report on Progress Against Cancer from the American Society of Clinical Oncology. J. Clin. Oncol. 2018, 36, 1020–1044. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.; Kim, S.; Kim, J. The 100 Most Influential Studies in Chimeric Antigen Receptor T-Cell: A Bibliometric Analysis. Front. Med. Technol. 2020, 2, 3. [Google Scholar] [CrossRef]
- Lundh, S.; Maji, S.; Melenhorst, J.J. Next-generation CAR T cells to overcome current drawbacks. Int. J. Hematol. 2021, 114, 532–543. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J., Jr.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Tahmasebi, S.; Elahi, R.; Khosh, E.; Esmaeilzadeh, A. Programmable and multi-targeted CARs: A new breakthrough in cancer CAR-T cell therapy. Clin. Transl. Oncol. 2021, 23, 1003–1019. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh Kozani, P.; Naseri, A.; Mirarefin, S.M.J.; Salem, F.; Nikbakht, M.; Evazi Bakhshi, S.; Safarzadeh Kozani, P. Nanobody-based CAR-T cells for cancer immunotherapy. Biomark. Res. 2022, 10, 24. [Google Scholar] [CrossRef]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, D.; Yang, M.H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019, 68, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.J.; Wang, X.; Wang, Z.; Zhao, L.; Li, S.; Zhang, Z.; Wei, X.; Yun, H.; Choi, S.H.; Liu, Z.; et al. Switchable CAR-T Cells Outperformed Traditional Antibody-Redirected Therapeutics Targeting Breast Cancers. ACS Synth. Biol. 2021, 10, 1176–1183. [Google Scholar] [CrossRef]
- Qi, J.; Tsuji, K.; Hymel, D.; Burke, T.R., Jr.; Hudecek, M.; Rader, C.; Peng, H. Chemically Programmable and Switchable CAR-T Therapy. Angew. Chem. Int. Ed. Engl. 2020, 59, 12178–12185. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Kuo, C.F.; Jenkins, K.; Hung, A.F.; Chang, W.C.; Park, M.; Aguilar, B.; Starr, R.; Hibbard, J.; Brown, C.; et al. Antibody-based redirection of universal Fabrack-CAR T cells selectively kill antigen bearing tumor cells. J. Immunother. Cancer 2022, 10, e003752. [Google Scholar] [CrossRef]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e1411. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biologics 2021, 15, 95–105. [Google Scholar] [CrossRef]
- Nguyen, A.; Johanning, G.; Shi, Y. Emerging Novel Combined CAR-T Cell Therapies. Cancers 2022, 14, 1403. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-zeta CAR T Cells Resist TGF-beta Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef] [Green Version]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459. [Google Scholar] [CrossRef]
- Brog, R.A.; Ferry, S.L.; Schiebout, C.T.; Messier, C.M.; Cook, W.J.; Abdullah, L.; Zou, J.; Kumar, P.; Sentman, C.L.; Frost, H.R.; et al. Superkine IL-2 and IL-33 Armored CAR T Cells Reshape the Tumor Microenvironment and Reduce Growth of Multiple Solid Tumors. Cancer. Immunol. Res. 2022, 10, 962–977. [Google Scholar] [CrossRef]
- Suarez, E.R.; Chang de, K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Narayan, V.; Barber-Rotenberg, J.S.; Jung, I.Y.; Lacey, S.F.; Rech, A.J.; Davis, M.M.; Hwang, W.T.; Lal, P.; Carpenter, E.L.; Maude, S.L.; et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat. Med. 2022, 28, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.; Xiong, Y.; Hu, P.; Wu, D.; Alabanza, L.; Orentas, R.J.; Dropulic, B.; Schneider, D. Self-driving armored CAR-T cells overcome a suppressive milieu and eradicate CD19(+) Raji lymphoma in preclinical models. Mol. Ther. 2021, 29, 2691–2706. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e488. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [Green Version]
- Kosti, P.; Opzoomer, J.W.; Larios-Martinez, K.I.; Henley-Smith, R.; Scudamore, C.L.; Okesola, M.; Taher, M.Y.M.; Davies, D.M.; Muliaditan, T.; Larcombe-Young, D.; et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep. Med. 2021, 2, 100227. [Google Scholar] [CrossRef]
- Jan, M.; Scarfo, I.; Larson, R.C.; Walker, A.; Schmidts, A.; Guirguis, A.A.; Gasser, J.A.; Slabicki, M.; Bouffard, A.A.; Castano, A.P.; et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci. Transl. Med. 2021, 13, eabb629. [Google Scholar] [CrossRef]
- Weber, E.W.; Parker, K.R.; Sotillo, E.; Lynn, R.C.; Anbunathan, H.; Lattin, J.; Good, Z.; Belk, J.A.; Daniel, B.; Klysz, D.; et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021, 372, 239. [Google Scholar] [CrossRef]
- Labanieh, L.; Majzner, R.G.; Klysz, D.; Sotillo, E.; Fisher, C.J.; Vilches-Moure, J.G.; Pacheco, K.Z.B.; Malipatlolla, M.; Xu, P.; Hui, J.H.; et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 2022, 185, 1745–1763.e1722. [Google Scholar] [CrossRef]
- Li, H.S.; Wong, N.M.; Tague, E.; Ngo, J.T.; Khalil, A.S.; Wong, W.W. High-performance multiplex drug-gated CAR circuits. Cancer Cell 2022, 40, 1294–1305.e4. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.K.; Lerman, B.J.; Barnes, J.I.; Boursiquot, B.C.; Tan, Y.J.; Robinson, A.Q.L.; Davis, K.L.; Owens, D.K.; Goldhaber-Fiebert, J.D. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Relapsed or Refractory Pediatric B-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 3192–3202. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Cheng, J.; Mu, W.; Zhou, J.; Zhu, L. Advances in Universal CAR-T Cell Therapy. Front. Immunol. 2021, 12, 744823. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Rezvani, K. Outlook for New CAR-Based Therapies with a Focus on CAR NK Cells: What Lies Beyond CAR-Engineered T Cells in the Race against Cancer. Cancer Discov. 2021, 11, 45–58. [Google Scholar] [CrossRef]
- Goldenson, B.H.; Hor, P.; Kaufman, D.S. iPSC-Derived Natural Killer Cell Therapies—Expansion and Targeting. Front. Immunol. 2022, 13, 841107. [Google Scholar] [CrossRef]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Rizwan, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2022, 22, 85–96. [Google Scholar] [CrossRef]
- Habib, S.; Tariq, S.M.; Tariq, M. Chimeric Antigen Receptor-Natural Killer Cells: The Future of Cancer Immunotherapy. Ochsner. J. 2019, 19, 186–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Leivas, A.; Valeri, A.; Cordoba, L.; Garcia-Ortiz, A.; Ortiz, A.; Sanchez-Vega, L.; Grana-Castro, O.; Fernandez, L.; Carreno-Tarragona, G.; Perez, M.; et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 146. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083–1089. [Google Scholar]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef]
- Geller, M.A.; Miller, J.S. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy 2011, 3, 1445–1459. [Google Scholar] [CrossRef] [Green Version]
- West, W.H.; Tauer, K.W.; Yannelli, J.R.; Marshall, G.D.; Orr, D.W.; Thurman, G.B.; Oldham, R.K. Constant-infusion recombinant interleukin-2 in adoptive immunotherapy of advanced cancer. N. Engl. J. Med. 1987, 316, 898–905. [Google Scholar] [CrossRef]
- Tornroos, H.; Hagerstrand, H.; Lindqvist, C. Culturing the Human Natural Killer Cell Line NK-92 in Interleukin-2 and Interleukin-15—Implications for Clinical Trials. Anticancer Res. 2019, 39, 107–112. [Google Scholar] [CrossRef]
- Morgan, M.A.; Buning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf”CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef]
- Carlsten, M.; Childs, R.W. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front. Immunol. 2015, 6, 266. [Google Scholar] [CrossRef] [Green Version]
- Guven, H.; Konstantinidis, K.V.; Alici, E.; Aints, A.; Abedi-Valugerdi, M.; Christensson, B.; Ljunggren, H.G.; Dilber, M.S. Efficient gene transfer into primary human natural killer cells by retroviral transduction. Exp. Hematol. 2005, 33, 1320–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streltsova, M.A.; Barsov, E.; Erokhina, S.A.; Kovalenko, E.I. Retroviral gene transfer into primary human NK cells activated by IL-2 and K562 feeder cells expressing membrane-bound IL-21. J. Immunol. Methods 2017, 450, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Badeti, S.; Kim, J.K.; Liu, D. Natural Killer (NK) and CAR-NK Cell Expansion Method using Membrane Bound-IL-21-Modified B Cell Line. J. Vis. Exp. 2022. [Google Scholar] [CrossRef]
- Papayannakos, C.; Daniel, R. Understanding lentiviral vector chromatin targeting: Working to reduce insertional mutagenic potential for gene therapy. Gene Ther. 2013, 20, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Portillo, A.L.; Hogg, R.; Ashkar, A.A. Production of human CAR-NK cells with lentiviral vectors and functional assessment in vitro. STAR Protoc. 2021, 2, 100956. [Google Scholar] [CrossRef]
- Bari, R.; Granzin, M.; Tsang, K.S.; Roy, A.; Krueger, W.; Orentas, R.; Schneider, D.; Pfeifer, R.; Moeker, N.; Verhoeyen, E.; et al. A Distinct Subset of Highly Proliferative and Lentiviral Vector (LV)-Transducible NK Cells Define a Readily Engineered Subset for Adoptive Cellular Therapy. Front. Immunol. 2019, 10, 2001. [Google Scholar] [CrossRef] [Green Version]
- Tomas, H.A.; Mestre, D.A.; Rodrigues, A.F.; Guerreiro, M.R.; Carrondo, M.J.T.; Coroadinha, A.S. Improved GaLV-TR Glycoproteins to Pseudotype Lentiviral Vectors: Impact of Viral Protease Activity in the Production of LV Pseudotypes. Mol. Ther. Methods Clin. Dev. 2019, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Albinger, N.; Pfeifer, R.; Nitsche, M.; Mertlitz, S.; Campe, J.; Stein, K.; Kreyenberg, H.; Schubert, R.; Quadflieg, M.; Schneider, D.; et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022, 12, 61. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Urena-Bailen, G.; Dobrowolski, J.M.; Hou, Y.; Dirlam, A.; Roig-Merino, A.; Schleicher, S.; Atar, D.; Seitz, C.; Feucht, J.; Antony, J.S.; et al. Preclinical Evaluation of CRISPR-Edited CAR-NK-92 Cells for Off-the-Shelf Treatment of AML and B-ALL. Int. J. Mol. Sci. 2022, 23, 12828. [Google Scholar] [CrossRef]
- Huang, R.S.; Shih, H.A.; Lai, M.C.; Chang, Y.J.; Lin, S. Enhanced NK-92 Cytotoxicity by CRISPR Genome Engineering Using Cas9 Ribonucleoproteins. Front. Immunol. 2020, 11, 1008. [Google Scholar] [CrossRef] [PubMed]
- Elmas, E.; Saljoughian, N.; de Souza Fernandes Pereira, M.; Tullius, B.P.; Sorathia, K.; Nakkula, R.J.; Lee, D.A.; Naeimi Kararoudi, M. CRISPR Gene Editing of Human Primary NK and T Cells for Cancer Immunotherapy. Front. Oncol. 2022, 12, 834002. [Google Scholar] [CrossRef] [PubMed]
- Naeimi Kararoudi, M.; Likhite, S.; Elmas, E.; Yamamoto, K.; Schwartz, M.; Sorathia, K.; de Souza Fernandes Pereira, M.; Sezgin, Y.; Devine, R.D.; Lyberger, J.M.; et al. Optimization and validation of CAR transduction into human primary NK cells using CRISPR and AAV. Cell Rep. Methods 2022, 2, 100236. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Pyykko, I. Size matters: Versatile use of PiggyBac transposons as a genetic manipulation tool. Mol. Cell Biochem. 2011, 354, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192 e185. [Google Scholar] [CrossRef] [Green Version]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017, 31, 186–194. [Google Scholar] [CrossRef]
- Lowe, E.; Truscott, L.C.; De Oliveira, S.N. In Vitro Generation of Human NK Cells Expressing Chimeric Antigen Receptor Through Differentiation of Gene-Modified Hematopoietic Stem Cells. Methods Mol. Biol. 2016, 1441, 241–251. [Google Scholar]
- Sabbah, M.; Jondreville, L.; Lacan, C.; Norol, F.; Vieillard, V.; Roos-Weil, D.; Nguyen, S. CAR-NK Cells: A Chimeric Hope or a Promising Therapy? Cancers 2022, 14, 3839. [Google Scholar] [CrossRef]
- Yan, Y.; Steinherz, P.; Klingemann, H.G.; Dennig, D.; Childs, B.H.; McGuirk, J.; O’Reilly, R.J. Antileukemia activity of a natural killer cell line against human leukemias. Clin. Cancer Res. 1998, 4, 2859–2868. [Google Scholar]
- Gong, J.H.; Maki, G.; Klingemann, H.G. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia 1994, 8, 652–658. [Google Scholar]
- Maki, G.; Klingemann, H.G.; Martinson, J.A.; Tam, Y.K. Factors regulating the cytotoxic activity of the human natural killer cell line, NK-92. J. Hematother. Stem Cell Res. 2001, 10, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Suck, G.; Odendahl, M.; Nowakowska, P.; Seidl, C.; Wels, W.S.; Klingemann, H.G.; Tonn, T. NK-92: An ‘off-the-shelf therapeutic’ for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef] [Green Version]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T.; et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xu, Y.; Mou, J.; Tang, K.; Fu, X.; Li, Y.; Xing, Y.; Rao, Q.; Xing, H.; Tian, Z.; et al. Irradiated chimeric antigen receptor engineered NK-92MI cells show effective cytotoxicity against CD19(+) malignancy in a mouse model. Cytotherapy 2020, 22, 552–562. [Google Scholar] [CrossRef]
- Schonfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bonig, H.; Kohl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genssler, S.; Schonfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, B.A.; Law, A.D.; Routy, B.; denHollander, N.; Gupta, V.; Wang, X.H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varani, M.; Auletta, S.; Signore, A.; Galli, F. State of the Art of Natural Killer Cell Imaging: A Systematic Review. Cancers 2019, 11, 967. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Li, X.J.; Kalimuthu, S.; Gangadaran, P.; Lee, H.W.; Oh, J.M.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; et al. Natural Killer Cell (NK-92MI)-Based Therapy for Pulmonary Metastasis of Anaplastic Thyroid Cancer in a Nude Mouse Model. Front. Immunol. 2017, 8, 816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangadaran, P.; Ahn, B.C. Molecular Imaging: A Useful Tool for the Development of Natural Killer Cell-Based Immunotherapies. Front. Immunol. 2017, 8, 1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottobrini, L.; Martelli, C.; Trabattoni, D.L.; Clerici, M.; Lucignani, G. In vivo imaging of immune cell trafficking in cancer. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 949–968. [Google Scholar] [CrossRef]
- Matera, L.; Galetto, A.; Bello, M.; Baiocco, C.; Chiappino, I.; Castellano, G.; Stacchini, A.; Satolli, M.A.; Mele, M.; Sandrucci, S.; et al. In vivo migration of labeled autologous natural killer cells to liver metastases in patients with colon carcinoma. J. Transl. Med. 2006, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Shamalov, K.; Meir, R.; Motiei, M.; Popovtzer, R.; Cohen, C.J. Noninvasive Tracking of Natural Killer Cells Using Gold Nanoparticles. ACS Omega 2021, 6, 28507–28514. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Paul, P.; Pedini, P.; Lyonnet, L.; Di Cristofaro, J.; Loundou, A.; Pelardy, M.; Basire, A.; Dignat-George, F.; Chiaroni, J.; Thomas, P.; et al. FCGR3A and FCGR2A Genotypes Differentially Impact Allograft Rejection and Patients’ Survival After Lung Transplant. Front. Immunol. 2019, 10, 1208. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Morillon, Y.M., 2nd; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X.; et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219–56232. [Google Scholar] [CrossRef] [Green Version]
- Pinz, K.G.; Yakaboski, E.; Jares, A.; Liu, H.; Firor, A.E.; Chen, K.H.; Wada, M.; Salman, H.; Tse, W.; Hagag, N.; et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget 2017, 8, 112783–112796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X.; et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 49. [Google Scholar] [CrossRef]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019, 9, 64–78. [Google Scholar]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef]
- Rafiq, S.; Purdon, T.; Schultz, L.; Klingemann, H.; Brentjens, R. NK-92 cells engineered with anti-CD33 chimeric antigen receptors (CAR) for the treatment of Acute Myeloid Leukemia (AML). Cytotherapy 2015, 17, S23. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W.; et al. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Daldrup-Link, H.E.; Meier, R.; Rudelius, M.; Piontek, G.; Piert, M.; Metz, S.; Settles, M.; Uherek, C.; Wels, W.; Schlegel, J.; et al. In vivo tracking of genetically engineered, anti-HER2/neu directed natural killer cells to HER2/neu positive mammary tumors with magnetic resonance imaging. Eur. Radiol. 2005, 15, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruschinski, A.; Moosmann, A.; Poschke, I.; Norell, H.; Chmielewski, M.; Seliger, B.; Kiessling, R.; Blankenstein, T.; Abken, H.; Charo, J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17481–17486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanski, A.; Uherek, C.; Bug, G.; Muller, T.; Rossig, C.; Kampfmann, M.; Krossok, N.; Hoelzer, D.; Seifried, E.; Wels, W.; et al. Re-Targeting of an NK Cell Line (NK92) with Specificity for CD19 Efficiently Kills Human B-Precursor Leukemia Cells. Blood 2004, 104, 2747. [Google Scholar] [CrossRef]
- Muller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Mathe, D.; Hegedus, N.; et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [Green Version]
- Luanpitpong, S.; Poohadsuan, J.; Klaihmon, P.; Issaragrisil, S. Selective Cytotoxicity of Single and Dual Anti-CD19 and Anti-CD138 Chimeric Antigen Receptor-Natural Killer Cells against Hematologic Malignancies. J. Immunol. Res. 2021, 2021, 5562630. [Google Scholar] [CrossRef]
- Rudek, L.S.; Zimmermann, K.; Galla, M.; Meyer, J.; Kuehle, J.; Stamopoulou, A.; Brand, D.; Sandalcioglu, I.E.; Neyazi, B.; Moritz, T.; et al. Generation of an NFkappaB-Driven Alpharetroviral “All-in-One” Vector Construct as a Potent Tool for CAR NK Cell Therapy. Front. Immunol. 2021, 12, 751138. [Google Scholar] [CrossRef]
- Kloss, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S.; et al. Optimization of Human NK Cell Manufacturing: Fully Automated Separation, Improved Ex Vivo Expansion Using IL-21 with Autologous Feeder Cells, and Generation of Anti-CD123-CAR-Expressing Effector Cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Luevano, M.; Daryouzeh, M.; Alnabhan, R.; Querol, S.; Khakoo, S.; Madrigal, A.; Saudemont, A. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum. Immunol. 2012, 73, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Sarvaria, A.; Jawdat, D.; Madrigal, J.A.; Saudemont, A. Umbilical Cord Blood Natural Killer Cells, Their Characteristics, and Potential Clinical Applications. Front. Immunol. 2017, 8, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hejazi, M.; Zhang, C.; Bennstein, S.B.; Balz, V.; Reusing, S.B.; Quadflieg, M.; Hoerster, K.; Heinrichs, S.; Hanenberg, H.; Oberbeck, S.; et al. CD33 Delineates Two Functionally Distinct NK Cell Populations Divergent in Cytokine Production and Antibody-Mediated Cellular Cytotoxicity. Front. Immunol. 2021, 12, 798087. [Google Scholar] [CrossRef] [PubMed]
- Topfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Muller, N.; Lindemann, D.; Bachmann, M.; Fussel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezvani, K.; Rouce, R.; Liu, E.; Shpall, E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017, 25, 1769–1781. [Google Scholar] [CrossRef]
- Passweg, J.R.; Tichelli, A.; Meyer-Monard, S.; Heim, D.; Stern, M.; Kuhne, T.; Favre, G.; Gratwohl, A. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia 2004, 18, 1835–1838. [Google Scholar] [CrossRef]
- Cany, J.; van der Waart, A.B.; Spanholtz, J.; Tordoir, M.; Jansen, J.H.; van der Voort, R.; Schaap, N.M.; Dolstra, H. Combined IL-15 and IL-12 drives the generation of CD34(+)-derived natural killer cells with superior maturation and alloreactivity potential following adoptive transfer. Oncoimmunology 2015, 4, e1017701. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Valamehr, B.; Tsutsui, H.; Ho, C.M.; Wu, H. Developing defined culture systems for human pluripotent stem cells. Regen. Med. 2011, 6, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.A.; Kaufman, D.S. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells 2016, 34, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Canedo, A.; Bonamino, M.H. Retroviral vectors and transposons for stable gene therapy: Advances, current challenges and perspectives. J. Transl. Med. 2016, 14, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dashtban, M.; Panchalingam, K.M.; Shafa, M.; Ahmadian Baghbaderani, B. Addressing Manufacturing Challenges for Commercialization of iPSC-Based Therapies. Methods Mol. Biol. 2021, 2286, 179–198. [Google Scholar] [PubMed]
- Valamehr, B.; Robinson, M.; Abujarour, R.; Rezner, B.; Vranceanu, F.; Le, T.; Medcalf, A.; Lee, T.T.; Fitch, M.; Robbins, D.; et al. Platform for induction and maintenance of transgene-free hiPSCs resembling ground state pluripotent stem cells. Stem Cell Reports 2014, 2, 366–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nit, K.; Tyszka-Czochara, M.; Bobis-Wozowicz, S. Oxygen as a Master Regulator of Human Pluripotent Stem Cell Function and Metabolism. J. Pers. Med. 2021, 11, 905. [Google Scholar] [CrossRef]
- Dakhore, S.; Nayer, B.; Hasegawa, K. Human Pluripotent Stem Cell Culture: Current Status, Challenges, and Advancement. Stem Cells Int. 2018, 2018, 7396905. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, T.; Arita, M.; Kuroda, H.; Suzuki, T.; Kawamata, S. Improving the differentiation potential of pluripotent stem cells by optimizing culture conditions. Sci. Rep. 2022, 12, 14147. [Google Scholar] [CrossRef]
- Ni, Y.; Zhao, Y.; Warren, L.; Higginbotham, J.; Wang, J. cGMP Generation of Human Induced Pluripotent Stem Cells with Messenger RNA. Curr. Protoc. Stem. Cell Biol. 2016, 39, 4A.6.1–4A.6.25. [Google Scholar] [CrossRef]
- Rivera, T.; Zhao, Y.; Ni, Y.; Wang, J. Human-Induced Pluripotent Stem Cell Culture Methods Under cGMP Conditions. Curr. Protoc. Stem. Cell Biol. 2020, 54, e117. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, P.; Kim, S.I. iPSC-Derived Natural Killer Cells for Cancer Immunotherapy. Mol. Cells 2021, 44, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Kailayangiri, S.; Altvater, B.; Wiebel, M.; Jamitzky, S.; Rossig, C. Overcoming Heterogeneity of Antigen Expression for Effective CAR T Cell Targeting of Cancers. Cancers 2020, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.I.; Grossenbacher, S.K.; Murphy, W.J.; Canter, R.J. Targeting Cancer Stem Cells with Natural Killer Cell Immunotherapy. Expert Opin. Biol. Ther. 2017, 17, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, T.; Wang, G.; He, S.; Liu, Q.; Sun, J.; Wang, Y. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 2016, 300, 41–45. [Google Scholar] [CrossRef]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [Green Version]
- Ferrari de Andrade, L.; Kumar, S.; Luoma, A.M.; Ito, Y.; Alves da Silva, P.H.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Wucherpfennig, K.W. Inhibition of MICA and MICB Shedding Elicits NK-Cell-Mediated Immunity against Tumors Resistant to Cytotoxic T Cells. Cancer Immunol. Res. 2020, 8, 769–780. [Google Scholar] [CrossRef]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef]
- Muller, N.; Michen, S.; Tietze, S.; Topfer, K.; Schulte, A.; Lamszus, K.; Schmitz, M.; Schackert, G.; Pastan, I.; Temme, A. Engineering NK Cells Modified with an EGFRvIII-specific Chimeric Antigen Receptor to Overexpress CXCR4 Improves Immunotherapy of CXCL12/SDF-1alpha-secreting Glioblastoma. J. Immunother. 2015, 38, 197–210. [Google Scholar] [CrossRef] [Green Version]
- Ng, Y.Y.; Tay, J.C.K.; Wang, S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice with Peritoneal Xenografts. Mol. Ther. Oncolytics 2020, 16, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, X.; Jin, T.; Tian, Y.; Dai, C.; Widarma, C.; Song, R.; Xu, F. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal Transduct. Target. Ther. 2020, 5, 250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Lu, C.; Guo, C.; Chen, H.; Zhang, H.; Zhi, L.; Lv, T.; Li, M.; Niu, Z.; Lu, P.; Zhu, W. A novel chimeric PD1-NKG2D-41BB receptor enhances antitumor activity of NK92 cells against human lung cancer H1299 cells by triggering pyroptosis. Mol. Immunol. 2020, 122, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Jimenez-Cortegana, C.; Tay, A.H.M.; Wickstrom, S.; Galluzzi, L.; Lundqvist, A. NK cells and solid tumors: Therapeutic potential and persisting obstacles. Mol. Cancer 2022, 21, 206. [Google Scholar] [CrossRef] [PubMed]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Chu, J.; Keung Chan, W.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genssler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci. Rep. 2020, 10, 2815. [Google Scholar] [CrossRef] [Green Version]
- Sahm, C.; Schonfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef]
- Cao, B.; Liu, M.; Wang, L.; Liang, B.; Feng, Y.; Chen, X.; Shi, Y.; Zhang, J.; Ye, X.; Tian, Y.; et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 524, 96–102. [Google Scholar] [CrossRef]
- Klapdor, R.; Wang, S.; Morgan, M.; Dork, T.; Hacker, U.; Hillemanns, P.; Buning, H.; Schambach, A. Characterization of a Novel Third-Generation Anti-CD24-CAR against Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, N.; Haopeng, P.; Gong, J.U.; Lu, J.; Chen, Z.; Zheng, Y.; Wang, Z.; Sun, Y.U.; Yang, Z.; Hoffman, R.M.; et al. Robo1-specific CAR-NK Immunotherapy Enhances Efficacy of (125)I Seed Brachytherapy in an Orthotopic Mouse Model of Human Pancreatic Carcinoma. Anticancer. Res. 2019, 39, 5919–5925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Yang, N.; Li, H.; Wang, Z. Robo1-specific chimeric antigen receptor natural killer cell therapy for pancreatic ductal adenocarcinoma with liver metastasis. J. Cancer Res. Ther. 2020, 16, 393–396. [Google Scholar]
- Ao, X.; Yang, Y.; Li, W.; Tan, Y.; Guo, W.; Ao, L.; He, X.; Wu, X.; Xia, J.; Xu, X.; et al. Anti-alphaFR CAR-engineered NK-92 Cells Display Potent Cytotoxicity Against alphaFR-positive Ovarian Cancer. J. Immunother. 2019, 42, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.E.; Ju, A.; Choi, H.W.; Kim, J.C.; Kim, E.E.; Kim, T.S.; Kang, H.J.; Kim, S.Y.; Jang, J.Y.; Ku, J.L.; et al. Rationally designed redirection of natural killer cells anchoring a cytotoxic ligand for pancreatic cancer treatment. J. Control Release 2020, 326, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, Z.Z.; Zhou, M.L.; Lin, J.W.; Chen, X.M.; Li, Z.; Gao, W.B.; Yu, Z.D.; Liu, T. Development of c-MET-specific chimeric antigen receptor-engineered natural killer cells with cytotoxic effects on human liver cancer HepG2 cells. Mol. Med. Rep. 2019, 20, 2823–2831. [Google Scholar] [CrossRef] [Green Version]
- Subrakova, V.G.; Kulemzin, S.V.; Belovezhets, T.N.; Chikaev, A.N.; Chikaev, N.A.; Koval, O.A.; Gorchakov, A.A.; Taranin, A.V. shp-2 gene knockout upregulates CAR-driven cytotoxicity of YT NK cells. Vavilovskii Zhurnal Genet. Sel. 2020, 24, 80–86. [Google Scholar] [CrossRef]
- Wrona, E.; Borowiec, M.; Potemski, P. CAR-NK Cells in the Treatment of Solid Tumors. Int. J. Mol. Sci. 2021, 22, 5899. [Google Scholar] [CrossRef]
- Geller, M.A.; Cooley, S.; Judson, P.L.; Ghebre, R.; Carson, L.F.; Argenta, P.A.; Jonson, A.L.; Panoskaltsis-Mortari, A.; Curtsinger, J.; McKenna, D.; et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy 2011, 13, 98–107. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Lim, O.; Kim, T.M.; Ahn, Y.O.; Choi, H.; Chung, H.; Min, B.; Her, J.H.; Cho, S.Y.; Keam, B.; et al. Phase I Study of Random Healthy Donor-Derived Allogeneic Natural Killer Cell Therapy in Patients with Malignant Lymphoma or Advanced Solid Tumors. Cancer Immunol. Res. 2016, 4, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Zhang, J. Reformation in chimeric antigen receptor based cancer immunotherapy: Redirecting natural killer cell. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wu, L.; Yin, L.; Shi, H.; Gu, Y.; Xing, N. Combined treatment with anti-PSMA CAR NK-92 cell and anti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin. Transl. Med. 2022, 12, e901. [Google Scholar] [CrossRef] [PubMed]
- Sengsayadeth, S.; Savani, B.N.; Oluwole, O.; Dholaria, B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. EJHaem 2022, 3, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, A.W.; Florian, S.; Printz, D.; Sotlar, K.; Krauth, M.T.; Fritsch, G.; Schernthaner, G.H.; Wacheck, V.; Selzer, E.; Sperr, W.R.; et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur. J. Clin. Investig. 2007, 37, 73–82. [Google Scholar] [CrossRef]
- Walter, R.B. The role of CD33 as therapeutic target in acute myeloid leukemia. Expert Opin. Ther. Targets 2014, 18, 715–718. [Google Scholar] [CrossRef]
- Jiang, Y.P.; Liu, B.Y.; Zheng, Q.; Panuganti, S.; Chen, R.; Zhu, J.; Mishra, M.; Huang, J.; Dao-Pick, T.; Roy, S.; et al. CLT030, a leukemic stem cell-targeting CLL1 antibody-drug conjugate for treatment of acute myeloid leukemia. Blood Adv. 2018, 2, 1738–1749. [Google Scholar] [CrossRef] [Green Version]
- Gurney, M.; O’Reilly, E.; Corcoran, S.; Brophy, S.; Hardwicke, D.; Krawczyk, J.; Hermanson, D.; Childs, R.; Szegezdi, E.; O’Dwyer, M.E. Tc Buster Transposon Engineered CLL-1 CAR-NK Cells Efficiently Target Acute Myeloid Leukemia. Blood 2021, 138, 1725. [Google Scholar] [CrossRef]
- Liu, F.; Cao, Y.; Pinz, K.; Ma, Y.; Wada, M.; Chen, K.; Ma, G.; Shen, J.; Tse, C.; Su, Y.; et al. First-in-Human CLL1-CD33 Compound CAR T Cell Therapy Induces Complete Remission in Patients with Refractory Acute Myeloid Leukemia: Update on Phase 1 Clinical Trial. Blood 2018, 132, 901. [Google Scholar] [CrossRef]
- Bras, A.E.; de Haas, V.; van Stigt, A.; Jongen-Lavrencic, M.; Beverloo, H.B.; Te Marvelde, J.G.; Zwaan, C.M.; van Dongen, J.J.M.; Leusen, J.H.W.; van der Velden, V.H.J. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytom. B Clin. Cytom. 2019, 96, 134–142. [Google Scholar] [CrossRef]
- Christodoulou, I.; Ho, W.J.; Marple, A.; Ravich, J.W.; Tam, A.; Rahnama, R.; Fearnow, A.; Rietberg, C.; Yanik, S.; Solomou, E.E.; et al. Engineering CAR-NK cells to secrete IL-15 sustains their anti-AML functionality but is associated with systemic toxicities. J. Immunother. Cancer 2021, 9, e003894. [Google Scholar] [CrossRef]
- Caruso, S.; De Angelis, B.; Del Bufalo, F.; Ciccone, R.; Donsante, S.; Volpe, G.; Manni, S.; Guercio, M.; Pezzella, M.; Iaffaldano, L.; et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J. Hematol. Oncol. 2022, 15, 163. [Google Scholar] [CrossRef] [PubMed]
- Colamartino, A.B.L.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Romero, H.; Selleri, S.; Beland, K.; Guiot, M.; et al. Efficient and Robust NK-Cell Transduction With Baboon Envelope Pseudotyped Lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szoor, A.; Velasquez, M.; Bonifant, C.; Vaidya, A.; Brunetti, L.; Gundry, M.; Parihar, R.; Goodell, M.; Gottschalk, S. Two-pronged Cell Therapy: Engineering NK cells to target CD22 and redirect bystander T cells to CD19 for the adoptive immunotherapy of B-cell malignancies. J. Immunol. 2017, 198, 6. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Du, Z.; Zhang, X.; Chng, W.J.; Wang, S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2022, 29, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Roex, G.; Campillo-Davo, D.; Flumens, D.; Shaw, P.A.G.; Krekelbergh, L.; De Reu, H.; Berneman, Z.N.; Lion, E.; Anguille, S. Two for one: Targeting BCMA and CD19 in B-cell malignancies with off-the-shelf dual-CAR NK-92 cells. J. Transl. Med. 2022, 20, 124. [Google Scholar] [CrossRef]
- Goebeler, M.E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients With Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results From a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Jiang, Z.; Qin, L.; Tang, Y.; Liao, R.; Shi, J.; He, B.; Li, S.; Zheng, D.; Cui, Y.; Wu, Q.; et al. Human induced-T-to-natural killer cells have potent anti-tumour activities. Biomark. Res. 2022, 10, 13. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Title | Trial Number (Number of Participants); Phase | Cancer Type | Treatment; Dosage † | Location |
|---|---|---|---|---|
| Intracranial Injection of NK-92/5.28.z Cells in Combination With Intravenous Ezabenlimab in Patients With Recurrent HER2-positive Glioblastoma * | NCT03383978 (42); 1 | Glioblastoma | CAR-NK-92/5.28.z Doses: 1 × 107–1 × 108 CAR-NK cells Ezabenlimab: 240 mg The standard therapy for glioblastoma (radiotherapy and alkylating chemotherapy) allowed until two weeks and Temozolomide allowed up to 48 h prior to the study | Germany |
| Clinical Research of Adoptive BCMA CAR-NK Cells on Relapse/Refractory MM + | NCT03940833 (20); 1,2 | Multiple Myeloma | BCMA CAR-NK 92 cells | China |
| CAR-pNK Cell Immunotherapy in CD7 Positive Leukemia and Lymphoma * | NCT02742727 (10); 1,2 | Myeloid Leukemia Acute Precursor T-Cell Lymphoblastic Leukemia-Lymphoma T-cell Prolymphocytic Leukemia | anti-CD7 CAR-pNK cells; The allogeneic NK-92 cell line engineered to contain anti-CD7 attached to TCRzeta, CD28 and 4-1BB signaling domains | China |
| CAR-pNK Cell Immunotherapy for Relapsed/Refractory CD33+ AML+ | NCT02944162 (10); 1,2 | Myeloid Leukemia Acute | CD33 CAR-NK NK92cells; chimeric antigen receptor NK92 cells transduced with the anti-CD33 vector anti-CD33 CAR-NK (coupled with CD28, CD137 and CD3 zeta signaling domains) | China |
| NKG2D CAR-NK Cell Therapy in Patients With Relapsed or Refractory Acute Myeloid Leukemia * | NCT05247957 (9); 1 | Myeloid Leukemia Relapsed/Refractory Acute | NKG2D ligand-specific umbilical cord blood CAR-NK cells Doses: 2 × 106/kg, 6 × 106/kg, 18 × 106/kg CAR-NK cells, Preconditioning: standard chemotherapy | China |
| NKG2D-CAR-NK92 Cells Immunotherapy for Solid Tumors * | NCT05528341 (20); 1 | Solid Tumors Relapsed/Refractory | NKG2D-CAR-NK92 cells; CAR-NK92 cells targeting NKG2D ligands, Doses: 0.5 × 106, 2 × 106/kg | China |
| NKG2D CAR-NK Cell Therapy in Patients With Refractory Metastatic Colorectal Cancer * | NCT05213195 (38); 1 | Refractory Metastatic Colorectal Cancer | NKG2DL-CAR-NK cells; CAR-NK cells targeting NKG2D ligands, | China |
| Pilot Study of NKG2D-Ligand Targeted CAR-NK Cells in Patients With Metastatic Solid Tumors + | NCT03415100 (30); 1 | Solid Tumors | NKG2DL-CAR-NK cells; CAR-NK cells targeting NKG2D ligands | China |
| NKX101, Intravenous Allogeneic CAR NK Cells, in Adults With AML or MDS * | NCT04623944 (90); 1 | AML, AML Relapsed/Refractory, Adult MDS | NKX101 allogeneic CAR NK cells targeting NKG2D ligands NK cells from haplo-matched related or unrelated donor Doses: 1 × 108 CAR-NK cells (2 × 106/kg for weight < 50 kg), 1.5 × 108 CAR-NK cells (3 × 106/kg for weight < 50 kg) Lymphodepletion: fludarabine/cyclophosphamide or fludarabine/cytarabine (ara-C) | United States |
| NKX019, Intravenous Allogeneic Chimeric Antigen Receptor Natural Killer Cells (CAR NK), in Adults With B-cell Cancers * | NCT05020678 (60); 1 | Lymphoma, Non-Hodgkin B-cell Acute Lymphoblastic Leukemia, Large B-cell Lymphoma | NKX019 allogeneic CAR NK product targeting CD19 on cells. Doses: 3 × 108 NK cells (2 × 106/kg for weight < 50 kg). Lymphodepletion: fludarabine/cyclophosphamide | United States |
| Anti-CD19 Universal CAR-NK Cells Therapy Combined With HSCT for B Cell Hematologic Malignancies | NCT05570188 (30); 1,2 (Withdrawn -the principal investigator decides to stop) | B-cell Lymphoma B-cell Leukemia | anti-CD19 UCAR-NK cells; Preconditioning: Hematopoietic Stem Cell Transplantation(HSCT) Doses: 5–10 × 106/kg, 1–2 × 107/kg, 2–5 × 107/kg | China |
| Anti-CD19 CAR-Engineered NK Cells in the Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia | NCT05563545 (Completed, November 28, 2023) (21); 1 | recurrent or refractory CD19 positive acute lymphoblastic leukemia | CAR-NK-CD19 Cells; Doses: 1.0 × 107, 2.0 × 107 and 3.0 × 107 cells /kg, Lymphodepletion: Fludarabine (25–30 mg/kg), Cyclophosphamide (250–300 mg/kg) | China |
| Anti-CD19 CAR-Engineered NK Cells in the Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia * | NCT05410041 (21); 1 | lymphocyte leukemia, B-cell non-Hodgkin’s lymphoma and chronic B-lymphocyte leukemia | CAR-NK-CD19 Cells; Doses: 1.0 × 107, 2.0 × 107 and 3.0 × 107 cells /kg, Lymphodepletion: Fludarabine (25–30 mg/kg), Cyclophosphamide (250–300 mg/kg) | China |
| Natural Killer (NK) Cell Therapy for B-Cell Malignancies * | NCT05379647 (24); 1 | B-cell Lymphoma B-cell Acute Lymphoblastic Leukemia | QN-019-a (allogeneic CAR-NK cells targeting CD19) as monotherapy or in combination with Rituximab, Lymphodepletion: Cyclophosphamid, Fludarabine, VP-16 | China |
| Study of Anti-CD19 CAR NK Cells in Relapsed and Refractory B Cell Lymphoma + | NCT03690310 (15); 1 | Refractory B-Cell Lymphoma | anti-CD19 CAR NK Cells; Doses: 50–600 103/kg | unknown |
| Clinical Study of HLA Haploidentical CAR-NK Cells Targeting CD19 in the Treatment of Refractory/Relapsed B-cell NHL * | NCT04887012 (25); 1 | B-cell Non-Hodgkin Lymphoma | anti-CD19 CAR-NK; lentiviral vector-transduced HLA haploidentical NK cells express anti-CD19 CAR | China |
| Anti-CD19 CAR NK Cell Therapy for R/R Non-Hodgkin Lymphoma # | NCT04639739 (9); 1 | Non-Hodgkin Lymphoma | anti-CD19 CAR-NK; Doses: 2 × 106 /kg, 6 × 106 /kg, 2 × 107/kg Lymphodepletion: Fludarabine (30 mg/m2) Cyclophosphamide (500 mg/m2) | China |
| Anti-CD19 CAR-Engineered NK Cells in the Treatment of Relapsed/Refractory B-cell Malignancies * | NCT05410041 NCT03690310 (9); 1 | Lymphocytic Leukemia Chronic/Acute Non-Hodgkin Lymphoma | CAR-NK-CD19 Cells; Derived from allogenic NK cells Doses: 1–3 × 107 /KG, Lymphodepletion: Fludarabine (25–30 mg/kg) Cyclophosphamide (250–300 mg/kg) | China |
| Allogeneic NK T-Cells Expressing CD19 Specific CAR in B-Cell Malignancies (ANCHOR2) * | NCT05487651 (36); 1 | Non-Hodgkin Lymphoma, Relapsed, Adult B-cell Lymphoma B-cell Leukemia | KUR-502 - transduced allogeneic natural killer T cells against CD19 (CD19.CAR-aNKT cells) Doses: 1 × 107/m2, 3 × 107/m2, 1 × 108/m2. Body surface area (BSA) capped at 2.4 m2. Lymphodepletion: cyclophosphamide (500 mg/m2/day) Fludarabine (30 mg/m2/day) | United States |
| Study of Anti-CD19/CD22 CAR NK Cells in Relapsed and Refractory B Cell Lymphoma + | NCT03824964 (10); 1 | Relapsed/Refractory B-Cell Lymphoma | Anti-CD19/CD22 CAR NK Cells; Doses: 50–600 103 cells/kg | unknown |
| Study of Anti-CD22 CAR NK Cells in Relapsed and Refractory B Cell Lymphoma + | NCT03692767 (9); 1 | Relapsed /Refractory B-Cell Lymphoma | Anti-CD22 CAR NK Cells Derived from allogenic NK cells Doses: 50–600 103 cells /kg | unknown |
| Study of Anti-CD33/CLL1 CAR-NK in Acute Myeloid Leukemia * | NCT05215015 (18); 1 | Myeloid Leukemia Acute | Anti-CD33/CLL1 CAR-NK Cells; Doses: 2.0 × 109, 3.0 × 109, 3.0 × 109 cells | China |
| Anti-CD33 CAR NK Cells in the Treatment of Relapsed/Refractory Acute Myeloid Leukemia * | NCT05008575 (27); 1 | Leukemia, Myeloid, Acute | anti-CD33 CAR NK cells; Doses: 6 × 108, 12 × 108, 18 × 108 cells/KG Lymphodepletion: Fludarabine (30 mg/m2) Cytoxan (300–500 mg/m2) | China |
| Study of Anti-5T4 CAR-NK Cell Therapy in Advanced Solid Tumors * | NCT05194709 (40); 1 | Solid Tumors Advanced | Anti-5T4 Oncofetal Trophoblast Glycoprotein (5T4) Conjugated Antibody Redirecting Natural Killer (CAR-NK) Cells Doses: 3.0 × 109, 4.0 × 109, 4.0 × 109 cells | China |
| Allogenic CD123-CAR-NK Cells in the Treatment of Refractory/Relapsed Acute Myeloid Leukemia # | NCT05574608 (12); 1 | Acute Myeloid Leukemia Refractory/ Recurrent | Allogenic CD123-CAR-NK cells; Doses: 1 × 106/kg, 5 × 106/kg, 2 × 107/kg Lymphodepletion: Fludarabine, Cyclophosphamide | unknown |
| Phase I/II Study of CAR.70- Engineered IL15-transduced Cord Blood-derived NK Cells in Conjunction With Lymphodepleting Chemotherapy for the Management of Relapse/Refractory Hematological Malignances * | NCT05092451 (94); 1,2 | Leukemia, Lymphoma, or Multiple Myeloma | CAR.70/IL15-transduced Cord blood NK cells; Lymphodepletion: Cyclophosphamide Fludarabine phosphate | United States |
| Anti-BCMA CAR-NK Cell Therapy for the Relapsed or Refractory Multiple Myeloma * | NCT05008536 (27); 1 | Multiple Myeloma, Refractory | Anti-BCMA CAR-NK, NK Cells from umbilical cord blood, Doses: 1–3 × 106/KG, 3–6 × 106/KG, 0.6–1.2 × 107/KG Lymphodepletion: Fludarabine (30 mg/m2), Cytoxan (300–500 mg/m2) | China |
| Cord Blood Derived Anti-CD19 CAR-Engineered NK Cells for B Lymphoid Malignancies * | NCT04796675 (27); 1 | Lymphocytic Leukemia Chronic/Acute Non-Hodgkin’s Lymphoma | Cord blood derived NK cells from healthy donor, transduced with a retroviral vector encoding the anti-CD19 CAR and interleukin-15.CAR-NK-CD19 Cells; Doses: 0.01 × 107, 0.1 × 107, 1.0 × 107/kg Lymphodepletion: fludarabine (30 mg/kg) cyclophosphamide (300 mg/kg) | China |
| Clinical Study of Cord Blood-derived CAR-NK Cells Targeting CD19 in the Treatment of Refractory/Relapsed B-cell NHL * | NCT05472558 (48); 1 | B-cell Non-Hodgkin Lymphoma | anti-CD19 CAR-NK; Doses: 2.5 × 108 cells,5 × 108 cells, 1 × 109 cells | China |
| Phase I/II Study of CD5 CAR Engineered IL15-Transduced Cord Blood-Derived NK Cells in Conjunction With Lymphodepleting Chemotherapy for the Management of Relapsed/Refractory Hematological Malignances # | NCT05110742 (48); 1,2 | Hematological Malignancy | CAR.5/IL15-transduced cord blood NK cells; Doses: 1 × 107, 1 × 108, 1 × 109, 1 × 1010 cells Lymphodepletion: Cyclophosphamide and Fludarabine or Fludarabine Phosphate | United States |
| Umbilical & Cord Blood (CB) Derived CAR-Engineered NK Cells for B Lymphoid Malignancies # | NCT03056339 (36); 1,2 | B-Lymphoid Malignancies Lymphocytic Leukemia Chronic/Acute Non-Hodgkin Lymphoma | CAR-NK cells CD19-CD28-zeta-2A-iCasp9-IL15-transduced cord blood natural killer (CB-NK) cells Lymphodepletion: Fludarabine Cyclophosphamide (AP1903 in case of graft-versus-host disease (GvHD) or cytokine release syndrome after the NK cell infusion) | United States |
| Study of Anti-Mesothelin Car NK Cells in Epithelial Ovarian Cancer + | NCT03692637 (30); 1 | Epithelial Ovarian Cancer | anti-Mesothelin CAR NK Cells; Doses: 0.5–3 × 106/kg cells | unknown |
| Study of Anti-PSMA CAR NK Cell (TABP EIC) in Metastatic Castration-Resistant Prostate Cancer * | NCT03692663 (9); 1 | Metastatic Castration-resistant Prostate Cancer | TABP EIC (anti-PSMA CAR NK cells) Doses: 0.5, 10, and 30 × 106 CAR NK cells Lymphodepletion: Cyclophosphamide 250 mg/m2, fludarabine 25 mg/m2 | China |
| CLDN6-CAR-NK Cell Therapy for Advanced Solid Tumors * | NCT05410717 (40); 1,2 | Stage IV Ovarian Cancer Testis Cancer, Refractory Endometrial Cancer Recurrent | CAR-NK cells from patients PMBC; (some CAR-NK cells are genetically engineered to express and secret IL7/CCL19 and/or SCFVs against PD1/CTLA4/Lag3) | China |
| Study of DLL3-CAR-NK Cells in the Treatment of Extensive Stage Small Cell Lung Cancer * | NCT05507593 (18); 1 | SCLC, Extensive Stage | DLL3-CAR-NK cells; Doses: 1 × 107, 1 × 108, 1 ×109 DLL3-CAR-NK cells | China |
| Irradiated PD-L1 CAR-NK Cells Plus Pembrolizumab Plus N-803 for Subjects With Recurrent/Metastatic Gastric or Head and Neck Cancer * | NCT04847466 (55); 2 | Gastroesophageal Junction (GEJ) Cancers Advanced HNSCC | Irradiated PD-L1 CAR-NK Cells Plus Pembrolizumab Plus N-803 Doses: 2 × 109 cells Pembrolizumab 400 mg N-803 (15 mcg/kg) | United States |
| FT576 in Subjects With Multiple Myeloma * (Allogenic CAR NK cells with BCMA expression) | NCT05182073 (168);1 | Multiple Myeloma Myeloma | FT576 as monotherapy and in combination with the monoclonal antibody daratumumab FT576: allogeneic natural killer (NK) cells, derived from a clonal, CD38-knockout, iPSC that expresses BCMA, CAR, high-affinity, non-cleavable CD16 (hnCD16), and IL-15/IL-15 receptor fusion protein (IL-15RF). Lymphodepletion: Cyclophosphamide Fludarabine | United States |
| Induced-T Cell Like NK Cellular Immunotherapy for Cancer Lack of MHC-I + | NCT03882840 (30); 1,2 | Anti-cancer Cell Immunotherapy T Cell and NK Cell | Induced-T cell-like NK cells; T-like NK cells (ITNK) from patient’s T cells, | China |
| Induced-T Cell Like NK Cells for B Cell Malignancies * | NCT04747093 (12); 1,2 | B Cell Leukemia B Cell Lymphoma B-cell Acute Lymphoblastic | CAR-ITNK cells; Induced-T cell-like NK cells with chimeric antigen receptor | China |
| Clinical Research of ROBO1 Specific BiCAR-NK Cells on Patients With Pancreatic Cancer + | NCT03941457 (9); 1,2 | Pancreatic Cancer | BiCAR-NK cells- (ROBO1 CAR-NK cells) Derived from allogenic NK cells | China |
| Clinical Research of ROBO1 Specific BiCAR-NK/T Cells on Patients With Malignant Tumor + | NCT03931720 (20); 1,2 | Malignant Tumor | BiCAR-NK/T cells (ROBO1 CAR-NK/T cells) Derived from allogenic NK cells | China |
| Clinical Research of ROBO1 Specific CAR-NK Cells on Patients With Solid Tumors + | NCT03940820 (20); 1,2 | Solid Tumor | ROBO1 CAR-NK cells; Derived from allogenic NK cells | China |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Włodarczyk, M.; Pyrzynska, B. CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer. Cancers 2023, 15, 117. https://doi.org/10.3390/cancers15010117
Włodarczyk M, Pyrzynska B. CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer. Cancers. 2023; 15(1):117. https://doi.org/10.3390/cancers15010117
Chicago/Turabian StyleWłodarczyk, Marta, and Beata Pyrzynska. 2023. "CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer" Cancers 15, no. 1: 117. https://doi.org/10.3390/cancers15010117
APA StyleWłodarczyk, M., & Pyrzynska, B. (2023). CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer. Cancers, 15(1), 117. https://doi.org/10.3390/cancers15010117