

Remarkable Synergy When Combining EZH2 Inhibitors with YM155 Is H3K27me3-Independent

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
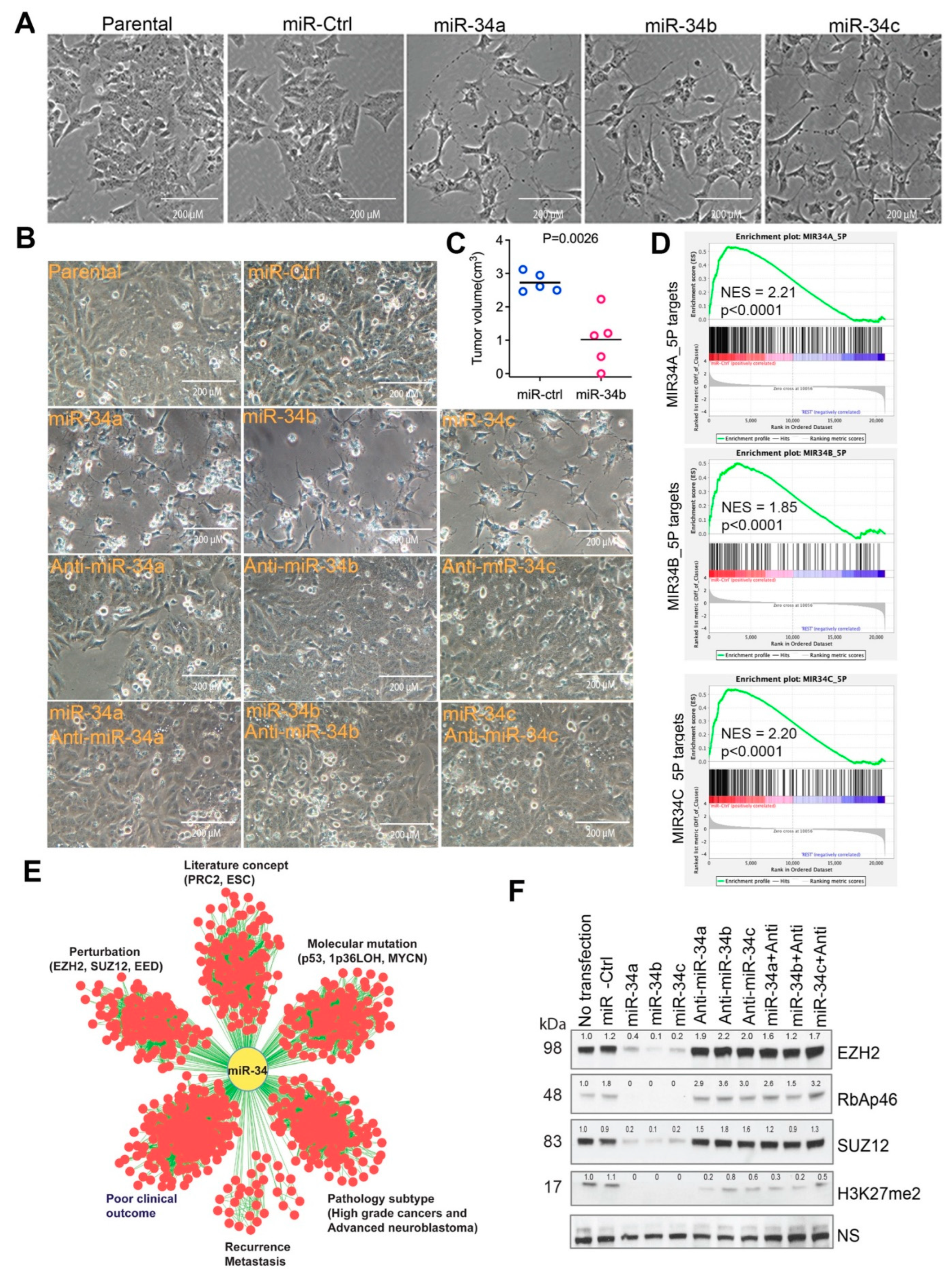
2.1. Tumor Suppressor miR-34 Regulates the Expression of EZH2
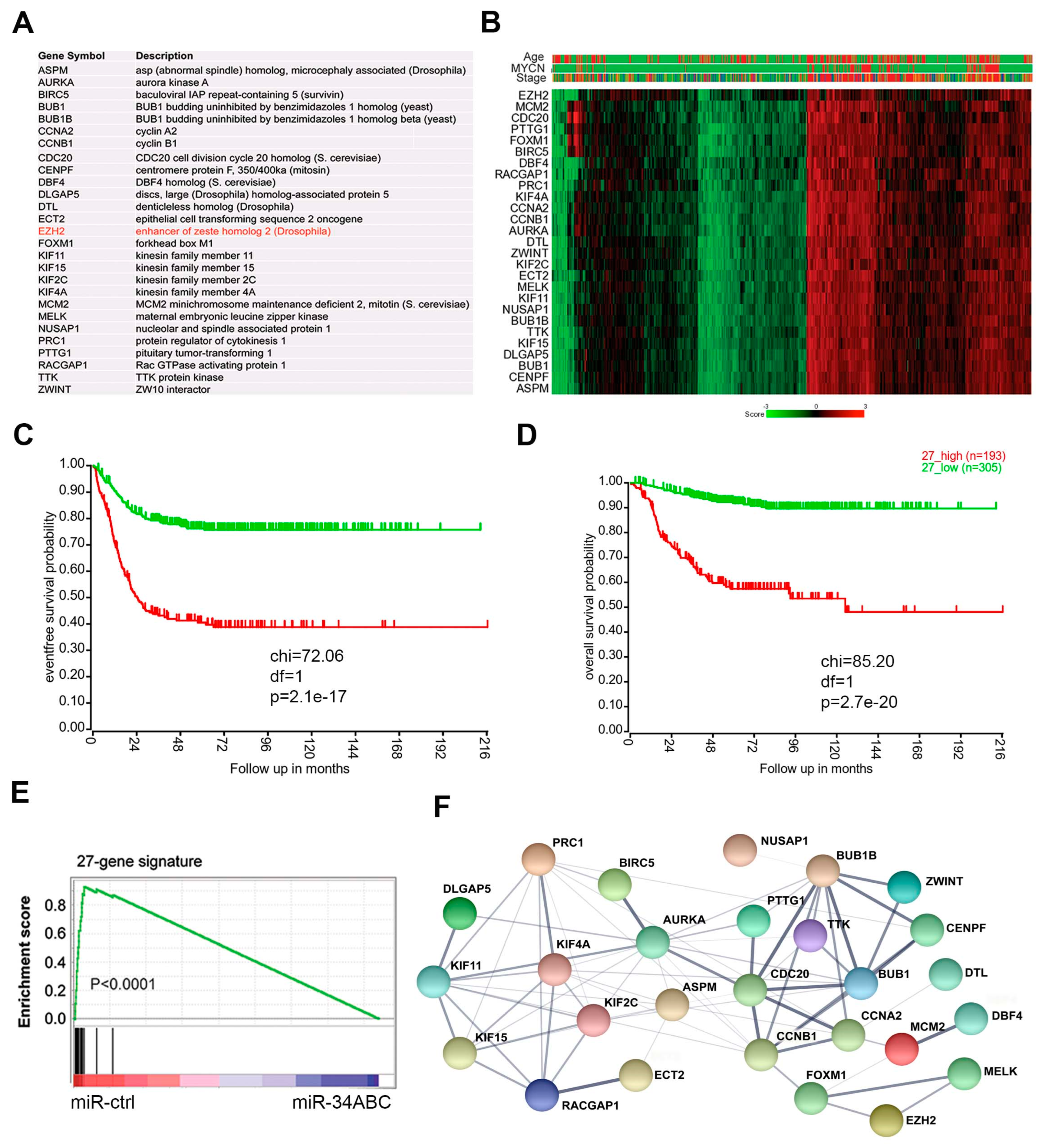
2.2. miR-34 Targets a 27-Gene Network Centered by EZH2
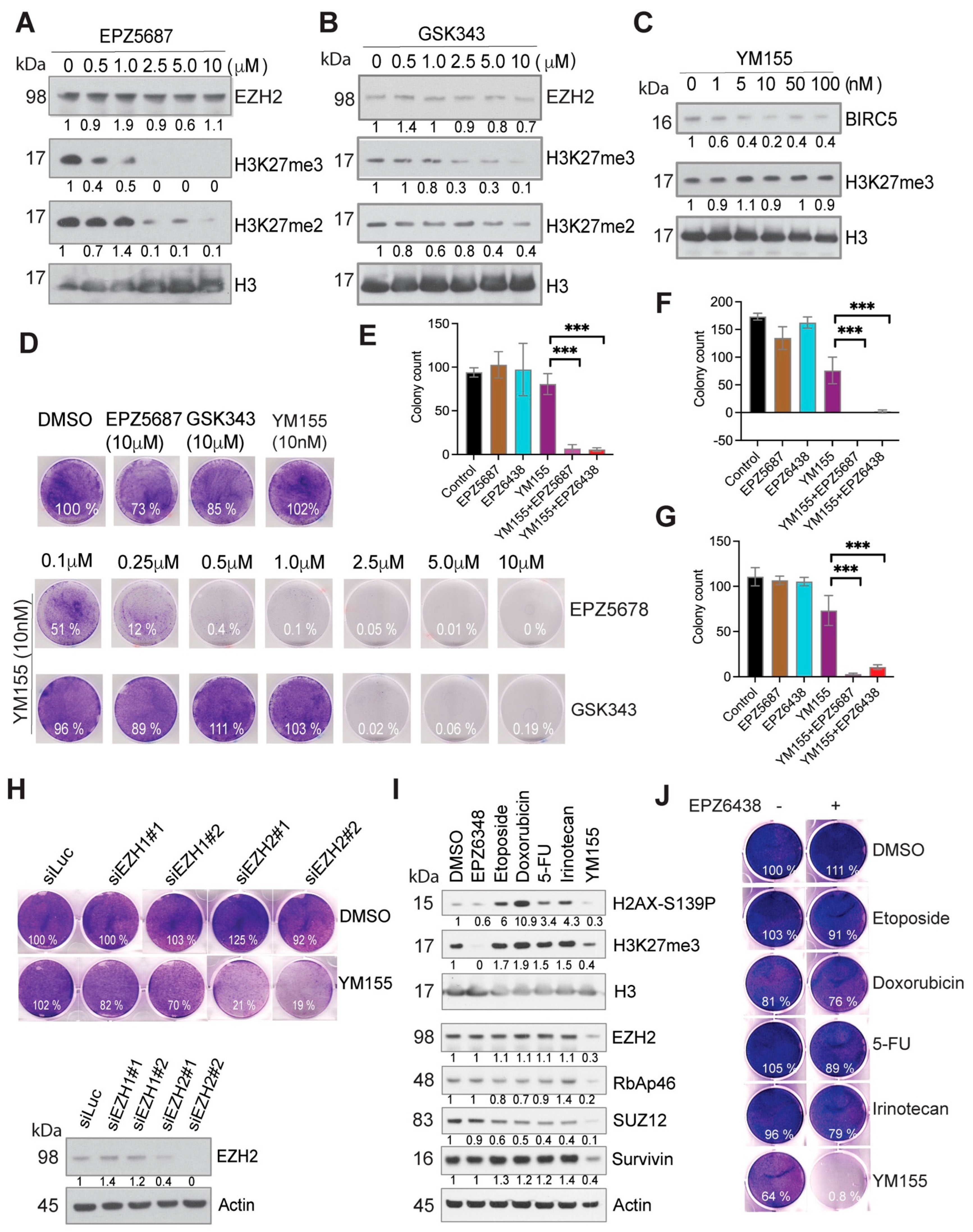
2.3. Pharmacologic Inhibition of EZH2 and BIRC5 Induces a Remarkable Synergistic Effect
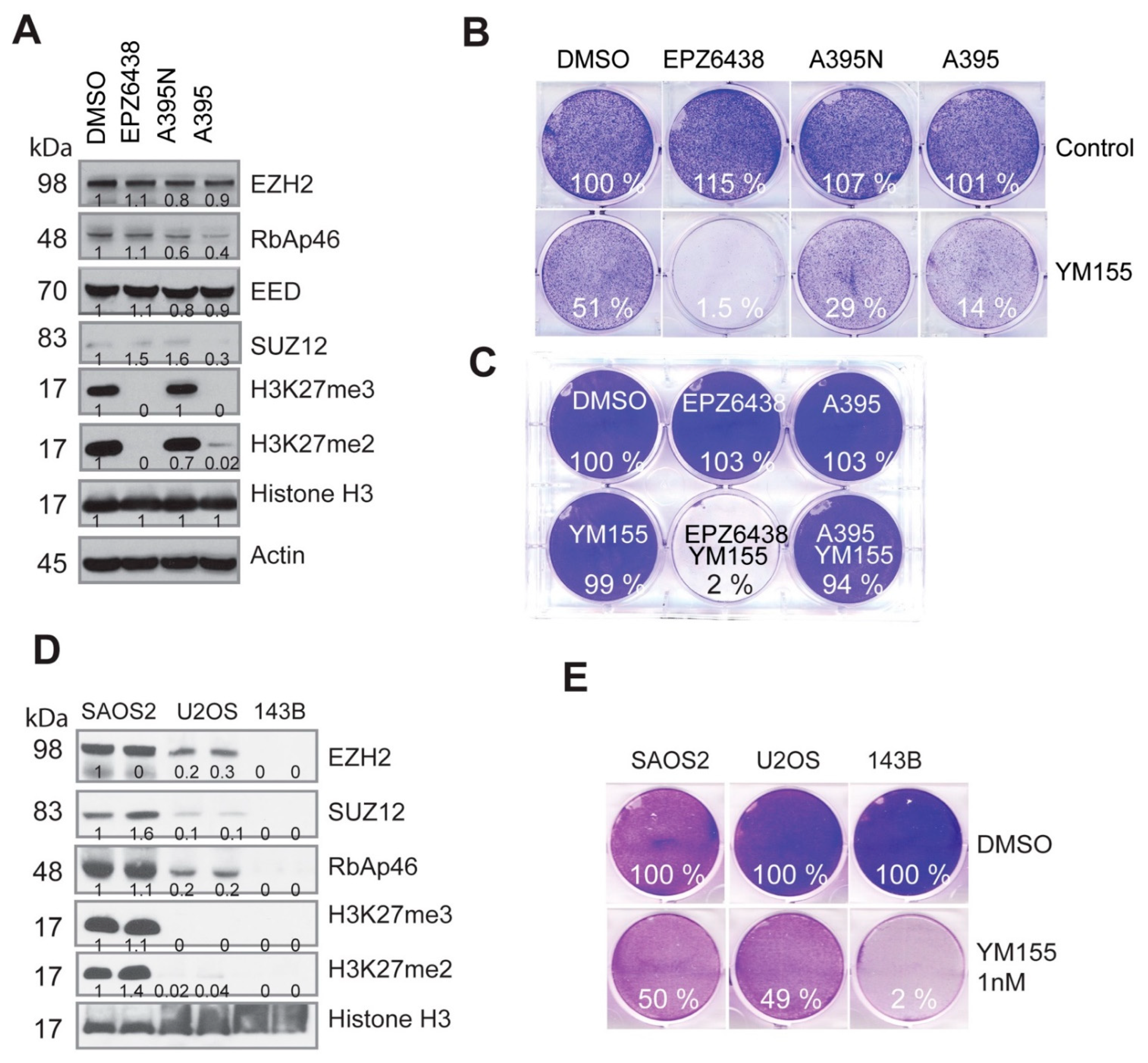
2.4. PRC2-Mediated H3K27 Methylation Is Dispensable for the Synthetic Lethality Induced by EZH2 Inhibition and YM155
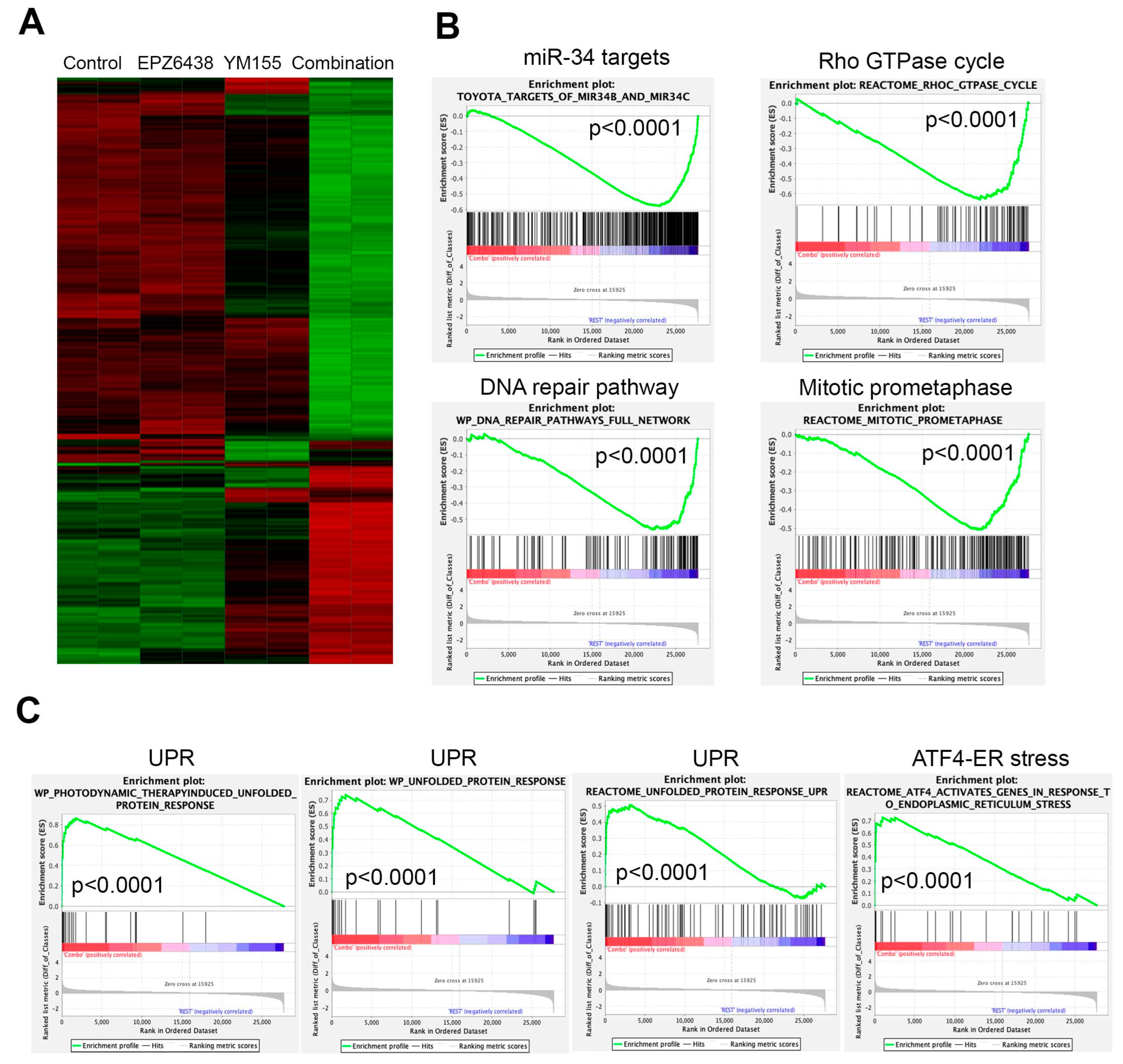
2.5. Combination of EZH2 Inhibitors and YM155 Induces Unfolded Protein Response and Downregulates Multiple Essential Signaling Pathways
3. Discussion
Limitations of This Study
4. Materials and Methods
5. Statistical Analysis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Yang, R.; Zhan, Y.; Lai, H.-D.; Ye, C.-J.; Yao, X.-Y.; Luo, W.-Q.; Cheng, X.-M.; Miao, J.-J.; Wang, J.-F.; et al. Single-Cell Characterization of Malignant Phenotypes and Developmental Trajectories of Adrenal Neuroblastoma. Cancer Cell 2020, 38, 716–733.e6. [Google Scholar] [CrossRef] [PubMed]
- Hanemaaijer, E.S.; Margaritis, T.; Sanders, K.; Bos, F.L.; Candelli, T.; Al-Saati, H.; van Noesel, M.M.; Meyer-Wentrup, F.A.G.; van de Wetering, M.; Holstege, F.C.P.; et al. Single-cell atlas of developing murine adrenal gland reveals relation of Schwann cell precursor signature to neuroblastoma phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2022350118. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, M.; Bachetti, T.; Corrias, M.V.; Brignole, C.; Pastorino, F.; Calarco, E.; Bensa, V.; Giusto, E.; Ceccherini, I.; Perri, P. Recent advances in the developmental origin of neuroblastoma: An overview. J. Exp. Clin. Cancer Res. 2022, 41, 92. [Google Scholar] [CrossRef] [PubMed]
- Kameneva, P.; Artemov, A.V.; Kastriti, M.E.; Faure, L.; Olsen, T.K.; Otte, J.; Erickson, A.; Semsch, B.; Andersson, E.R.; Ratz, M.; et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat. Genet. 2021, 53, 694–706. [Google Scholar] [CrossRef]
- Jansky, S.; Sharma, A.K.; Körber, V.; Quintero, A.; Toprak, U.H.; Wecht, E.M.; Gartlgruber, M.; Greco, A.; Chomsky, E.; Grünewald, T.G.P.; et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat. Genet. 2021, 53, 683–693. [Google Scholar] [CrossRef]
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2015, 122, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef]
- Gartlgruber, M.; Sharma, A.K.; Quintero, A.; Dreidax, D.; Jansky, S.; Park, Y.-G.; Kreth, S.; Meder, J.; Doncevic, D.; Saary, P.; et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat. Cancer 2020, 2, 114–128. [Google Scholar] [CrossRef]
- Van Groningen, T.; Koster, J.; Valentijn, L.J.; Zwijnenburg, D.A.; Akogul, N.; Hasselt, N.E.; Broekmans, M.; Haneveld, F.; Nowakowska, N.E.; Bras, J.; et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat. Genet. 2017, 49, 1261–1266. [Google Scholar] [CrossRef]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugène, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Attiyeh, E.F.; London, W.B.; Mossé, Y.P.; Wang, Q.; Winter, C.; Khazi, D.; McGrady, P.W.; Seeger, R.C.; Look, A.T.; Shimada, H.; et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N. Engl. J. Med. 2005, 353, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; Van Sluis, P.; Valentijn, L.J.; Van Der Ploeg, I.; Hamdi, M.; Van Nes, J.; Westerman, B.A.; Van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Quarni, W.; Goralski, M.; Wan, S.; Jin, H.; Van de Velde, L.-A.; Fang, J.; Wu, Q.; Abu-Zaid, A.; Wang, T.; et al. Targeting the spliceosome through RBM39 degradation results in exceptional responses in high-risk neuroblastoma models. Sci. Adv. 2021, 7, eabj5405. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, A.; Sikka, A.; Yogev, O.; Herendi, L.; Balcells, C.; Ma, Y.; Poon, E.; Eckold, C.; Valbuena, G.N.; Xu, Y.; et al. Indisulam targets RNA splicing and metabolism to serve as a therapeutic strategy for high-risk neuroblastoma. Nat. Commun. 2022, 13, 1380. [Google Scholar] [CrossRef]
- Chipumuro, E.; Marco, E.; Christensen, C.L.; Kwiatkowski, N.; Zhang, T.; Hatheway, C.M.; Abraham, B.J.; Sharma, B.; Yeung, C.; Altabef, A.; et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 2014, 159, 1126–1139. [Google Scholar] [CrossRef] [Green Version]
- D’Oto, A.; Fang, J.; Jin, H.; Xu, B.; Singh, S.; Mullasseril, A.; Jones, V.; Abu-Zaid, A.; von Buttlar, X.; Cooke, B.; et al. KDM6B promotes activation of the oncogenic CDK4/6-pRB-E2F pathway by maintaining enhancer activity in MYCN-amplified neuroblastoma. Nat. Commun. 2021, 12, 7204. [Google Scholar] [CrossRef]
- Poon, E.; Liang, T.; Jamin, Y.; Walz, S.; Kwok, C.; Hakkert, A.; Barker, K.; Urban, Z.; Thway, K.; Zeid, R.; et al. Orally bioavailable CDK9/2 inhibitor shows mechanism-based therapeutic potential in MYCN-driven neuroblastoma. J. Clin. Investig. 2020, 130, 5875–5892. [Google Scholar] [CrossRef]
- Szydzik, J.; Lind, D.E.; Arefin, B.; Kurhe, Y.; Umapathy, G.; Siaw, J.T.; Claeys, A.; Gabre, J.L.; Eynden, J.V.D.; Hallberg, B.; et al. ATR inhibition enables complete tumour regression in ALK-driven NB mouse models. Nat. Commun. 2021, 12, 6813. [Google Scholar] [CrossRef] [PubMed]
- Roeschert, I.; Poon, E.; Henssen, A.G.; Garcia, H.D.; Gatti, M.; Giansanti, C.; Jamin, Y.; Ade, C.P.; Gallant, P.; Schülein-Völk, C.; et al. Combined inhibition of Aurora-A and ATR kinases results in regression of MYCN-amplified neuroblastoma. Nat. Rev. Cancer 2021, 2, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Heitzeneder, S.; Bosse, K.R.; Zhu, Z.; Zhelev, D.; Majzner, R.G.; Radosevich, M.T.; Dhingra, S.; Sotillo, E.; Buongervino, S.; Pascual-Pasto, G.; et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell 2021, 40, 53–69.e9. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Helin, K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer 2009, 9, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, A.; Shen, X.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Wang, G.; Marquez, V.E.; Orkin, S.H.; Pu, W.T. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012, 26, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanulli, S.; Justin, N.; Teissandier, A.; Ancelin, K.; Portoso, M.; Caron, M.; Michaud, A.; Lombard, B.; da Rocha, S.T.; Offer, J.; et al. Jarid2 Methylation via the PRC2 Complex Regulates H3K27me3 Deposition during Cell Differentiation. Mol. Cell 2015, 57, 769–783. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Lee, J.S.; Kim, H.; Kim, K.; Park, H.; Kim, J.-Y.; Lee, S.H.; Kim, I.S.; Kim, J.; Lee, M.; et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol. Cell 2012, 48, 572–586. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Kim, M.; Woo, D.-H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Gunawan, M.; Venkatesan, N.; Loh, J.T.; Wong, J.F.; Berger, H.; Neo, W.H.; Li, L.Y.J.; La Win, M.K.; Yau, Y.H.; Guo, T.; et al. The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat. Immunol. 2015, 16, 505–516. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoloski, G.; Langemeijer, S.M.C.; Kuiper, R.P.; Knops, R.; Massop, M.; Tönnissen, E.R.L.T.M.; Van Der Heijden, A.; Scheele, T.N.; Vandenberghe, P.; De Witte, T.; et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat. Genet. 2010, 42, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Granada, C.N.P.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Roberts, K.H.K.C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Alexe, G.; Dharia, N.; Ross, L.; Iniguez, A.B.; Conway, A.S.; Wang, E.J.; Veschi, V.; Lam, N.; Qi, J.; et al. CRISPR-Cas9 screen reveals a MYCN-amplified neuroblastoma dependency on EZH2. J. Clin. Investig. 2017, 128, 446–462. [Google Scholar] [CrossRef]
- Bownes, L.V.; Williams, A.P.; Marayati, R.; Stafman, L.L.; Markert, H.; Quinn, C.H.; Wadhwani, N.; Aye, J.M.; Stewart, J.E.; Yoon, K.J.; et al. EZH2 inhibition decreases neuroblastoma proliferation and in vivo tumor growth. PLoS ONE 2021, 16, e0246244. [Google Scholar] [CrossRef]
- Li, Z.; Takenobu, H.; Setyawati, A.N.; Akita, N.; Haruta, M.; Satoh, S.; Shinno, Y.; Chikaraishi, K.; Mukae, K.; Akter, J.; et al. EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene 2018, 37, 2714–2727. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chen, C.; Song, Z.; Wang, H.; Ye, M.; Wang, D.; Kang, W.; Liu, H.; Qing, G. EZH2 depletion potentiates MYC degradation inhibiting neuroblastoma and small cell carcinoma tumor formation. Nat. Commun. 2022, 13, 12. [Google Scholar] [CrossRef]
- Wang, C.; Liu, Z.; Woo, C.-W.; Li, Z.; Wang, L.; Wei, J.S.; Marquez, V.E.; Bates, S.E.; Jin, Q.; Khan, J.; et al. EZH2 mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res 2012, 72, 315–324. [Google Scholar] [CrossRef]
- Bracken, A.; Pasini, D.; Capra, M.; Prosperini, E.; Colli, E.; Helin, K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003, 22, 5323–5335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varambally, S.; Cao, Q.; Mani, R.-S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chen, Y.-H.; Li, L.-Y.; Lang, J.; Yeh, S.-P.; Shi, B.; Yang, C.-C.; Yang, J.-Y.; Lin, C.-Y.; Lai, C.-C.; et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nature 2010, 13, 87–94. [Google Scholar] [CrossRef]
- Kaneko, S.; Li, G.; Son, J.; Xu, C.-F.; Margueron, R.; Neubert, T.A.; Reinberg, D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010, 24, 2615–2620. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Chen, S.; Huang, H. Phosphorylation of EZH2 by CDK1 and CDK2: A possible regulatory mechanism of transmission of the H3K27me3 epigenetic mark through cell divisions. Cell Cycle 2011, 10, 579–583. [Google Scholar] [CrossRef]
- Cabezas-Wallscheid, N.; Eichwald, V.; De Graaf, J.; Löwer, M.; Lehr, H.; Kreft, A.; Eshkind, L.; Hildebrandt, A.; Abassi, Y.; Heck, R.; et al. Instruction of haematopoietic lineage choices, evolution of transcriptional landscapes and cancer stem cell hierarchies derived from an AML 1- ETO mouse model. EMBO Mol. Med. 2013, 5, 1804–1820. [Google Scholar] [CrossRef]
- Hongquan, Z.; Lu, D.; Jiang, H.; Chi, X.; Zhang, H. EZH2 is required for mouse oocyte meiotic maturation by interacting with and stabilizing spindle assembly checkpoint protein BubRI. Nucleic Acids Res. 2016, 44, 7659–7672. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.; Sharp, P.A. MicroRNA functions in stress responses. Mol. Cell 2010, 40, 205–215. [Google Scholar] [CrossRef]
- Mendell, J.T.; Olson, E.N. MicroRNAs in stress signaling and human disease. Cell 2012, 148, 1172–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, A.G. miR-34—A microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, K.A.; Attiyeh, E.F.; Mosse, Y.P.; Laquaglia, M.J.; Diskin, S.J.; Brodeur, G.M.; Maris, J.M. A functional screen identifies miR-34a as a candidate neuroblastoma tumor suppressor gene. Mol. Cancer Res. 2008, 6, 735–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivnan, A.; Tracey, L.; Buckley, P.G.; Alcock, L.C.; Davidoff, A.M.; Stallings, R.L. MicroRNA-34a is a potent tumor suppressor molecule in vivo in neuroblastoma. BMC Cancer 2011, 11, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocak, H.; Ackermann, S.; Hero, B.; Kahlert, Y.; Oberthuer, A.; Juraeva, D.; Roels, F.; Theissen, J.; Westermann, F.; Deubzer, H.; et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013, 4, e586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramani, A.K.; Li, Z.; Hart, G.T.; Carlson, M.W.; Boutz, D.R.; Marcotte, E.M. A map of human protein interactions derived from co-expression of human mRNAs and their orthologs. Mol. Syst. Biol. 2008, 4, 180. [Google Scholar] [CrossRef] [PubMed]
- Tannous, B.A.; Kerami, M.; Van der Stoop, P.M.; Kwiatkowski, N.; Wang, J.; Zhou, W.; Kessler, A.F.; Lewandrowski, G.; Hiddingh, L.; Sol, N.; et al. Effects of the selective MPS1 inhibitor MPS1-IN-3 on glioblastoma sensitivity to antimitotic drugs. Gynecol. Oncol. 2013, 105, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.-C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective Inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [Green Version]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Iii, A.D.P.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Feng, H.; Tillman, H.; Wu, G.; Davidoff, A.M.; Yang, J. Frequent epigenetic alterations in polycomb repressive complex 2 in osteosarcoma cell lines. Oncotarget 2018, 9, 27087–27091. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Bohrer, L.R.; Rai, A.N.; Pan, Y.; Gan, L.; Zhou, X.; Bagchi, A.; Simon, J.A.; Huang, H. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. Nat. Cell Biol. 2010, 12, 1108–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bown, N.; Cotterill, S.; Lastowska, M.; O’Neill, S.; Pearson, A.D.J.; Plantaz, D.; Meddeb, M.; Danglot, G.; Brinkschmidt, C.; Christiansen, H.; et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 1999, 340, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaros, T.G.; Stockwin, L.H.; Mullendore, M.E.; Smith, B.; Morrison, B.L.; Newton, D.L. The “survivin suppressants” NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother. Pharmacol. 2012, 70, 207–212. [Google Scholar] [CrossRef]
- Voges, Y.; Michaelis, M.; Rothweiler, F.; Schaller, T.; Schneider, C.; Politt, K.; Mernberger, M.; Nist, A.; Stiewe, T.; Wass, M.; et al. Effects of YM155 on survivin levels and viability in neuroblastoma cells with acquired drug resistance. Cell Death Dis. 2016, 7, e2410. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Davidoff, A.M. Remarkable Synergy When Combining EZH2 Inhibitors with YM155 Is H3K27me3-Independent. Cancers 2023, 15, 208. https://doi.org/10.3390/cancers15010208
Yang J, Davidoff AM. Remarkable Synergy When Combining EZH2 Inhibitors with YM155 Is H3K27me3-Independent. Cancers. 2023; 15(1):208. https://doi.org/10.3390/cancers15010208
Chicago/Turabian StyleYang, Jun, and Andrew M. Davidoff. 2023. "Remarkable Synergy When Combining EZH2 Inhibitors with YM155 Is H3K27me3-Independent" Cancers 15, no. 1: 208. https://doi.org/10.3390/cancers15010208
APA StyleYang, J., & Davidoff, A. M. (2023). Remarkable Synergy When Combining EZH2 Inhibitors with YM155 Is H3K27me3-Independent. Cancers, 15(1), 208. https://doi.org/10.3390/cancers15010208