1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a common digestive system malignancy with high invasiveness and metastasis. The latest report on cancer showed that both the incidence and mortality of PDAC had been increasing during 1990–2017 worldwide [
1], and it has been the third leading cause of cancer-related deaths in the United States [
2]. Despite the development of preventive and therapeutic strategies, the prognosis of PDAC is still unsatisfactory, with a 5-year survival rate of less than 9%. It is attributed to the diagnosis at advanced stages in most PDAC patients and the high recurrence rate after resection, as well as the resistance to drug therapy [
3,
4]. Therefore, it is imperative to elucidate the molecular mechanisms of the occurrence and progression of PDAC to develop novel therapeutic strategies for improving their survival outcomes.
The S100 calcium-binding protein family (S100s) is a class of small molecular proteins expressed only in vertebrates and consists of 21 family members with a similar EF-hand motif structure [
5]. As a unique member of S100s, S100A10 has similar EF-hand motifs, but its key amino acids in the motifs have been substituted or deleted, which maintains it in a continuously activated state and is insensitive to Ca
2+ [
6]. Additionally, S100A10 often interacts with annexin A2 (ANXA2) to form an ANXA2-S100A10 complex to play various biological regulatory roles [
7]. Up to now, S100A10 has been reported to be up-regulated in several cancers, including lung cancer [
8], gall bladder adenocarcinoma [
9], colorectal adenocarcinoma [
10], and hepatocellular carcinoma [
11], which correlated with worse survival outcomes. The significant up-regulation of S100A10 has also been revealed in PDAC tissues, compared with that in normal pancreas tissues or pancreatitis tissues [
12,
13]. Moreover, it is demonstrated that S100A10 is regulated by the oncogene KRAS in PDAC [
14]. However, little is known about the functional roles of S100A10 in PDAC.
In this study, we compared the expression of S100A10 between PDAC tumor tissues and pancreas non-tumor tissues and further analyzed the correlation between S100A10 expression and clinicopathological characteristics to evaluate its prognostic value for PDAC patients. Furthermore, a series of in vitro functional experiments were performed to investigate the role of S100A10 in the progression of PDAC and identify the genes activated by S100A10. An orthotopic xenograft model was also performed to evaluate the role of S100A10 on pancreatic tumor growth in vivo.
2. Materials and Methods
2.1. Oncomine Database Analysis
The transcriptional expressions of S100s were analyzed in twenty types of human cancers, including PDAC and corresponding normal tissues in Oncomine database (
www.Oncomine.org, accessed on 17 October 2021), an online public database that integrates the DNA and RNA sequencing data from multiple human cancers. The difference in mRNA expression was compared by Student’s
t-test (cutoff
p-value: 0.01, cutoff fold-change: 1.5).
2.2. GEPIA Database Analysis
The mRNA expressions of S100s were compared between 179 PDAC tissues and 171 normal pancreas tissues in Gene Expression Profiling Interactive Analysis database (GEPIA,
http://gepia.cancer-pku.cn/, accessed on 21 October 2021), based on the data derived from The Cancer Genome Atlas (TCGA) and Genotype Tissue Expression project (GTEx). The cutoff of
p-value and fold-change were 0.01 and 2, respectively. The correlation analyses between two genes in PDAC were also performed in GEPIA database by using Pearson correlation coefficient, with
p-value < 0.05 defined as statistically significant.
2.3. Kaplan–Meier Plotter Database Analysis
According to their median expression levels in PDAC tissues, the mRNA expressions of S100s were divided into two groups (Low expression group vs. High expression group) and further used to make survival analyses to overall survival (OS) and recurrence-free survival (RFS) for PDAC patients in Kaplan–Meier plotter database (
https://kmplot.com/, accessed on 21 October 2021), an online public database, which can be used to make survival analysis on the expression of 54,675 genes in 21 types of human cancers. The logrank
p < 0.05 is defined as statistically significant.
2.4. UALCAN Database Analysis
UALCAN database (
http://ualcan.path.uab.edu/, accessed on 21 October 2021) is an interactive website providing analysis of cancer OMICS data. The correlation analyses between the mRNA expression of S100A2/10 in PDAC tissues and clinicopathological characteristics, including grade and stage, were performed in UALCAN database based on the data derived from The Cancer Genome Atlas (TCGA). The comparison was made by Student’s
t-test, with
p-value < 0.05 regarded as statistically significant.
2.5. Human Protein Atlas Database (HPA) Analysis
The Human Protein Atlas database (HPA,
http://www.proteinatlas.org, accessed on 25 October 2021) is an online public website, which provides data on the proteome from 17 different human cancer in ten aspects, including The Tissue Atlas, The Brain Atlas, The Single Cell Type Atlas, The Cell Type Atlas, The Pathology Atlas and so on. To evaluate differences in S100A10, ITGA2, LAMB3, and LAMC2 expression at the protein level, immunohistochemistry images of S100A10, ITGA2, LAMB3, and LAMC2 protein expressions in normal pancreas tissues and PDAC tissues, were downloaded from The Tissue Atlas and The Pathology Atlas in HPA, respectively.
2.6. cBioPortal Database Analysis
cBioPortal database (
http://www.cbioportal.org/, accessed on 31 October 2021), an online tool that integrates multidimensional cancer genomics data, was used to acquire the genes significantly associated with S100A10 in PDAC tissues in the QCMG project (Nature 2016, 456 samples) and TCGA project (PanCancer Atlas, 184 samples), with
q-value < 0.05 and
q-value < 0.01 regarded as statistically significant, respectively.
2.7. DEGs Analysis in GEO and UCSC Xena Database
In the Gene Expression Omnibus database (GEO,
http://www.ncbi.nlm.nih.gov/geo, accessed on 31 October 2021), the public microarray dataset GSE62452 was analyzed with an online tool GEO2R to acquire the significantly different expression genes (DEGs) between 69 PDAC samples and 61 adjacent non-tumor tissue samples (
padj-value < 0.05, |log2-fold change| > 1). The DEGSeq2 standardized transcriptional data in the TCGA-TARGET-GTEx cohort were downloaded from the public online website: UCSC Xena (
https://xenabrowser.net/, accessed on 31 October 2021). Statistical software R (version 4.1.0,
https://cran.r-project.org/, accessed on 10 September 2021) was used to conduct significance analysis of DEGs between 179 PDAC samples and 171 normal pancreas samples. DEGSeq2 package (version 1.32.0,
http://www.bioconductor.org/, accessed on 10 September 2021) was used to select significant DEGs, where the standard was
padj-value < 0.05 and |log2-fold change| > 2.
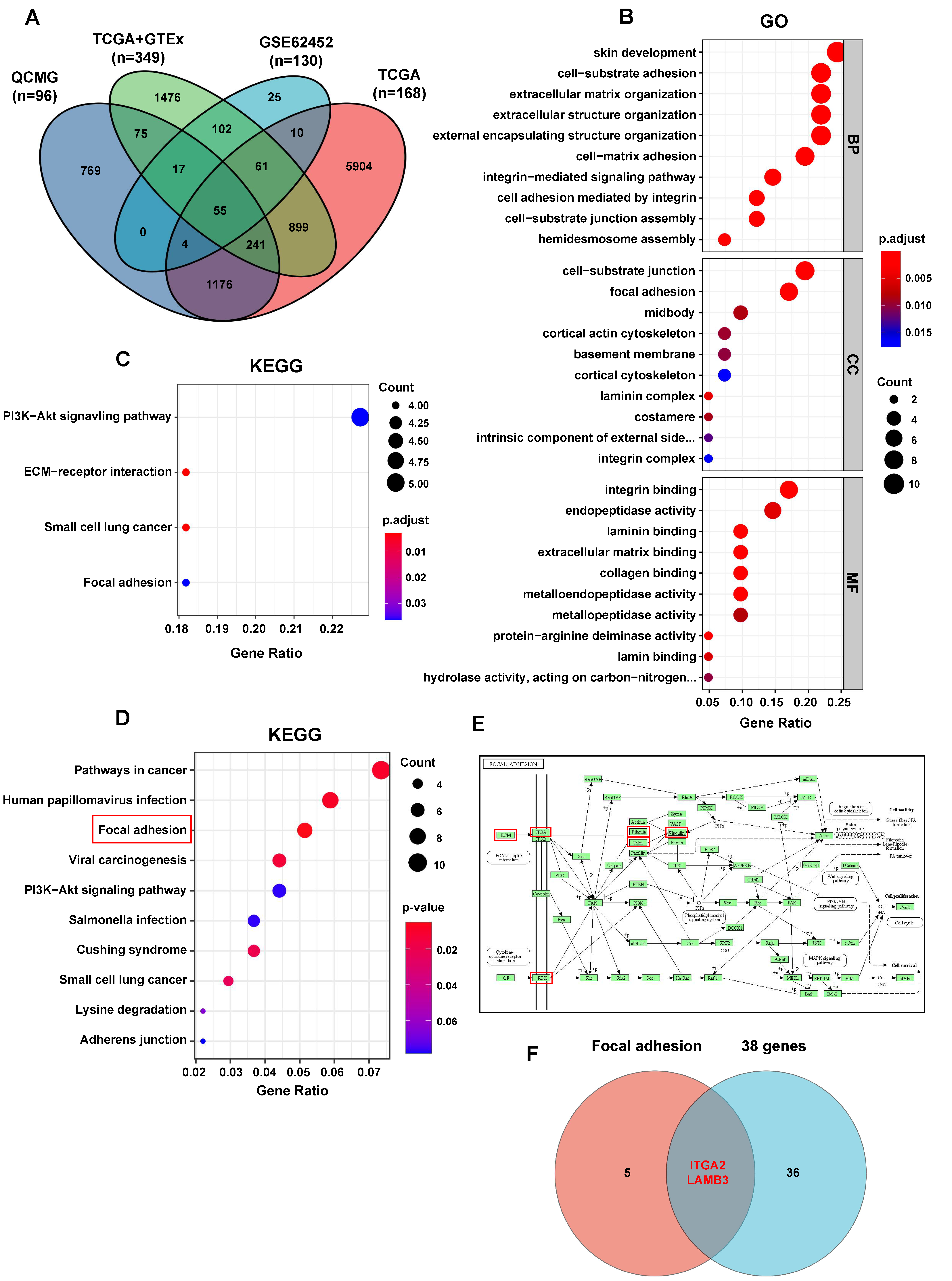
2.8. Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis
After the intersection of the genes identified to be significantly associated with S100A10 in PDAC in cBioPortal database analysis and the significant DEGs identified in GEO and UCSC Xena database analyses, 55 genes were acquired. Survival analyses for these 55 genes were further performed in GEPIA database, and only 38 genes were identified to be significantly associated with the OS for PDAC patients (logrank
p-value < 0.05). After gene ID conversion, these 38 genes were used for Gene ontology (GO) term enrichment analysis (including biological process, cellular component, and molecular function) and KEGG pathway enrichment analysis with the Statistical software R (version 4.1.0,
https://cran.r-project.org/, accessed on 10 September 2021) and cluster Profile package (version 4.0.5,
http://www.bioconductor.org/, accessed on 10 September 2021), where the standard was
p-value < 0.05.
2.9. Cells and Reagents
Six PDAC cell lines (including PANC-1, CFPAC-1, HPAF II, MIAPaCa-2, AsPC-1 and Bx-PC3) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C in a humidified atmosphere containing 5% CO2. The JNK inhibitor SP600125 was purchased from Selleck (Houston, TX, USA).
2.10. Transient Transfection, Lentiviral Transduction and Generation of Stable Cell Lines
Two siRNAs against S100A10 (siS100A10-1, siS100A10-2), one siRNA against LAMB3 (siLAMB3), and corresponding control siRNAs (siNC) were purchased from RIBOBIO (Guangzhou, China). A plasmid containing full-length S100A10 cDNA (NM_002966) (pS100A10) and another plasmid containing full-length LAMB3 cDNA (NM_000228.3) (pLAMB3) as well as corresponding control plasmids (pNC) were purchased from MiaoLing (Wuhan, China). PDAC cells were seeded into 6-well plates at 5 × 10
5 cells/well density and incubated 24 h to reach 50–60% confluence for transfection. Transfections were performed with a Lipofectamine-3000 transfection kit (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. After transfection for appropriate time, cells were harvested and used for further in vitro experiments. After transfection with pNC or pS100A10 for 24 h, PDAC cells were exposed to a JNK inhibitor (SP600125) at 60 umol concentration for 48 h, or transfected with a siRNA against JNK1 (siJNK1) or control siRNA (siNC) for 48 h, followed by western blotting analysis to measure the protein expressions of interesting genes. The target sequences used in the siRNA were summarized in
Table S1. S100A10 stably knocked-down (shS100A10) and negative control (shNC) PANC-1 and AsPC-1 cells were generated by using lentiviral constructs expressing shS100A10 and shNC, which were designed by Genechem (Shanghai, China). Briefly, PANC-1 and AsPC-1 cells were seeded into 6-well plates at 2 × 10
5 cells/well density and incubated overnight, followed by lentiviral infection (MOI = 10) for 24 h. Subsequently, the shNC and shS100A10 PANC-1 and AsPC-1 cells were selected with puromycin (MP, Santa Ana, CA, USA) at 5 μg/mL concentration for 7 days.
2.11. RNA Sequencing (RANseq) and Database for Annotation, Visualization and Integrated Discovery (DAVID) Analysis
PANC-1 cells were seeded into 6-well plates at 5 × 10
5 cells/well density and incubated 24 h to reach 50–60% confluence, followed by transfected with a siRNA against S100A10 (siS100A10-1) or control siRNA (siNC), a plasmid containing full-length S100A10 cDNA (pS100A10) or control plasmid (pNC) for 72 h. The transfection concentrations of siRNAs and plasmids were 100 nM and 2.5 ng/uL, respectively. Subsequently, the cells were collected and washed 2 times with 1× PBS. A total of 1ml Trizol was added to each sample (siNC, siS100A10-1, pNC, and pS100A10) and sent to GENEWIZ Corporation (Suzhou, China) for RNAseq. RNAseq was performed by using the Illumina HiSeq X platform. Briefly, total RNA was isolated from each sample, followed by quality control. Subsequently, cDNA was synthesized and further amplified and purified. Htseq Software (V 0.6.1,
https://anaconda.org/bcbio/htseq, accessed on 10 October 2021) and Fragments Per Kilo bases per Million reads (FPKM) method were used to calculate gene expression. When analyzing the RNAseq data (read count) of 4 groups (including siNC, siS100A10-1, pNC, and pS100A10), we first filtered the low-expressed genes (read count ≤ 25) and further set the screening thresholds for each gene as: “siS100A10-1/siNC ≤ 0.5 and pS100A10/pNC ≥ 2” or “siS100A10-1/siNC ≥ 2 and pS100A10/pNC ≤ 0.5”. One-hundred-thirty-four genes were identified and further used for KEGG pathway enrichment analysis in the Database for Annotation, Visualization and Integrated Discovery (DAVID) (
https://david.ncifcrf.gov/, accessed on 24 January 2022), with
p-value < 0.05 defined as statistical significance.
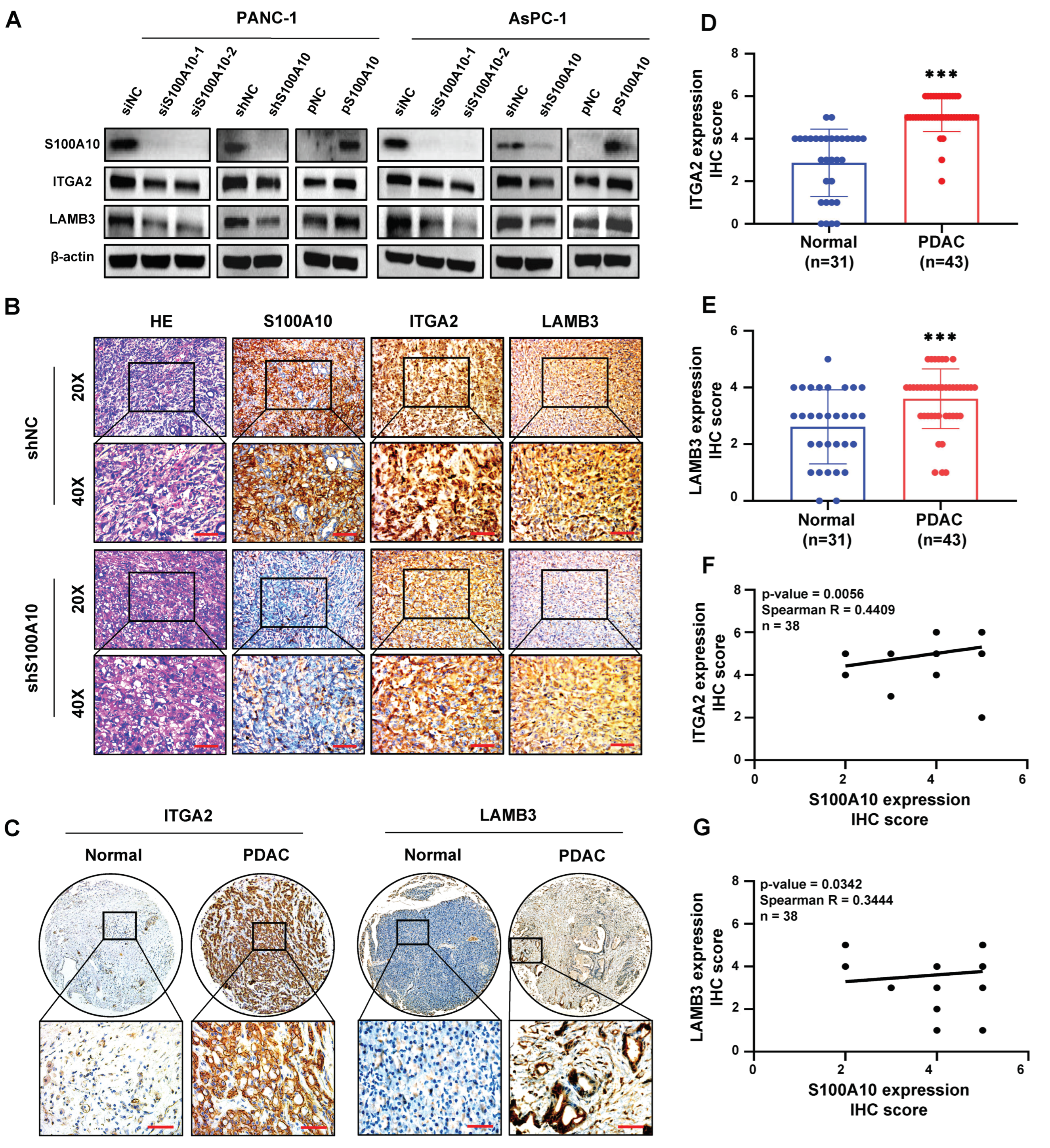
2.12. The Human PDAC Tissue Samples Analysis and Immunohistochemistry (IHC)
PDAC specimens (including 43 PDAC tissues and 31 adjacent non-tumor tissues) were collected from patients who received an operation at the Fifth Affiliated Hospital of Sun Yat-sen University from October 2004 to May 2020. The clinicopathological characteristics of these 43 PDAC patients were summarized in
Table S2. The protocol was approved by the Medical Ethics Committee of the Fifth Affiliated Hospital of Sun Yat-sen University, and the informed consent was written. These specimens were embedded with paraffin and subsequently used for immunohistochemistry. The immunohistochemistry was performed by using an IHC staining kit (SA1022, Boster, Wuhan, China) according to the manufactory protocol on paraffin-embedded tissue. In brief, after deparaffinization, rehydration, antigen retrieval, endogenous peroxidase blocking, and 5% BSA blocking, the specimens were incubated with primary antibodies overnight at 4 °C, and subsequently biotinylated with a secondary antibody for 1 h at 37 °C, followed by visualized using a DAB reagent (KIT-9710-6 ml, MXB, Fuzhou, China) and counterstained with Mayer’s hematoxylin (DH0005, LEAGENE, Beijing, China) for 10 min. The primary antibodies against S100A10, LAMB3, and ITGA2 were used at 1/100, 1/300, and 1/500 dilution in 1×PBS, respectively. Finally, two independent pathologists who were not aware of the clinicopathological data of 43 PDAC patients interpreted the results. The immunoreactive score (IRS) was calculated by the quantity and intensity of the staining. The number of positive cells was scored as follows: 0 points (<10% cell staining), 1 point (10–25% cell staining), 2 points (26–75% cell staining) and 3 points (>75% cell staining). The intensity of staining was calculated as follows: 0 points (negative), 1 point (weak), 2 points (moderate), and 3 points (strong). Images were captured by a microscope (Leica, Wetzlar, Hessen, Germany) at 50×, 200× or 400× magnification. Antibodies’ information for IHC is listed in
Table S5.
2.13. Real-Time Quantitative PCR (RT-qPCR) for Measurement of Gene Expression
Total RNA was extracted by using an RNA-Quick Purification Kit (RN001, ES Science, Beijing, China) according to the manufacturer’s protocol. Subsequently, cDNA was synthesized from total RNA by using a PrimeScript™ RT Master Mix reagent kit (TaKaRa, Beijing, China). Real-time quantitative polymerase chain reaction (RT-qPCR) was performed to compare the relative mRNA expression level of interesting genes by using Hieff UNICON
® Univeral Blue qPCR SYBR Green Master Mix (11184ES08, YEASEN, Shanghai, China) on the Agilent AriaMx Real-time PCR System (Agilent, Palo alto, California, USA). The sequences of primers for RT-qPCR are listed in
Table S3.
2.14. Western Blotting
Protein samples were prepared by using RIPA lysis buffers (CWBIO, Jiangsu, China) containing 1× protease inhibitors (Selleck, Houston, TX, USA) and 1× phosphatase inhibitors (Selleck, Houston, TX, USA). Subsequently, western blotting was performed to detect the protein expression levels of interesting genes. Briefly, SDS-PAGE on 10% or 15% gel was used for protein separation, and the separated proteins were transferred to a PVDF membrane (Merck millipore, Billerica, MA, USA) and blocked with 5% non-fat milk in 1× PBST for 1 h, followed by incubated with primary antibodies overnight at 4 °C. After the incubation of HRP-conjugated secondary antibodies (1:5000~10,000) in 1× PBST at room temperature for 1 h, the signals of protein levels were detected with a Chemistar ECL Western Blotting Substrate (180–5001, Tanon, Shanghai, China) by using a Tanon-5200 multi-imaging system (Tanon, Shanghai, China). The primary antibodies against S100A10 (1/1000), LAMB3 (1/1000), ITGA2 (1/2500), β-actin (1/2500), Flag (1/1000), p105/NF-kB (1/1000), p-p105/NF-kB (1/1000), p65/NF-kB (1/1000), p-p65/NF-kB (1/1000), AKT (1/1000), p-AKT (1/1000), SAPK/JNK (1/1000), p-SAPK/JNK (1/1000), LAMC2 (1/500) were diluted in primary antibody diluent (Beyotime, Shanghai, China) according to corresponding manufacturers’ instructions. Antibodies’ information for western blotting is listed in
Table S5.
2.15. Cell Count Kit-8 (CCK-8) Assay
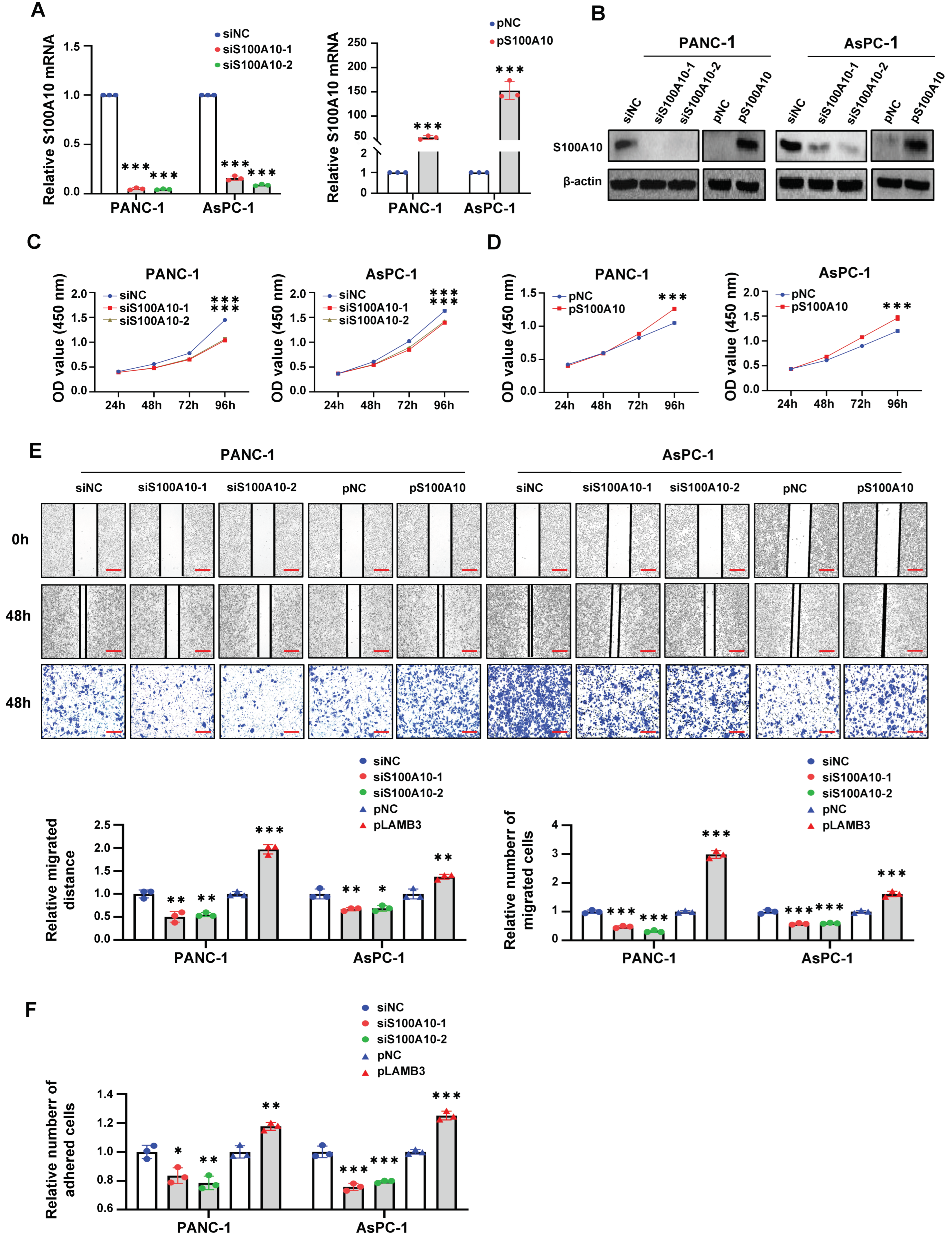
After transfection for 48 h, cells were seeded into 96-well plates at 5 × 103 cells/well density and incubated in a 37 °C humidified atmosphere containing 5% CO2. When incubated for 24 h, 48 h, 72 h, and 96 h, the culture medium was discarded, and subsequently, 10 μL of CCK-8 Cell Counting Kit (A311-02, Vazyme, Nangjing, China) and 90 μL DMEM medium containing 10% FBS were added to each well and incubated in an incubator at 37 °C for 2–3 h before the measure at 450 nm with a SpectraMax® Absorbance Reader CMax Plus (Molecular Devices, Silicon Valley, CA, USA).
2.16. Cell Scratch Assay
Cells were seeded in six-well plates at 8 × 105 cells/well density and incubated 24 h, followed by transfection for 48 h. When cells were incubated to about 90–100% confluence, a sterile plastic micropipette tip was used to make artificial scratches, and the cells were washed 3 times with 1× PBS to remove the floating cells, followed by incubated with serum-free medium for 48 h. The images of each artificial scratch at 0 h and 48 h were acquired with microscopy at 100× magnification. The migration distance of cells was calculated by the distance of each scratch at 0 h and 48 h. The results were expressed as relative migration distances.
2.17. Transwell Migration Assay
After transfection for 48 h, cells were harvested and diluted in serum-free DMEM medium. Subsequently, cells (5 × 104, 100 uL) were seeded into the upper chamber (8.0 μm pore size, corning Life Sciences, Corning, NY, USA), with the lower chamber filled with 600 μL DMEM medium containing 20% FBS. After incubation for 48 h, the migrated cells attached on the underside of the membrane were fixed with 4% paraformaldehyde for 20 min and stained with 0.1% crystal violet for 20 min. The cells above the membrane were wiped with a cotton ball. The migrated cells attached on the underside of the membrane were imaged with an inverted microscope (Leica, Wetzlar, Hessen, Germany) and the total cell numbers were quantified from three random fields at 100× magnification. The results were expressed as relative migration cells.
2.18. Cell Adhesion Assay
Matrigel (100 μL) (356234, corning Life Sciences, Corning, NY, USA) was diluted in 900 μL serum-free DMEM medium. Ninety six-well plates were pre-coated with matrigel solution (50 μL/well) and air-dried in incubator at 37 °C overnight and then washed 3 times with serum-free DMEM medium containing 0.1% bovine serum albumin (BSA) (Solarbio, Beijing, China) and blocked for 1 h with serum-free DMEM medium containing 0.5% BSA. After transfection for 72 h, cells (104) were seeded with 100 uL DMEM medium containing 10% FBS into each well of the pre-coated 96-well plate and incubated for 40 min, followed by gently washing 3 times with 1× PBS. Subsequently, CCK-8 assay was performed to detect the number of adhered cells. The results were expressed as the relative number of adhesive cells.
2.19. Co-Immunoprecipitation (Co-IP), Liquid Chromatography Tandem Mass Spectrometry (LC-MS) and Protein–Protein Interaction (PPI) Analysis
Cells were transfected with plasmids (concentration: 2.5 ng/uL) for 72 h. Cell lysates were prepared by using IP lysis buffers (Beyotime, Shanghai, China) and further diluted in 1×PBS to a concentration of 1 μg/uL. Subsequently, 500 ug of the total protein samples were incubated with 5 μg mouse anti-FLAG monoclonal antibody and 50 μL BeyoMagTM Protein A+G Magnetic beads (P2108, Beyotime, Shanghai, China) at 4 °C for 24 h. The immunoprecipitants were washed 3 times with cold 1× PBS and dissolved with 50 μL 1× loading buffer, followed by boiling at 100 °C for 10 min. Western blotting assay was performed according to the protocol above. In addition, 30 μL of the immunoprecipitant samples were sent to Wininnovate Bio company (Shenzhen, China) for Liquid Chromatography Tandem Mass Spectrometry (LC-MS) analysis. Briefly, the immunoprecipitant samples were analyzed by western blotting. For MS analysis, the gels were firstly cut into small pieces and decolorized with certain solution, followed by dried with 100% acetonitrile. Subsequently, the gels were digested, and the peptides were obtained from them. The peptides were detected by LC-MS on a ThermoFisher Q Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). The LC-MS data were analyzed for protein identification and quantification by using PEAKS Studio 8.5 (Bioinformatics Solutions Inc., Waterloo, ON, Canada). The local false discovery rate at PSM was 1.0% after searching against Homo sapiens database with a maximum of two missed cleavages. The following settings were selected: Oxidation (M), Acetylation (Protein N-term), Deamidation (NQ), Pyro-glu from E, Pyro-glu from Q for variable modifications as well as fixed Carbamidomethylation of cysteine. Precursor and fragment mass tolerance were set to 10 ppm and 0.05 Da, respectively. Proteins identified from LC-MS results were submitted to STRING database (STRING,
https://cn.string-db.org, accessed on 11 September 2022) for PPI analysis to acquire the proteins interacting with LAMB3, followed by visualized with the Cytoscape software (version 3.9.1,
https://cytoscape.org/, accessed on 2 October 2022). For endogenous protein interaction between LAMB3 and LAMC2, 500 ug of the total protein sample derived from PANC-1 or AsPC-1 cells were incubated with 1 ug rabbit anti-LAMB3 polyclonal antibody and 20 uL BeyoMagTM Protein A+G Magnetic beads at 4 °C for 24 h. Antibodies’ information for Co-IP is listed in
Table S5.
2.20. Cell Immunofluorescence Assay
After transfected with a plasmid containing full-length LAMB3 cDNA (pLAMB3) at 2.5 ng/uL concentration for 48 h, PANC-1 cells were seeded into dishes (801002-1, NEST, Wuxi, China) at 2 × 10
5 cells/dish density and incubated for 48 h. After fixed with 4% paraformaldehyde for 15 min, permeabilized in immunostaining permeabilization buffers (Beyotime, Shanghai, China) for 15 min, and blocked with 5% BSA for 1 h at room temperature, the cells were added with primary antibodies against LAMC2 and Flag at 1:250 dilution in 1× PBS and incubated overnight at 4 °C. The next morning, after washed 3 times with cold 1× PBS, the cells were added with Alexa Fluor
®488, and Alexa Fluor
TM594 conjugated secondary antibodies at a 1:300 dilution in 1× PBS and further incubated at room temperature for 1 h in a dark condition. After washed 3 times with cold 1× PBS, the cells were incubated with DAPI at 1:1000 dilution in 1× PBS for 1 min in a dark condition, followed by washed with cold 1× PBS for 5 min and added with 100 uL fluorescence anti-fade reagent (Boster, Wuhan, China). Images were taken with a Laser confocal microscope (LSM880, Zeiss, Oberkochen, Baden-Wurtberg, Germany) at 600× magnification. Antibodies’ information for cell immunofluorescence is listed in
Table S5.
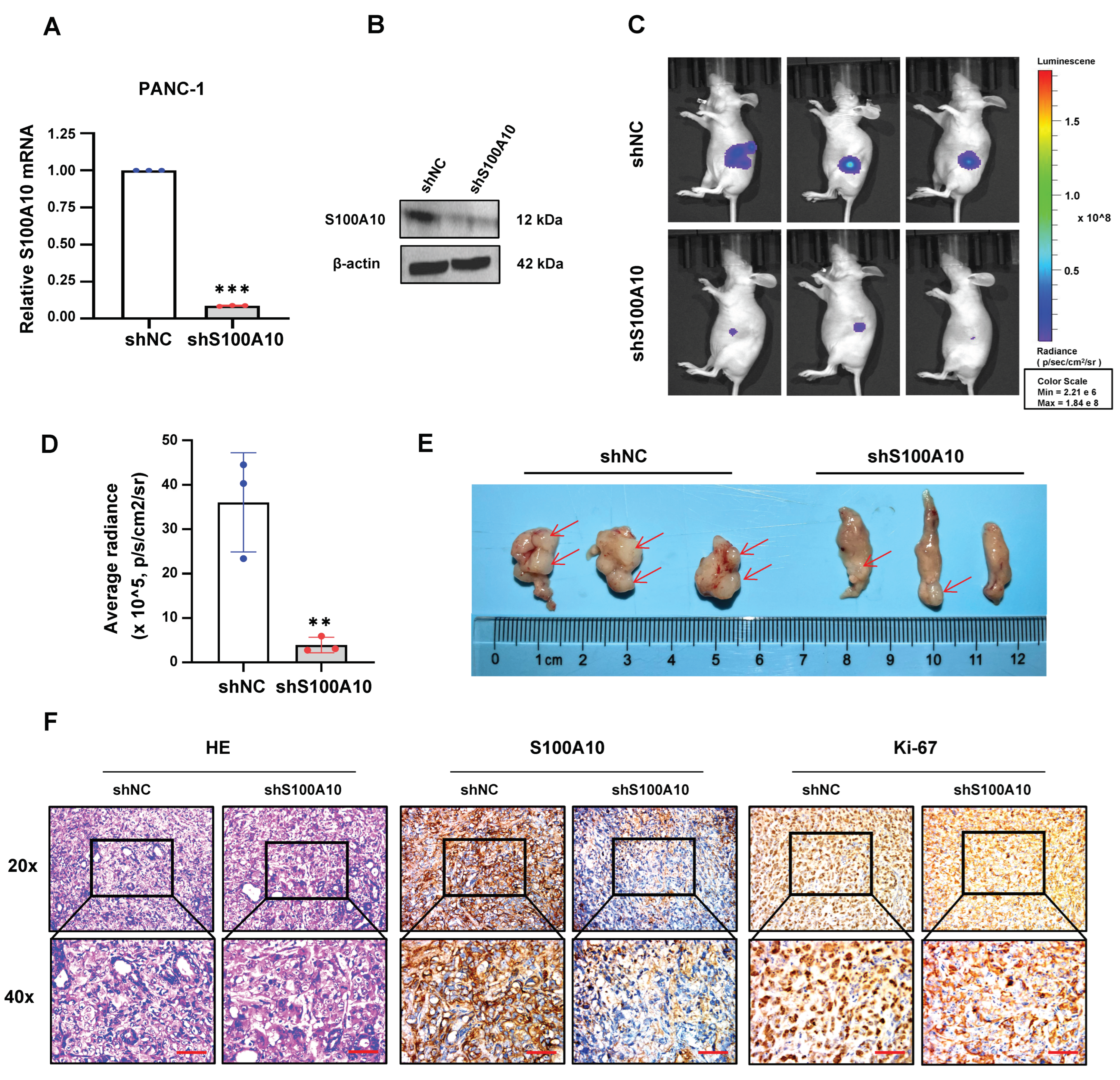
2.21. Mouse Pancreatic Orthotopic Xenograft Model and In Vivo Imaging Analysis
BALB/c nude mice (female, 4–6 weeks) were purchased from Guangdong Medical Laboratory Animal Center (
www.gdmlac.com.cn, accessed on 7 July 2022). All animal experiments were approved by the Experimental Animal Ethics Committee of the Fifth Affiliated Hospital of Sun Yat-sen University. After anesthesia, the abdominal cavity of mice was opened, and the pancreas was exposed, then S100A10 stably knocked-down (shS100A10) and negative control (shNC) PANC-1 cells (2 × 10
6) were injected with 10 uL 1×PBS into the tissue under the pancreatic capsule by using a micro syringe. The weight of mice was measured weekly. Two months after inoculation, in vivo imaging analysis was performed to detect the bioluminescence signals of pancreatic orthotopic tumors by using appropriate amount of VivoGlo
TM Luciferin (150 mg/kg) (P1042, Promega, Madison, WI, USA) with an IVIS Lumina III device (PerkinElmer, Waltham, MA, USA). Subsequently, the mice were sacrificed, and their pancreatic tumor tissues were dissected, followed by fixed with 4% paraformaldehyde. The tumor tissue sections were used for H&E staining or Immunohistochemistry. H&E staining was used for the routine histopathological examination.
2.22. Statistical Analysis
All analyses were conducted by using GraphPad Prism 8.0 software (GraphPad Software, San Diego, CA, USA). Mann–Whitney test was used to compare the protein expression level of interesting genes in human PDAC specimens. The correlation between target proteins in human PDAC specimens were analyzed with Spearman’s p statistic. Data from qRT-PCR, cell experiments and animal experiments were analyzed by Student’s t-test, and the results are represented as mean ± standard deviation (SD) of three independent experiments. p < 0.05 was considered statistically significant.
4. Discussion
S100A10 has been demonstrated to have a certain value in the diagnosis, treatment, and prognosis assessment of a variety of cancers since being first identified in 1985 [
16]. In lung squamous cell carcinoma, the expression of S100A10 in tumor tissue was positively correlated with tumor size, TMN stage, and lymphatic metastasis [
8], while elevated S100A10 in lung adenocarcinoma indicated poorer tumor differentiation, higher TMN stage, more frequent intratumoral vascular invasion and a poorer prognosis [
17]. What is more, the serum S100A10 levels in lung cancer were not only significantly higher than that in patients with benign lung nodules and healthy cancer-free controls but also positively associated with TNM stage and lymphatic metastasis [
18], indicating that S100A10 is not only a prognostic marker but also a potential diagnostic maker for lung cancer. Similarly, Yi Tan et al. reported that S100A10 might serve as a potential biomarker for early detection and a potential therapeutic target for gallbladder cancer [
9]. In addition, higher S100A10 expression is also reported to be correlated with shorter survival rates in gastric cancer [
19] and breast cancer [
7]. In the present study, S100A10 was demonstrated to be significantly up-regulated in PDAC tissues by integrated bioinformatic and human PDAC sample analyses, and high S100A10 expression is associated with advanced clinical stage, poorer differentiation, and shorter survival rate. It is implied that S1000A10 has the potential to be a prognostic biomarker for PDAC patients.
Sustained proliferative signaling is one of the hallmarks of cancer. It is reported that decreased S100A10 expression not only inhibits ovarian cancer cell proliferation and colony formation in vitro but also remarkably suppresses ovary tumor growth in vivo [
20]. Likewise, the knockdown of S100A10 significantly reduced the proliferation and metastasis capacity of colorectal cancer cells [
10]. In hepatocellular carcinoma, the over-expression of S100A10 accelerates cell proliferation in Hep3B and Huh-7 cells, while decreased S100A10 expression shows the opposite effect on the proliferation capacity of SK-Hep-1 and HepG2 cells [
11]. Additionally, it is demonstrated that S100A10 promotes the malignant growth of cancer cells by activating the AKT/mTOR signaling pathway in osteosarcoma [
21] and the Src/ANXA2/AKT/mTOR signaling pathway in gastric cancer [
19]. Collectively, S100A10 serves as a positive regulator in cell proliferation in multiple cancers. In this study, we revealed that S100A10 not only promotes PDAC cell proliferation in vitro but also accelerates pancreatic tumor growth in vivo, which is consistent with the in vivo results in a previous study [
14]. However, Moamen Bydoun et al. reported that similar proliferation rates were observed between S100A10 stably depleted PANC-1 cells and scramble control cells in vitro [
14], which is inconsistent with our in vitro results. Further studies are needed to elucidate the effect of S100A10 on cell proliferation in vitro.
Activation of invasion and metastasis is another hallmark of cancer. S100A10 has been reported to promote tumor progression and metastasis by regulating multiple signaling pathways related to cell migration, invasion, metastasis, and angiogenesis [
22]. S100A10 is known for its role in interacting with ANXA2 to form the ANXA2/S100A10 heterotetrameric complex (AIIt), which is identified as one of the oncogenic plasminogen receptors [
23]. The activation of plasminogen by AIIt not only activates pro-matrix metalloproteinases (pro-MMPs) but also induces the angiogenesis of cancer stroma via ECM-associated proangiogenic growth factors, which both play an important role in the invasion and metastasis of cancer [
7]. Choi KS et al. [
24] reported that the depletion of S100A10 significantly reduced the lung metastases of fibrosarcoma cells in vivo, while Zhang et al. [
25] revealed that stable suppression of S100A10 in colorectal adenocarcinoma CCL-222 cells without ANXA2 expression resulted in a complete loss of plasminogen-dependent invasiveness. It is indicated that S100A10 may promote the invasiveness and metastasis of these cancer cells through an ANXA2-independent pathway. In addition, S100A10 also plays an important role in cell migration. Kyle D. Phipps et al. [
26] reported that S100A10 is crucial for tumor-promoting macrophage migration to tumor sites. As a regulator of the filamentous actin network, the depletion of S100A10 impairs cell motility and further inhibits cell migration in squamous carcinoma A431 cells [
27]. Our results reveal that S100A10 enhances the migration capacity of PDAC cell lines in vitro. In addition, the genes significantly associated with S100A10 and significantly differentially expressed in PDAC are enriched in the cellular components, including cortical actin cytoskeleton and cortical cytoskeleton. It is reported that S100A10 participates in the organization of actin stress fibers and the formation of focal adhesions by activating Rac1, promoting cell migration and spreading [
28]. Furthermore, SUMO1-modified S100A10 can translocate into nuclei to up-regulate the expression of genes associated with actin dynamics and cytoskeleton remodeling, followed by the enhanced migration capacity of polyploid tumor giant cells and their daughter cells [
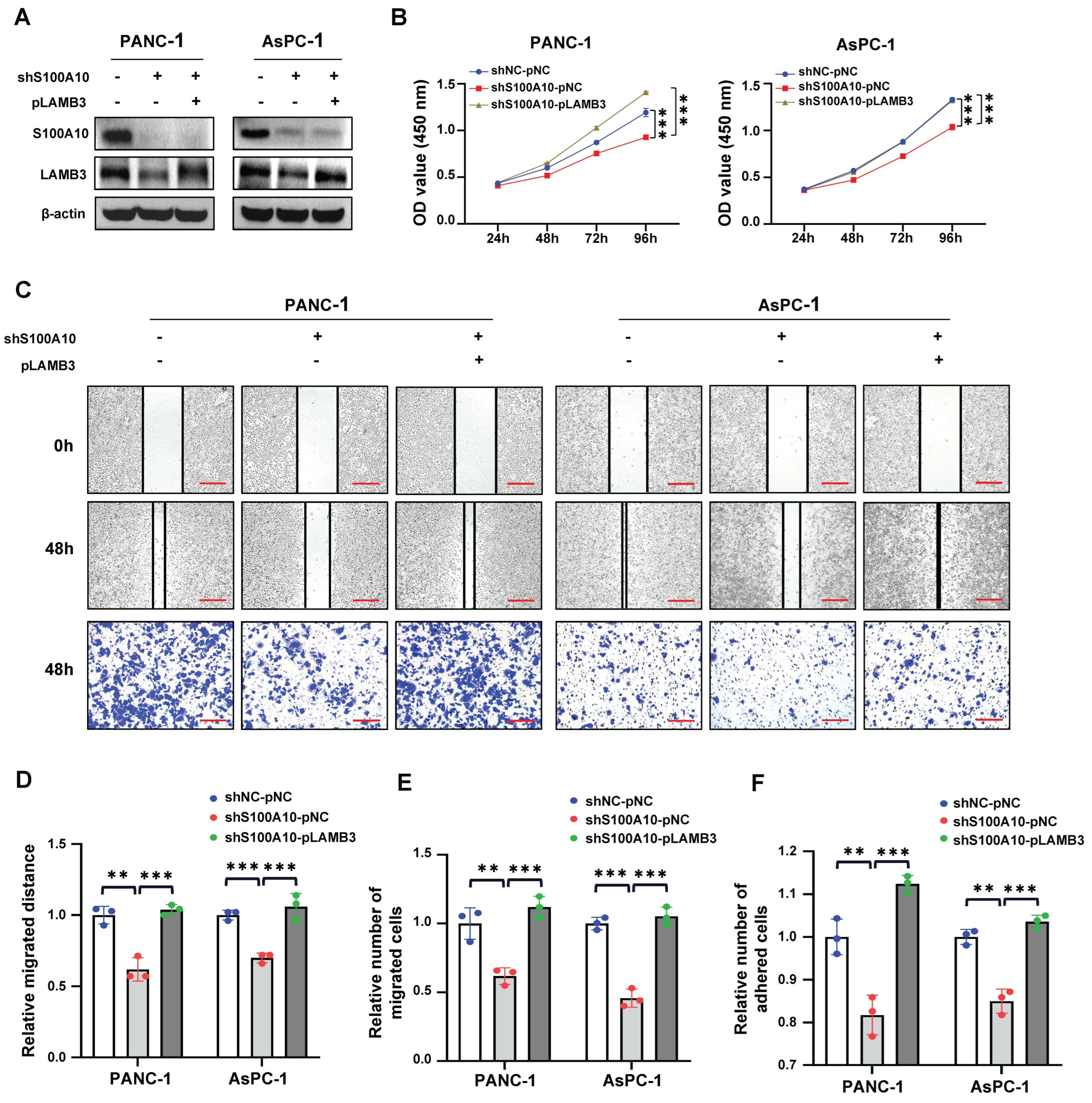
29]. Based on these results, we speculated that S100A10 might participate in the cytoskeleton remodeling of PDAC cells and thus enhance cell motility and migration ability.
Focal adhesion is an integrin-based structure that mediates the adhesion of cells to ECM and involves in the signaling transmission between cytoplasm and ECM, playing an important role in cell proliferation, adhesion, migration, and invasion [
30]. We also found that the genes (including LAMB3, LAMC2, ITGA2, FLNA, EGFR, TLN2, and VCL) activated by S100A10 in PDAC were mainly enriched in the focal adhesion pathway by RNAseq and KEGG pathway enrichment analyses. Subsequently, ITGA2 and LAMB3 were further proved to be activated by S100A10 in PDAC, which both validated to be not only up-expressed in PDAC tissues but also associated with the prognosis of PDAC patients [
31]. In addition, ITGA2 has also been demonstrated to promote PDAC progression via the focal adhesion pathway [
32], and LAMB3 is also identified as an oncogene related to the focal adhesion pathway in cervical squamous cell carcinoma [
33]. In vitro, S100A10 was further demonstrated to enhance the adhesion capacity of PDAC cells to the extracellular matrix in our study. In a previous study, the activation of focal adhesion signaling pathways has been reported to promote cell adhesion, migration, and invasion in breast cancer [
34]. As a signal transducer between ECM and cytoskeleton, focal adhesion also influences cytoskeleton remodeling and cell motility in cancer [
35,
36]. Therefore, It is indicated that the enhanced cell adhesion may be one of the reasons why S100A10 promotes PDAC cell migration in vitro.
LAMB3 is emerging as a potential therapeutic target for cancer due to its oncogenic role. LAMB3 was reported to be up-regulated in thyroid cancer and promote papillary thyroid cancer cell migration and invasion by activating the c-MET/AKT signaling pathway [
37]. In HNSCC, high LAMB3 expression is not only correlated with unfavorable prognosis in HNSCC patients but also associated the cisplatin cytotoxicity to HNSCC cells [
38]. In gastric cancer, the up-regulation of LAMB3 plays an important role in cancer progression [
39]. Similarly, our results indicated that higher LAMB3 expression was detected in PDAC tissues than in matched pancreas normal tissues, whose expression was inversely associated with the survival rates of PDAC patients. Furthermore, LAMB3 was identified as an oncogene to promote PDAC cell proliferation, migration, and adhesion in vitro. In line with our results, LAMB3 has been proven to be up-regulated in PDAC [
15,
31] and enhance PDAC malignant phenotypes through PI3K/AKT axis [
40]. In previous studies, LAMB3 was also reported to promote tumor progression and chemoresistance through the AKT-FOXO3/4 axis and be transcriptionally regulated by the BRD2/acetylated ELK4 complex [
41] and polymeric immunoglobulin receptor [
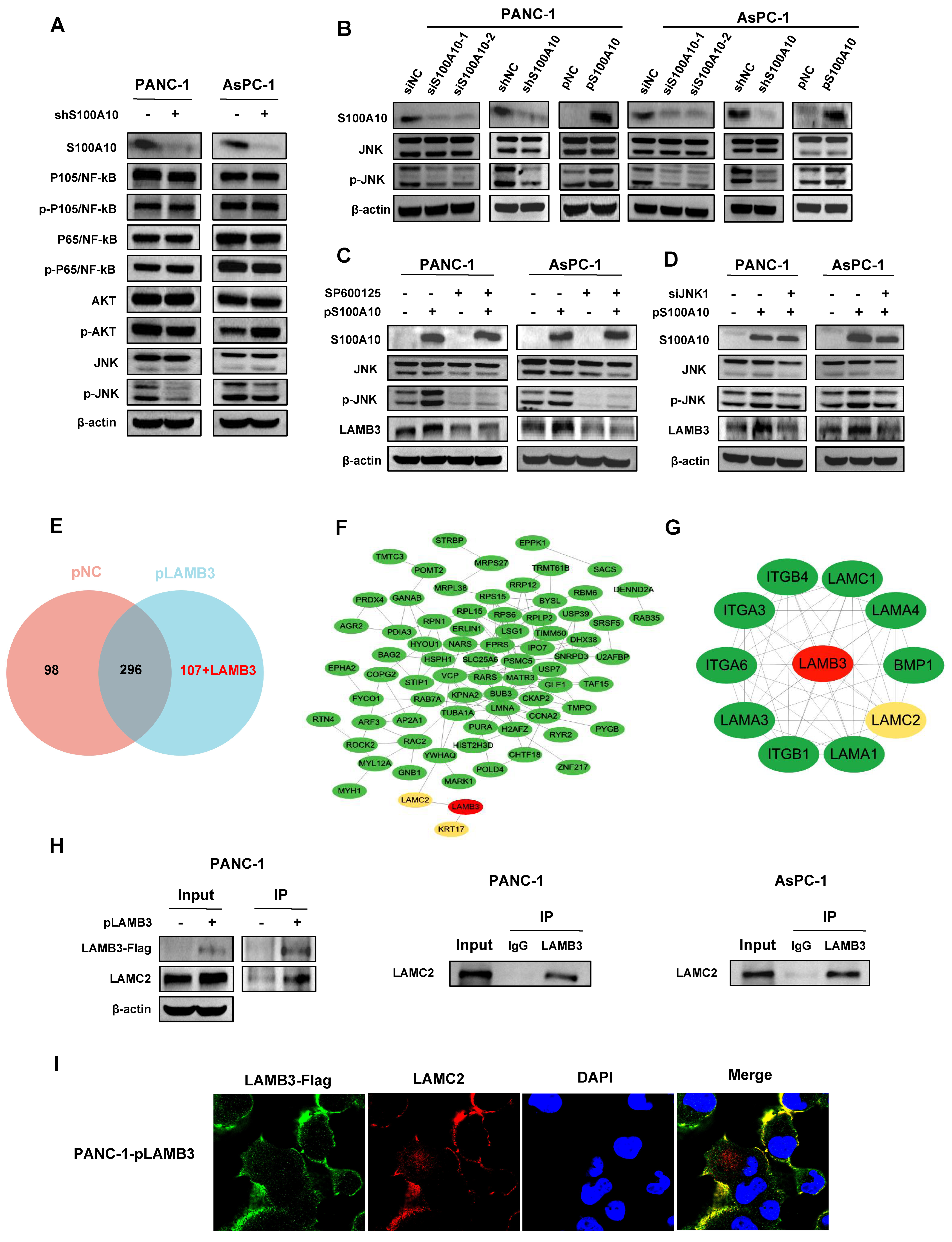
42] in colorectal cancer. In the present study, we also found that LAMB3 was activated by S100A10 in PDAC cells, and its expression was positively correlated with S100A10 expression in human PDAC tissues. Additionally, S100A10-mediated expression of LAMB3 was dependent on the JNK pathway, while LAMB3 has been reported to be modulated by miR-24-3p to influence the progression of PDAC [
43]. Collectively, S100A10 promoted the progression of PDAC via JNK/LAMB3 axis.
Laminin-332 (LM-332) is an important adhesive molecule of basement membranes and is involved in regulating multiple biological functions related to cell adhesion, migration, and invasion in normal tissues [
44]. As a unique subunit of LM-332, LAMC2 has been reported to be up-regulated in esophageal squamous cell carcinoma [
45] and ovarian carcinoma [
46]. In PDAC, the elevated expression of LAMC2 has been detected not only in tumor tissues [
47] but also in serum [
48]. In addition, LAMC2 has also been identified as an oncogene to promote PDAC progression by modulating EMT- and ATP-binding assiette transporters [
49] or EGFR/ERK
1/2/AKT/mTOR axis [
50]. It is indicated that LAMC2 may be served as a candidate biomarker for PDAC diagnosis and treatment. In our study, the expression of LAMC2 was significantly up-regulated in PDAC tissues and showed a strong correlation with the expression of LAMB3, indicating a co-expression pattern of these two genes in PDAC. Consistent with our results, LAMC2 has been reported to be frequently co-expressed with LAMB3 in colorectal cancer, squamous cell carcinoma of the tongue [
51], gastric cancer [
39], prostate cancer [
52] and PDAC [
53], whose intracellular retention at the invasive front of cancer cells has been considered as a more invasive phenotype. We also revealed an interaction between LAMB3 and LAMC2 in PDAC cells, and these two proteins co-localized in the cytoplasm of PDAC cells. In previous studies, LAMB3 has been reported to interact with LAMC2 to form the β3γ2 heterodimer in the cytoplasm of colorectal cancer cells and thus contribute to cancer cell budding [
54,
55]. Based on these results, we postulated that the interaction between LAMB3 and LAMC2 may be a significant step in the progression of PDAC. Further studies are needed to confirm this hypothesis in the future.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







