
Current State of Immunotherapy and Mechanisms of Immune Evasion in Ewing Sarcoma and Osteosarcoma
 , and
, and 
Abstract
:Simple Summary
Abstract
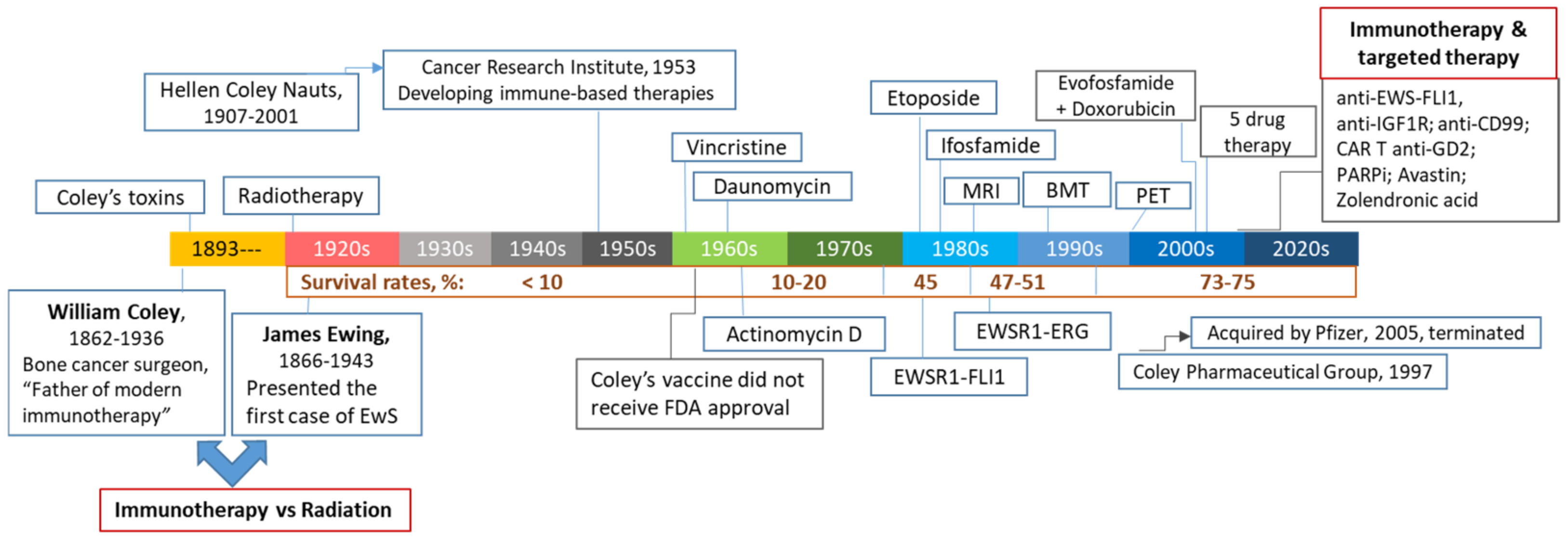
1. Historical Perspectives and Modern Immunotherapy in Pediatric Sarcomas
1.1. William Bradley Coley (1862–1936): The First Focused Effort at Cancer Immunotherapy
1.2. Conventional and Targeted Therapeutics in Treating EwS and OS
1.3. Immunotherapy of Pediatric Bone Cancers

{kind=link}
{kind=link}
| Approach | Goal | Target | Therapeutic Agent | Major Obstacles | Refs |
|---|---|---|---|---|---|
| Immune checkpoint inhibitors | Reactivating and amplifying preexisting antitumor immunity | PD-1 | Nivolumab/OPDIVO® Pembrolizumab/ Keytruda® | Low expression, low mutational burden, Immunological cold TME | [19,43,44,45,46,47,48,49,50,51,52,53,54,55] |
| PD-L1 | Atezolizumab/Tecentriq® | ||||
| CTLA-4 | IpilimumabYERVOY® | ||||
| Tumor specific antigens (TSAs) | Direct tumor targeting | GD2 | Dinutuximab/Unituxin® anti-GD2 CAR-T cells anti-GD2 CAR-engineered NK cells | Variable expression in EwS and OS Upregulation of HLA-G checkpoint | [28,29,30,56] |
| IGF1R | Ganitumab, Dalotuzumab | Activation of compensatory mechanisms, Toxicity | [57] | ||
| HER2 | Trastuzumab/Herceptin® | Not expressed in EwS, no clinical benefit for OS | [18,45] | ||
| B7-H3 | Anti-B7-H3 CAR T cells | [27] | |||
| Antitumor vaccines | Direct tumor targeting | Tumor TSAs or proteins | Dendritic cell vaccine | Need for autologous DCs, Labor-intensive and costly cell isolation | [58] |
| Activation of DC responses | Multiple tumor antigens | Attenuated tumor cells, could be pulsed with GM-CSF, IL-2 or IL-7 or siRNAs | Immunosuppressive TME, low tumor immunogenicity | [22] | |
| Oncolytic viruses | Increase tumor immunogenicity Induce immunogenic cell death | Tumor | Vaccinia virus/Pexa-Vec Reovirus/Reolysin HSV-1/HSV1716 Adenovirus X-Vir | Antiviral immunity, Low delivery efficacy, Immunosuppression, T cell exhaustion | [34,59,60,61,62] |
| Targeting immunosuppressive TME | Macrophage activation | TME | L-MTP-PE/Mifamurtide, BCG, Coley’s toxins, oncolytic viruses | [63,64] | |
| Macrophage polarization | TME | All-trans retinoic acid (ATRA) | Low delivery efficacy | [65] | |
| Macrophage/MDSC depletion | TME | All-trans retinoic acid (ATRA) Trabectedin | Toxicity | [66,67,68,69] |
2. Mechanisms of Immune Escape
2.1. Lack of Tumor-Specific Antigens (TSAs)
2.2. Low Expression of MHC-I and Upregulation of Immune Checkpoints
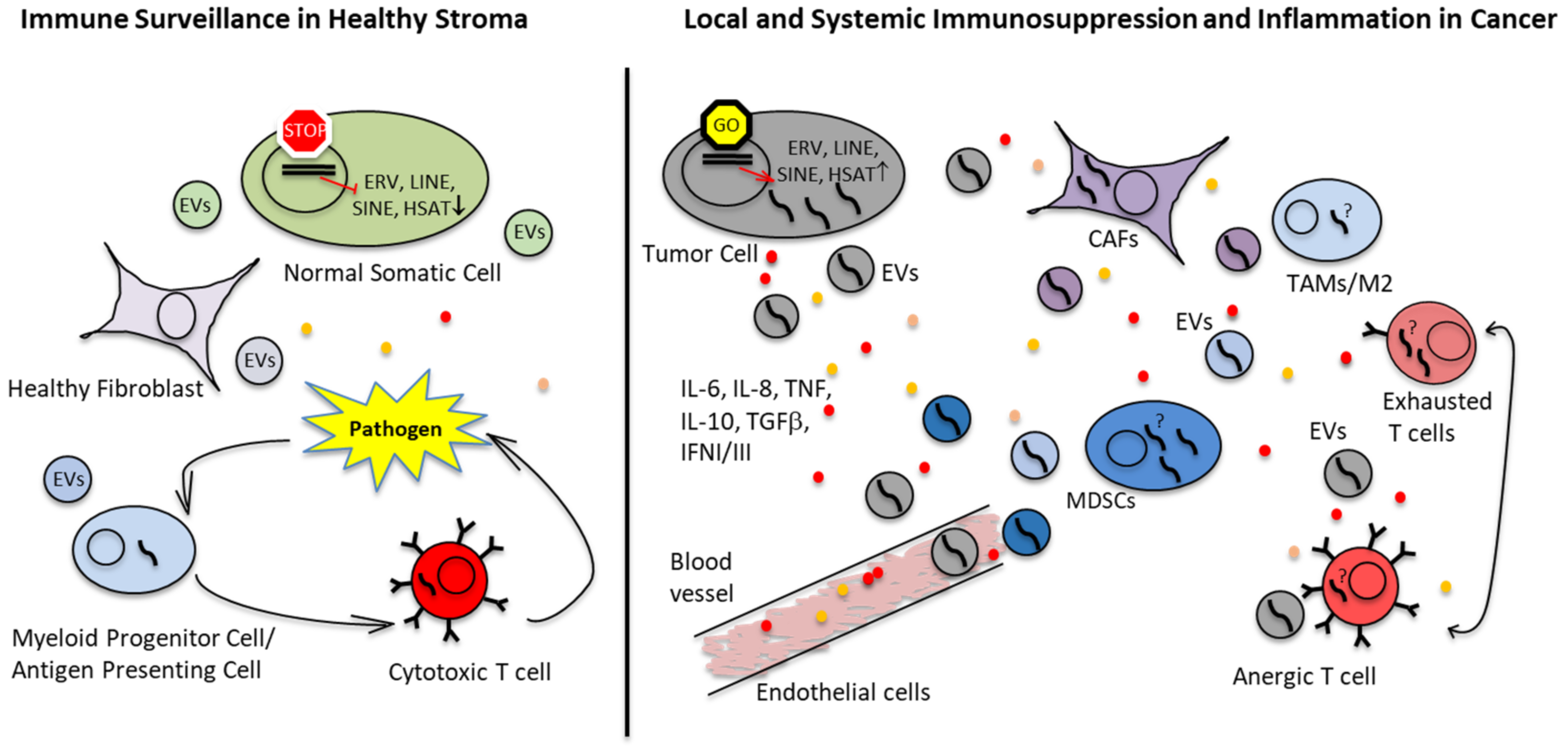
2.3. Immunosuppressive TME in EwS and OS
2.3.1. Improving CD8+ T Cell Infiltration and Antitumor Activity
2.3.2. Targeting Tumor-Associated Macrophages
- Macrophage activation using liposome-encapsulated muramyl tripeptide phosphatidyl ethanolamine (L-MTP-PE or mifamurtide), a constituent of the Mycobacterium cell wall originally purified from the attenuated Mycoblasma bovis, also known as Bacille Calmette-Guerin (BCG). BCG vaccine is currently used for treatment of certain types of cancer and may have a mechanism of action similar to Coley’s toxins. The synthetic L-MTP-PE was shown to stimulate macrophages and to improve survival in OS [63,64,128,129]. Another approach to activate TAMs are oncolytic viruses, which are capable of inducing immunogenic tumor cell death (ICD). This is accompanied by release of TSAs and danger- and pathogen-associated molecular patterns (DAMPs and PAMPs), switching TAMs to antitumorigenic M1 macrophages [130].
- Depleting TAMs and MDSCs using chemotherapeutic agents. For example, trabectedin, which is a natural product from sea squirt shown to inhibit transcription factor bindings such as FUS-CHOP in myxoid liposarcoma or EWS-FLI1 in EwS [131], enhanced CD3+ T cell infiltration in OS and other cancers as well as oncolytic virotherapy against EwS xenograft in a mouse model [67,68,69]. Trabectedin is currently tested in a clinical trial for EwS (NCT04067115).
- Other therapeutic options include block of recruitment and reprogramming metabolic switches.

2.4. Immunogenicity and Response to Immunotherapy of EwS and OS in the Context of Bone and Soft Tissue Sarcomas
2.5. Extracellular Vesicles (EVs) as Means of Immune Escape
3. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ewing, J. Classics in oncology. Diffuse endothelioma of bone. James Ewing. Proceedings of the New York Pathological Society, 1921. CA Cancer J. Clin. 1972, 22, 95–98. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Coley, W.B. The therapeutic value of the mixed toxins of the streptococcus of erysipelas and bacillus prodigiosus in the treatment of inoperable malignant tumors: With a report of one hundred and sixty cases. Am. J. Med. Sci. 1896, 112, 251. [Google Scholar] [CrossRef] [Green Version]
- Coley, W.B. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauts, H.C.; Fowler, G.A.; Bogatko, F.H. A review of the influence of bacterial infection and of bacterial products (Coley’s toxins) on malignant tumors in man; a critical analysis of 30 inoperable cases treated by Coley’s mixed toxins, in which diagnosis was confirmed by microscopic examination selected for special study. Acta Med. Scandinavica Suppl. 1953, 276, 1–103. [Google Scholar]
- Johnston, B.J.; Novales, E.T. Clinical effect of Coley’s toxin. II. A seven-year study. Cancer Chemother. Rep. 1962, 21, 43–68. [Google Scholar]
- Coley, B.L. Neoplasms of Bone; Medical Book Departmnent of Harper & Brothers: New York, NY, USA, 1949; pp. 565–570. [Google Scholar]
- Karbach, J.; Neumann, A.; Brand, K.; Wahle, C.; Siegel, E.; Maeurer, M.; Ritter, E.; Tsuji, T.; Gnjatic, S.; Old, L.J.; et al. Phase I clinical trial of mixed bacterial vaccine (Coley’s toxins) in patients with NY-ESO-1 expressing cancers: Immunological effects and clinical activity. Clin. Cancer Res. 2012, 18, 5449–5459. [Google Scholar] [CrossRef] [Green Version]
- Sedighi, M.; Zahedi Bialvaei, A.; Hamblin, M.R.; Ohadi, E.; Asadi, A.; Halajzadeh, M.; Lohrasbi, V.; Mohammadzadeh, N.; Amiriani, T.; Krutova, M.; et al. Therapeutic bacteria to combat cancer; current advances, challenges, and opportunities. Cancer Med. 2019, 8, 3167–3181. [Google Scholar] [CrossRef] [Green Version]
- Lilienthal, I.; Herold, N. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies. Int. J. Mol. Sci. 2020, 21, 6885. [Google Scholar] [CrossRef]
- Harrison, D.J.; Geller, D.S.; Gill, J.D.; Lewis, V.O.; Gorlick, R. Current and future therapeutic approaches for osteosarcoma. Expert Rev. Anticancer Ther. 2018, 18, 39–50. [Google Scholar] [CrossRef]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat Rev Dis Prim. 2018, 4, 5. [Google Scholar] [CrossRef]
- Riggi, N.; Suvà, M.L.; Stamenkovic, I. Ewing’s Sarcoma. N. Engl. J. Med. 2021, 384, 154–164. [Google Scholar] [CrossRef]
- Grünewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef]
- Zöllner, S.K.; Amatruda, J.F.; Bauer, S.; Collaud, S.; de Álava, E.; DuBois, S.G.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. 2021, 10, 1685. [Google Scholar] [CrossRef]
- Ginsberg, J.P.; Goodman, P.; Leisenring, W.; Ness, K.K.; Meyers, P.A.; Wolden, S.L.; Smith, S.M.; Stovall, M.; Hammond, S.; Robison, L.L.; et al. Long-term survivors of childhood Ewing sarcoma: Report from the childhood cancer survivor study. J. Natl. Cancer Inst. 2010, 102, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging novel agents for patients with advanced Ewing sarcoma: A report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Research 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Cripe, T.P. Immunotherapies for Pediatric Solid Tumors: A Targeted Update. Paediatr. Drugs 2022, 24, 1–12. [Google Scholar] [CrossRef]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Dyson, K.A.; Stover, B.D.; Grippin, A.; Mendez-Gomez, H.R.; Lagmay, J.; Mitchell, D.A.; Sayour, E.J. Emerging trends in immunotherapy for pediatric sarcomas. J. Hematol. Oncol. 2019, 12, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696. [Google Scholar] [CrossRef]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Dobrenkov, K.; Ostrovnaya, I.; Gu, J.; Cheung, I.Y.; Cheung, N.K. Oncotargets GD2 and GD3 are highly expressed in sarcomas of children, adolescents, and young adults. Pediatr. Blood Cancer 2016, 63, 1780–1785. [Google Scholar] [CrossRef] [Green Version]
- Poon, V.I.; Roth, M.; Piperdi, S.; Geller, D.; Gill, J.; Rudzinski, E.R.; Hawkins, D.S.; Gorlick, R. Ganglioside GD2 expression is maintained upon recurrence in patients with osteosarcoma. Clin. Sarcoma Res. 2015, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Kailayangiri, S.; Altvater, B.; Meltzer, J.; Pscherer, S.; Luecke, A.; Dierkes, C.; Titze, U.; Leuchte, K.; Landmeier, S.; Hotfilder, M.; et al. The ganglioside antigen GD2 is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br. J. Cancer 2012, 106, 1123–1133. [Google Scholar] [CrossRef]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef]
- Morales, E.; Olson, M.; Iglesias, F.; Dahiya, S.; Luetkens, T.; Atanackovic, D. Role of immunotherapy in Ewing sarcoma. J. Immunother. Cancer 2020, 8, e000653. [Google Scholar] [CrossRef]
- Roberts, S.S.; Chou, A.J.; Cheung, N.K. Immunotherapy of Childhood Sarcomas. Front. Oncol. 2015, 5, 181. [Google Scholar] [CrossRef] [Green Version]
- Corre, I.; Verrecchia, F.; Crenn, V.; Redini, F.; Trichet, V. The Osteosarcoma Microenvironment: A Complex But Targetable Ecosystem. Cells 2020, 9, 976. [Google Scholar] [CrossRef] [Green Version]
- Morales, E.; Olson, M.; Iglesias, F.; Luetkens, T.; Atanackovic, D. Targeting the tumor microenvironment of Ewing sarcoma. Immunotherapy 2021, 13, 1439–1451. [Google Scholar] [CrossRef]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef]
- Haworth, K.B.; Leddon, J.L.; Chen, C.Y.; Horwitz, E.M.; Mackall, C.L.; Cripe, T.P. Going back to class I: MHC and immunotherapies for childhood cancer. Pediatr. Blood Cancer 2015, 62, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Buzas, E.I. The roles of extracellular vesicles in the immune system. Nat. Rev. Immunol. 2022, 1–15. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Zeng, F.; Morelli, A.E. Extracellular vesicle-mediated MHC cross-dressing in immune homeostasis, transplantation, infectious diseases, and cancer. Semin. Immunopathol. 2018, 40, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Keung, E.Z.; Burgess, M.; Salazar, R.; Parra, E.R.; Rodrigues-Canales, J.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Attia, S.; Riedel, R.F.; et al. Correlative Analyses of the SARC028 Trial Reveal an Association Between Sarcoma-Associated Immune Infiltrate and Response to Pembrolizumab. Clin. Cancer Res. 2020, 26, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Gorlick, R. Advancing therapy for osteosarcoma. Nat. Rev. Clin. Oncol. 2021, 18, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Sundara, Y.T.; Kostine, M.; Cleven, A.H.; Bovée, J.V.; Schilham, M.W.; Cleton-Jansen, A.M. Increased PD-L1 and T-cell infiltration in the presence of HLA class I expression in metastatic high-grade osteosarcoma: A rationale for T-cell-based immunotherapy. Cancer Immunol. Immunother. CII 2017, 66, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Machado, I.; López-Guerrero, J.A.; Scotlandi, K.; Picci, P.; Llombart-Bosch, A. Immunohistochemical analysis and prognostic significance of PD-L1, PD-1, and CD8+ tumor-infiltrating lymphocytes in Ewing’s sarcoma family of tumors (ESFT). Virchows Arch. Int. J. Pathol. 2018, 472, 815–824. [Google Scholar] [CrossRef]
- Koirala, P.; Roth, M.E.; Gill, J.; Piperdi, S.; Chinai, J.M.; Geller, D.S.; Hoang, B.H.; Park, A.; Fremed, M.A.; Zang, X.; et al. Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma. Sci. Rep. 2016, 6, 30093. [Google Scholar] [CrossRef] [Green Version]
- Spurny, C.; Kailayangiri, S.; Jamitzky, S.; Altvater, B.; Wardelmann, E.; Dirksen, U.; Hardes, J.; Hartmann, W.; Rossig, C. Programmed cell death ligand 1 (PD-L1) expression is not a predominant feature in Ewing sarcomas. Pediatr. Blood Cancer 2018, 65. [Google Scholar] [CrossRef]
- Pinto, N.; Park, J.R.; Murphy, E.; Yearley, J.; McClanahan, T.; Annamalai, L.; Hawkins, D.S.; Rudzinski, E.R. Patterns of PD-1, PD-L1, and PD-L2 expression in pediatric solid tumors. Pediatr. Blood Cancer 2017, 64, e26613. [Google Scholar] [CrossRef]
- Raj, S.; Bui, M.; Gonzales, R.; Letson, D.; Antonia, S.J. Impact of Pdl1 Expression on Clinical Outcomes in Subtypes of Sarcoma. Ann. Oncol. 2014, 25, iv498. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Davis, K.L.; Fox, E.; Merchant, M.S.; Reid, J.M.; Kudgus, R.A.; Liu, X.; Minard, C.G.; Voss, S.; Berg, S.L.; Weigel, B.J.; et al. Nivolumab in children and young adults with relapsed or refractory solid tumours or lymphoma (ADVL1412): A multicentre, open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020, 21, 541–550. [Google Scholar] [CrossRef]
- Van Erp, A.E.M.; Versleijen-Jonkers, Y.M.H.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Gorris, M.A.J.; van Dam, L.S.; Mentzel, T.; Weidema, M.E.; Savci-Heijink, C.D.; Desar, I.M.E.; et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8(+) lymphocytes in primary sarcomas is subtype dependent. Oncotarget 2017, 8, 71371–71384. [Google Scholar] [CrossRef] [Green Version]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [Green Version]
- Amin, H.M.; Morani, A.C.; Daw, N.C.; Lamhamedi-Cherradi, S.E.; Subbiah, V.; Menegaz, B.A.; Vishwamitra, D.; Eskandari, G.; George, B.; Benjamin, R.S.; et al. IGF-1R/mTOR Targeted Therapy for Ewing Sarcoma: A Meta-Analysis of Five IGF-1R-Related Trials Matched to Proteomic and Radiologic Predictive Biomarkers. Cancers 2020, 12, 1768. [Google Scholar] [CrossRef]
- Merchant, M.S.; Bernstein, D.; Amoako, M.; Baird, K.; Fleisher, T.A.; Morre, M.; Steinberg, S.M.; Sabatino, M.; Stroncek, D.F.; Venkatasan, A.M.; et al. Adjuvant Immunotherapy to Improve Outcome in High-Risk Pediatric Sarcomas. Clin. Cancer Res. 2016, 22, 3182–3191. [Google Scholar] [CrossRef] [Green Version]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Kolb, E.A.; Sampson, V.; Stabley, D.; Walter, A.; Sol-Church, K.; Cripe, T.; Hingorani, P.; Ahern, C.H.; Weigel, B.J.; Zwiebel, J.; et al. A phase I trial and viral clearance study of reovirus (Reolysin) in children with relapsed or refractory extra-cranial solid tumors: A Children’s Oncology Group Phase I Consortium report. Pediatr. Blood Cancer 2015, 62, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Streby, K.A.; Currier, M.A.; Triplet, M.; Ott, K.; Dishman, D.J.; Vaughan, M.R.; Ranalli, M.A.; Setty, B.; Skeens, M.A.; Whiteside, S.; et al. First-in-Human Intravenous Seprehvir in Young Cancer Patients: A Phase 1 Clinical Trial. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 1930–1938. [Google Scholar] [CrossRef] [Green Version]
- Koch, J.; Schober, S.J.; Hindupur, S.V.; Schöning, C.; Klein, F.G.; Mantwill, K.; Ehrenfeld, M.; Schillinger, U.; Hohnecker, T.; Qi, P.; et al. Targeting the Retinoblastoma/E2F repressive complex by CDK4/6 inhibitors amplifies oncolytic potency of an oncolytic adenovirus. Nat. Commun. 2022, 13, 4689. [Google Scholar] [CrossRef] [PubMed]
- Meyers, P.A.; Schwartz, C.L.; Krailo, M.D.; Healey, J.H.; Bernstein, M.L.; Betcher, D.; Ferguson, W.S.; Gebhardt, M.C.; Goorin, A.M.; Harris, M.; et al. Osteosarcoma: The addition of muramyl tripeptide to chemotherapy improves overall survival—A report from the Children’s Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Pahl, J.H.W.; Kwappenberg, K.M.C.; Varypataki, E.M.; Santos, S.J.; Kuijjer, M.L.; Mohamed, S.; Wijnen, J.T.; van Tol, M.J.D.; Cleton-Jansen, A.-M.; Egeler, R.M.; et al. Macrophages inhibit human osteosarcoma cell growth after activation with the bacterial cell wall derivative liposomal muramyl tripeptide in combination with interferon-γ. J. Exp. Clin. Cancer Res. 2014, 33, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Xian, M.; Xiang, S.; Xiang, D.; Shao, X.; Wang, J.; Cao, J.; Yang, X.; Yang, B.; Ying, M.; et al. All-Trans Retinoic Acid Prevents Osteosarcoma Metastasis by Inhibiting M2 Polarization of Tumor-Associated Macrophages. Cancer Immunol. Res. 2017, 5, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, A.H.; Highfill, S.L.; Cui, Y.; Smith, J.P.; Walker, A.J.; Ramakrishna, S.; El-Etriby, R.; Galli, S.; Tsokos, M.G.; Orentas, R.J.; et al. Reduction of MDSCs with All-trans Retinoic Acid Improves CAR Therapy Efficacy for Sarcomas. Cancer Immunol. Res. 2016, 4, 869–880. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Xu, Y.; Fu, J.; Zhu, X.; Chen, H.; Xu, H.; Wang, G.; Song, Y.; Song, G.; Lu, J.; et al. Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci. 2020, 111, 1899–1909. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Denton, N.L.; Chen, C.-Y.; Hutzen, B.; Currier, M.A.; Scott, T.; Nartker, B.; Leddon, J.L.; Wang, P.-Y.; Srinivas, R.; Cassady, K.A.; et al. Myelolytic Treatments Enhance Oncolytic Herpes Virotherapy in Models of Ewing Sarcoma by Modulating the Immune Microenvironment. Mol. Ther.—Oncolytics 2018, 11, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Carter, R.A.; Li, Y.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar] [CrossRef]
- Thiel, U.; Schober, S.J.; Einspieler, I.; Kirschner, A.; Thiede, M.; Schirmer, D.; Gall, K.; Blaeschke, F.; Schmidt, O.; Jabar, S.; et al. Ewing sarcoma partial regression without GvHD by chondromodulin-I/HLA-A*02:01-specific allorestricted T cell receptor transgenic T cells. Oncoimmunology 2017, 6, e1312239. [Google Scholar] [CrossRef]
- Staege, M.S.; Gorelov, V.; Bulankin, A.; Fischer, U.; Dumon, K.; Hohndorf, L.; Hattenhorst, U.; Kramm, C.; Burdach, S. Stable transgenic expression of IL-2 and HSV1-tk by single and fusion tumor cell lines bearing EWS/FLI-1 chimeric genes. Pediatr. Hematol. Oncol. 2003, 20, 119–140. [Google Scholar] [CrossRef]
- Biele, E.; Schober, S.J.; Prexler, C.; Thiede, M.; Heyking, K.V.; Gassmann, H.; Eck, J.; Xue, B.; Burdach, S.; Thiel, U. Monocyte Maturation Mediators Upregulate CD83, ICAM-1 and MHC Class 1 Expression on Ewing’s Sarcoma, Enhancing T Cell Cytotoxicity. Cells 2021, 10, 3070. [Google Scholar] [CrossRef]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, P.; Alcazer, V.; Mutez, V.; Tonon, L.; Martin, J.; Chuvin, N.; Michel, E.; Boulos, R.E.; Estornes, Y.; Valladeau-Guilemond, J.; et al. Identification of shared tumor epitopes from endogenous retroviruses inducing high-avidity cytotoxic T cells for cancer immunotherapy. Sci. Adv. 2022, 8, eabj3671. [Google Scholar] [CrossRef]
- Ouspenskaia, T.; Law, T.; Clauser, K.R.; Klaeger, S.; Sarkizova, S.; Aguet, F.; Li, B.; Christian, E.; Knisbacher, B.A.; Le, P.M.; et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol. 2022, 40, 209–217. [Google Scholar] [CrossRef]
- Camp, F.A.; Slansky, J.E. Implications of Antigen Selection on T Cell-Based Immunotherapy. Pharmaceuticals 2021, 14, 993. [Google Scholar] [CrossRef]
- Ishak, C.A.; Carvalho, D.D.D. Reactivation of Endogenous Retroelements in Cancer Development and Therapy. Annu. Rev. Cancer Biol. 2020, 4, 159–176. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. TIG 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Perry, J.A.; Seong, B.K.A.; Stegmaier, K. Biology and Therapy of Dominant Fusion Oncoproteins Involving Transcription Factor and Chromatin Regulators in Sarcomas. Annu. Rev. Cancer Biol. 2019, 3, 299–321. [Google Scholar] [CrossRef]
- Vibert, J.; Saulnier, O.; Collin, C.; Petit, F.; Borgman, K.J.E.; Vigneau, J.; Gautier, M.; Zaidi, S.; Pierron, G.; Watson, S.; et al. Oncogenic chimeric transcription factors drive tumor-specific transcription, processing, and translation of silent genomic regions. Mol. Cell 2022, 82, 2458–2471.e2459. [Google Scholar] [CrossRef]
- Berghuis, D.; de Hooge, A.S.; Santos, S.J.; Horst, D.; Wiertz, E.J.; van Eggermond, M.C.; van den Elsen, P.J.; Taminiau, A.H.; Ottaviano, L.; Schaefer, K.L.; et al. Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: Implications for immune recognition. J. Pathol. 2009, 218, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Yabe, H.; Tsukahara, T.; Kawaguchi, S.; Wada, T.; Torigoe, T.; Sato, N.; Terai, C.; Aoki, M.; Hirose, S.; Morioka, H.; et al. Prognostic significance of HLA class I expression in Ewing’s sarcoma family of tumors. J. Surg. Oncol. 2011, 103, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Kawaguchi, S.; Torigoe, T.; Asanuma, H.; Nakazawa, E.; Shimozawa, K.; Nabeta, Y.; Kimura, S.; Kaya, M.; Nagoya, S.; et al. Prognostic significance of HLA class I expression in osteosarcoma defined by anti-pan HLA class I monoclonal antibody, EMR8-5. Cancer Sci. 2006, 97, 1374–1380. [Google Scholar] [CrossRef]
- Nada, O.H.; Ahmed, N.S.; Abou Gabal, H.H. Prognostic significance of HLA EMR8-5 immunohistochemically analyzed expression in osteosarcoma. Diagn. Pathol. 2014, 9, 72. [Google Scholar] [CrossRef]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef] [Green Version]
- Cornel, A.M.; Mimpen, I.L.; Nierkens, S. MHC Class I Downregulation in Cancer: Underlying Mechanisms and Potential Targets for Cancer Immunotherapy. Cancers 2020, 12, 1760. [Google Scholar] [CrossRef]
- Borowski, A.; van Valen, F.; Ulbrecht, M.; Weiss, E.H.; Blasczyk, R.; Jorgens, H.; Gobel, U.; Schneider, E.M. Monomorphic HLA class I-(non-A, non-B) expression on Ewing’s tumor cell lines, modulation by TNF-α and IFN-γ. Immunobiology 1999, 200, 1–20. [Google Scholar] [CrossRef]
- Spurny, C.; Kailayangiri, S.; Altvater, B.; Jamitzky, S.; Hartmann, W.; Wardelmann, E.; Ranft, A.; Dirksen, U.; Amler, S.; Hardes, J.; et al. T cell infiltration into Ewing sarcomas is associated with local expression of immune-inhibitory HLA-G. Oncotarget 2018, 9, 6536–6549. [Google Scholar] [CrossRef] [Green Version]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- Rübe, C.E.; van Valen, F.; Wilfert, F.; Palm, J.; Schuck, A.; Willich, N.; Winkelmann, W.; Jürgens, H.; Rübe, C. Ewing’s sarcoma and peripheral primitive neuroectodermal tumor cells produce large quantities of bioactive tumor necrosis factor-alpha (TNF-alpha) after radiation exposure. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 1414–1425. [Google Scholar] [CrossRef]
- Lam, E.; Stein, S.; Falck-Pedersen, E. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol. 2014, 88, 974–981. [Google Scholar] [CrossRef] [Green Version]
- Rouas-Freiss, N.; Gonçalves, R.M.; Menier, C.; Dausset, J.; Carosella, E.D. Direct evidence to support the role of HLA-G in protecting the fetus from maternal uterine natural killer cytolysis. Proc. Natl. Acad. Sci. USA 1997, 94, 11520–11525. [Google Scholar] [CrossRef] [Green Version]
- Altvater, B.; Kailayangiri, S.; Pérez Lanuza, L.F.; Urban, K.; Greune, L.; Flügge, M.; Meltzer, J.; Farwick, N.; König, S.; Görlich, D.; et al. HLA-G and HLA-E Immune Checkpoints Are Widely Expressed in Ewing Sarcoma but Have Limited Functional Impact on the Effector Functions of Antigen-Specific CAR T Cells. Cancers 2021, 13, 2857. [Google Scholar] [CrossRef]
- Loustau, M.; Anna, F.; Dréan, R.; Lecomte, M.; Langlade-Demoyen, P.; Caumartin, J. HLA-G Neo-Expression on Tumors. Front. Immunol. 2020, 11, 1685. [Google Scholar] [CrossRef]
- Chowdhury, F.; Dunn, S.; Mitchell, S.; Mellows, T.; Ashton-Key, M.; Gray, J.C. PD-L1 and CD8+PD1+ lymphocytes exist as targets in the pediatric tumor microenvironment for immunomodulatory therapy. Oncoimmunology 2015, 4, e1029701. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; Larousserie, F.; Entz-Werlé, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 trial. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Wu, C.-C.; Beird, H.C.; Andrew Livingston, J.; Advani, S.; Mitra, A.; Cao, S.; Reuben, A.; Ingram, D.; Wang, W.-L.; Ju, Z.; et al. Immuno-genomic landscape of osteosarcoma. Nat. Commun. 2020, 11, 1008. [Google Scholar] [CrossRef] [Green Version]
- Brohl, A.S.; Sindiri, S.; Wei, J.S.; Milewski, D.; Chou, H.-C.; Song, Y.K.; Wen, X.; Kumar, J.; Reardon, H.V.; Mudunuri, U.S.; et al. Immuno-transcriptomic profiling of extracranial pediatric solid malignancies. Cell Rep. 2021, 37, 110047. [Google Scholar] [CrossRef]
- Trieb, K.; Lechleitner, T.; Lang, S.; Windhager, R.; Kotz, R.; Dirnhofer, S. Evaluation of HLA-DR expression and T-lymphocyte infiltration in osteosarcoma. Pathol. Res. Pract. 1998, 194, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, D.; Santos, S.J.; Baelde, H.J.; Taminiau, A.H.M.; Maarten Egeler, R.; Schilham, M.W.; Hogendoorn, P.C.W.; Lankester, A.C. Pro-inflammatory chemokine–chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8+ T-lymphocyte infiltration and affect tumour progression. J. Pathol. 2011, 223, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Stahl, D.; Gentles, A.J.; Thiele, R.; Gütgemann, I. Prognostic profiling of the immune cell microenvironment in Ewing’s Sarcoma Family of Tumors. Oncoimmunology 2019, 8, e1674113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- House, I.G.; Savas, P.; Lai, J.; Chen, A.X.Y.; Oliver, A.J.; Teo, Z.L.; Todd, K.L.; Henderson, M.A.; Giuffrida, L.; Petley, E.V.; et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin. Cancer Res. 2020, 26, 487–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumwalt, T.J.; Arnold, M.; Goel, A.; Boland, C.R. Active secretion of CXCL10 and CCL5 from colorectal cancer microenvironments associates with GranzymeB+ CD8+ T-cell infiltration. Oncotarget 2015, 6, 2981–2991. [Google Scholar] [CrossRef] [Green Version]
- Cillo, A.R.; Mukherjee, E.; Bailey, N.G.; Onkar, S.; Daley, J.; Salgado, C.; Li, X.; Li, D.; Ranganathan, S.; Burgess, M.; et al. Ewing sarcoma and osteosarcoma have distinct immune signatures and intercellular communication networks. Clin. Cancer Res. 2022, 28, 4968–4982. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef]
- Li, X.; Lu, M.; Yuan, M.; Ye, J.; Zhang, W.; Xu, L.; Wu, X.; Hui, B.; Yang, Y.; Wei, B.; et al. CXCL10-armed oncolytic adenovirus promotes tumor-infiltrating T-cell chemotaxis to enhance anti-PD-1 therapy. Oncoimmunology 2022, 11, 2118210. [Google Scholar] [CrossRef]
- Barreira da Silva, R.; Laird, M.E.; Yatim, N.; Fiette, L.; Ingersoll, M.A.; Albert, M.L. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat. Immunol. 2015, 16, 850–858. [Google Scholar] [CrossRef]
- Decalf, J.; Tarbell, K.V.; Casrouge, A.; Price, J.D.; Linder, G.; Mottez, E.; Sultanik, P.; Mallet, V.; Pol, S.; Duffy, D.; et al. Inhibition of DPP4 activity in humans establishes its in vivo role in CXCL10 post-translational modification: Prospective placebo-controlled clinical studies. EMBO Mol. Med. 2016, 8, 679–683. [Google Scholar] [CrossRef]
- Ligon, J.A.; Choi, W.; Cojocaru, G.; Fu, W.; Hsiue, E.H.-C.; Oke, T.F.; Siegel, N.; Fong, M.H.; Ladle, B.; Pratilas, C.A.; et al. Pathways of immune exclusion in metastatic osteosarcoma are associated with inferior patient outcomes. J. ImmunoTherapy Cancer 2021, 9, e001772. [Google Scholar] [CrossRef]
- Altvater, B.; Kailayangiri, S.; Theimann, N.; Ahlmann, M.; Farwick, N.; Chen, C.; Pscherer, S.; Neumann, I.; Mrachatz, G.; Hansmeier, A.; et al. Common Ewing sarcoma-associated antigens fail to induce natural T cell responses in both patients and healthy individuals. Cancer Immunol. Immunother. CII 2014, 63, 1047–1060. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Fukushi, J.; Yamamoto, S.; Matsumoto, Y.; Setsu, N.; Oda, Y.; Yamada, H.; Okada, S.; Watari, K.; Ono, M.; et al. Macrophage infiltration predicts a poor prognosis for human ewing sarcoma. Am. J. Pathol. 2011, 179, 1157–1170. [Google Scholar] [CrossRef]
- Vakkila, J.; Jaffe, R.; Michelow, M.; Lotze, M.T. Pediatric cancers are infiltrated predominantly by macrophages and contain a paucity of dendritic cells: A major nosologic difference with adult tumors. Clin. Cancer Res. 2006, 12, 2049–2054. [Google Scholar] [CrossRef] [Green Version]
- Handl, M.; Hermanova, M.; Hotarkova, S.; Jarkovsky, J.; Mudry, P.; Shatokhina, T.; Vesela, M.; Sterba, J.; Zambo, I. Clinicopathological correlation of tumor-associated macrophages in Ewing sarcoma. Biomed. Pap. 2018, 162, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.J.; Bürger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.W.; Lankester, A.C.; et al. Tumor-Infiltrating Macrophages Are Associated with Metastasis Suppression in High-Grade Osteosarcoma: A Rationale for Treatment with Macrophage Activating Agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [Green Version]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.-Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Dumars, C.; Ngyuen, J.-M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.-F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ségaliny, A.I.; Mohamadi, A.; Dizier, B.; Lokajczyk, A.; Brion, R.; Lanel, R.; Amiaud, J.; Charrier, C.; Boisson-Vidal, C.; Heymann, D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 2015, 137, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Guo, W.; Ren, T.; Huang, Y.; Wang, S.; Liu, K.; Zheng, B.; Yang, K.; Zhang, H.; Liang, X. Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis. Cancer Lett. 2019, 440–441, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, A.J.; Maloney, C.; Behr, C.A.; Edelman, M.C.; Glick, R.D.; Al-Abed, Y.; Symons, M.; Soffer, S.Z.; Steinberg, B.M. The Macrophage Inhibitor CNI-1493 Blocks Metastasis in a Mouse Model of Ewing Sarcoma through Inhibition of Extravasation. PLoS ONE 2016, 10, e0145197. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Guo, W.; Ren, T.; Huang, Y.; Sun, K.; Zhang, H.; Yu, Y.; Wang, W.; Niu, J. Macrophages reduce the sensitivity of osteosarcoma to neoadjuvant chemotherapy drugs by secreting Interleukin-1 beta. Cancer Lett. 2020, 480, 4–14. [Google Scholar] [CrossRef]
- Han, Q.; Shi, H.; Liu, F. CD163+ M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int. Immunopharmacol. 2016, 34, 101–106. [Google Scholar] [CrossRef]
- MacEwen, E.G.; Kurzman, I.D.; Rosenthal, R.C.; Smith, B.W.; Manley, P.A.; Roush, J.K.; Howard, P.E. Therapy for osteosarcoma in dogs with intravenous injection of liposome-encapsulated muramyl tripeptide. J. Natl. Cancer Inst. 1989, 81, 935–938. [Google Scholar] [CrossRef]
- Kurzman, I.D.; Shi, F.; Vail, D.M.; MacEwen, E.G. In vitro and in vivo enhancement of canine pulmonary alveolar macrophage cytotoxic activity against canine osteosarcoma cells. Cancer Biother. Radiopharm. 1999, 14, 121–128. [Google Scholar] [CrossRef]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. J. Am. Soc. Gene Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia 2011, 13, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Healey, J.; Ogura, K.; Yoshida, A.; Kondo, H.; Hata, T.; Kure, M.; Tazawa, H.; Nakata, E.; Kunisada, T.; et al. Role of Tumor-Associated Macrophages in Sarcomas. Cancers 2021, 13, 1086. [Google Scholar] [CrossRef]
- Koo, J.; Hayashi, M.; Verneris, M.R.; Lee-Sherick, A.B. Targeting Tumor-Associated Macrophages in the Pediatric Sarcoma Tumor Microenvironment. Front. Oncol. 2020, 10, 581107. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I Clinical Trial of Ipilimumab in Pediatric Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [Green Version]
- Heymann, M.-F.; Schiavone, K.; Heymann, D. Bone sarcomas in the immunotherapy era. Br. J. Pharmacol. 2021, 178, 1955–1972. [Google Scholar] [CrossRef]
- WHO. WHO Editorial Board WHO Classification of Tumours: Soft Tissue and Bone Tumours, 5th ed.; WHO: Geneva, Switzerland, 2020; ISBN 978-92-8324502-5. [Google Scholar]
- Smolle, M.A.; Herbsthofer, L.; Goda, M.; Granegger, B.; Brcic, I.; Bergovec, M.; Scheipl, S.; Prietl, B.; El-Heliebi, A.; Pichler, M.; et al. Influence of tumor-infiltrating immune cells on local control rate, distant metastasis, and survival in patients with soft tissue sarcoma. Oncoimmunology 2021, 10, 1896658. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Setsu, N.; Gao, D.; Blay, J.-Y.; Thomas, D.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Expression of lymphocyte immunoregulatory biomarkers in bone and soft-tissue sarcomas. Mod. Pathol. 2019, 32, 1772–1785. [Google Scholar] [CrossRef]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e928. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orth, M.F.; Buecklein, V.L.; Kampmann, E.; Subklewe, M.; Noessner, E.; Cidre-Aranaz, F.; Romero-Pérez, L.; Wehweck, F.S.; Lindner, L.; Issels, R.; et al. A comparative view on the expression patterns of PD-L1 and PD-1 in soft tissue sarcomas. Cancer Immunol. Immunother. CII 2020, 69, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Boxberg, M.; Steiger, K.; Lenze, U.; Rechl, H.; von Eisenhart-Rothe, R.; Wörtler, K.; Weichert, W.; Langer, R.; Specht, K. PD-L1 and PD-1 and characterization of tumor-infiltrating lymphocytes in high grade sarcomas of soft tissue—Prognostic implications and rationale for immunotherapy. Oncoimmunology 2018, 7, e1389366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolini, G.; Bergamaschi, L.; Ferrari, A.; Renne, S.L.; Collini, P.; Gardelli, C.; Barisella, M.; Centonze, G.; Chiaravalli, S.; Paolino, C.; et al. PD-L1 assessment in pediatric rhabdomyosarcoma: A pilot study. BMC Cancer 2018, 18, 652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siozopoulou, V.; Domen, A.; Zwaenepoel, K.; Van Beeck, A.; Smits, E.; Pauwels, P.; Marcq, E. Immune Checkpoint Inhibitory Therapy in Sarcomas: Is There Light at the End of the Tunnel? Cancers 2021, 13, 360. [Google Scholar] [CrossRef]
- Saerens, M.; Brusselaers, N.; Rottey, S.; Decruyenaere, A.; Creytens, D.; Lapeire, L. Immune checkpoint inhibitors in treatment of soft-tissue sarcoma: A systematic review and meta-analysis. Eur. J. Cancer 2021, 152, 165–182. [Google Scholar] [CrossRef]
- Somaiah, N.; Conley, A.P.; Parra, E.R.; Lin, H.; Amini, B.; Solis Soto, L.; Salazar, R.; Barreto, C.; Chen, H.; Gite, S.; et al. Durvalumab plus tremelimumab in advanced or metastatic soft tissue and bone sarcomas: A single-centre phase 2 trial. Lancet Oncol. 2022, 23, 1156–1166. [Google Scholar] [CrossRef]
- Nakata, E.; Fujiwara, T.; Kunisada, T.; Ito, T.; Takihira, S.; Ozaki, T. Immunotherapy for sarcomas. Jpn. J. Clin. Oncol. 2021, 51, 523–537. [Google Scholar] [CrossRef]
- Nathenson, M.J.; Conley, A.P.; Sausville, E. Immunotherapy: A New (and Old) Approach to Treatment of Soft Tissue and Bone Sarcomas. Oncol. 2017, 23, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell 2019, 49, 347–360. [Google Scholar] [CrossRef]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Pan, B.T.; Johnstone, R.M. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 1983, 33, 967–978. [Google Scholar] [CrossRef]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Carollo, E.; Melling, G.E.; Carter, D.R.F. Extracellular Vesicles: Emerging Modulators of Cancer Drug Resistance. Cancers 2021, 13, 749. [Google Scholar] [CrossRef]
- Ab Razak, N.S.; Ab Mutalib, N.S.; Mohtar, M.A.; Abu, N. Impact of Chemotherapy on Extracellular Vesicles: Understanding the Chemo-EVs. Front. Oncol. 2019, 9, 1113. [Google Scholar] [CrossRef] [Green Version]
- Paskeh, M.D.A.; Entezari, M.; Mirzaei, S.; Zabolian, A.; Saleki, H.; Naghdi, M.J.; Sabet, S.; Khoshbakht, M.A.; Hashemi, M.; Hushmandi, K.; et al. Emerging role of exosomes in cancer progression and tumor microenvironment remodeling. J. Hematol. Oncol. 2022, 15, 83. [Google Scholar] [CrossRef]
- Cappariello, A.; Rucci, N. Tumour-Derived Extracellular Vesicles (EVs): A Dangerous “Message in A Bottle” for Bone. Int. J. Mol. Sci. 2019, 20, 4805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chicon-Bosch, M.; Tirado, O.M. Exosomes in Bone Sarcomas: Key Players in Metastasis. Cells 2020, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Pachva, M.C.; Lai, H.; Jia, A.; Rouleau, M.; Sorensen, P.H. Extracellular Vesicles in Reprogramming of the Ewing Sarcoma Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 726205. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Hu, X.; Wen, Y.; Tu, C.; Hornicek, F.; Duan, Z.; Min, L. Exosomes in the tumor microenvironment of sarcoma: From biological functions to clinical applications. J. Nanobiotechnol. 2022, 20, 403. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, L.; De Luca, A.; Gallo, A.; Costa, V.; Russelli, G.; Cuscino, N.; Manno, M.; Raccosta, S.; Carina, V.; Bellavia, D.; et al. Osteosarcoma cell-derived exosomes affect tumor microenvironment by specific packaging of microRNAs. Carcinogenesis 2020, 41, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, A.; Urdinez, J.; Boro, A.; Migliavacca, J.; Arlt, M.J.E.; Muff, R.; Fuchs, B.; Snedeker, J.G.; Gvozdenovic, A. Osteosarcoma-Derived Extracellular Vesicles Induce Lung Fibroblast Reprogramming. Int. J. Mol. Sci. 2020, 21, 5451. [Google Scholar] [CrossRef]
- Urciuoli, E.; Giorda, E.; Scarsella, M.; Petrini, S.; Peruzzi, B. Osteosarcoma-derived extracellular vesicles induce a tumor-like phenotype in normal recipient cells. J. Cell. Physiol. 2018, 233, 6158–6172. [Google Scholar] [CrossRef]
- Mannerström, B.; Kornilov, R.; Abu-Shahba, A.G.; Chowdhury, I.M.; Sinha, S.; Seppänen-Kaijansinkko, R.; Kaur, S. Epigenetic alterations in mesenchymal stem cells by osteosarcoma-derived extracellular vesicles. Epigenetics 2019, 14, 352–364. [Google Scholar] [CrossRef] [Green Version]
- Sarhadi, V.K.; Daddali, R.; Seppänen-Kaijansinkko, R. Mesenchymal Stem Cells and Extracellular Vesicles in Osteosarcoma Pathogenesis and Therapy. Int. J. Mol. Sci. 2021, 22, 1035. [Google Scholar] [CrossRef]
- Baglio, S.R.; Lagerweij, T.; Pérez-Lanzón, M.; Ho, X.D.; Léveillé, N.; Melo, S.A.; Cleton-Jansen, A.M.; Jordanova, E.S.; Roncuzzi, L.; Greco, M.; et al. Blocking Tumor-Educated MSC Paracrine Activity Halts Osteosarcoma Progression. Clin. Cancer Res. 2017, 23, 3721–3733. [Google Scholar] [CrossRef] [Green Version]
- Lissat, A.; Joerschke, M.; Shinde, D.A.; Braunschweig, T.; Meier, A.; Makowska, A.; Bortnick, R.; Henneke, P.; Herget, G.; Gorr, T.A.; et al. IL6 secreted by Ewing sarcoma tumor microenvironment confers anti-apoptotic and cell-disseminating paracrine responses in Ewing sarcoma cells. BMC Cancer 2015, 15, 552. [Google Scholar] [CrossRef]
- Dolan, B.P.; Gibbs, K.D., Jr.; Ostrand-Rosenberg, S. Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J. Immunol. 2006, 177, 6018–6024. [Google Scholar] [CrossRef]
- Gassmann, H.; Schneider, K.; Evdokimova, V.; Ruzanov, P.; Schober, S.J.; Xue, B.; von Heyking, K.; Thiede, M.; Richter, G.H.S.; Pfaffl, M.W.; et al. Ewing Sarcoma-Derived Extracellular Vesicles Impair Dendritic Cell Maturation and Function. Cells 2021, 10, 2081. [Google Scholar] [CrossRef]
- Arkhypov, I.; Lasser, S.; Petrova, V.; Weber, R.; Groth, C.; Utikal, J.; Altevogt, P.; Umansky, V. Myeloid Cell Modulation by Tumor-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 6319. [Google Scholar] [CrossRef]
- Miller, I.V.; Raposo, G.; Welsch, U.; Prazeres da Costa, O.; Thiel, U.; Lebar, M.; Maurer, M.; Bender, H.U.; von Luettichau, I.; Richter, G.H.; et al. First identification of Ewing’s sarcoma-derived extracellular vesicles and exploration of their biological and potential diagnostic implications. Biol. Cell 2013, 105, 289–303. [Google Scholar] [CrossRef]
- Tsugita, M.; Yamada, N.; Noguchi, S.; Yamada, K.; Moritake, H.; Shimizu, K.; Akao, Y.; Ohno, T. Ewing sarcoma cells secrete EWS/Fli-1 fusion mRNA via microvesicles. PLoS ONE 2013, 8, e77416. [Google Scholar] [CrossRef] [Green Version]
- Villasante, A.; Marturano-Kruik, A.; Ambati, S.R.; Liu, Z.; Godier-Furnemont, A.; Parsa, H.; Lee, B.W.; Moore, M.A.; Vunjak-Novakovic, G. Recapitulating the Size and Cargo of Tumor Exosomes in a Tissue-Engineered Model. Theranostics 2016, 6, 1119–1130. [Google Scholar] [CrossRef]
- Wei, Z.; Batagov, A.O.; Schinelli, S.; Wang, J.; Wang, Y.; El Fatimy, R.; Rabinovsky, R.; Balaj, L.; Chen, C.C.; Hochberg, F.; et al. Coding and noncoding landscape of extracellular RNA released by human glioma stem cells. Nat. Commun. 2017, 8, 1145. [Google Scholar] [CrossRef] [Green Version]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [Green Version]
- Evdokimova, V.; Ruzanov, P.; Gassmann, H.; Zaidi, S.H.; Peltekova, V.; Heisler, L.E.; McPherson, J.D.; Orlic-Milacic, M.; Specht, K.; Steiger, K.; et al. Exosomes transmit retroelement RNAs to drive inflammation and immunosuppression in Ewing Sarcoma. bioRxiv 2019, 806851. [Google Scholar] [CrossRef] [Green Version]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nat. Rev. Immunol. 2016, 16, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evdokimova, V.; Gassmann, H.; Radvanyi, L.; Burdach, S.E.G. Current State of Immunotherapy and Mechanisms of Immune Evasion in Ewing Sarcoma and Osteosarcoma. Cancers 2023, 15, 272. https://doi.org/10.3390/cancers15010272
Evdokimova V, Gassmann H, Radvanyi L, Burdach SEG. Current State of Immunotherapy and Mechanisms of Immune Evasion in Ewing Sarcoma and Osteosarcoma. Cancers. 2023; 15(1):272. https://doi.org/10.3390/cancers15010272
Chicago/Turabian StyleEvdokimova, Valentina, Hendrik Gassmann, Laszlo Radvanyi, and Stefan E. G. Burdach. 2023. "Current State of Immunotherapy and Mechanisms of Immune Evasion in Ewing Sarcoma and Osteosarcoma" Cancers 15, no. 1: 272. https://doi.org/10.3390/cancers15010272
APA StyleEvdokimova, V., Gassmann, H., Radvanyi, L., & Burdach, S. E. G. (2023). Current State of Immunotherapy and Mechanisms of Immune Evasion in Ewing Sarcoma and Osteosarcoma. Cancers, 15(1), 272. https://doi.org/10.3390/cancers15010272