
Correlation of Transcriptomics and FDG-PET SUVmax Indicates Reciprocal Expression of Stemness-Related Transcription Factor and Neuropeptide Signaling Pathways in Glucose Metabolism of Ewing Sarcoma

 , , ,
, , ,  ,
, 

Abstract
:Simple Summary
Abstract
1. Introduction
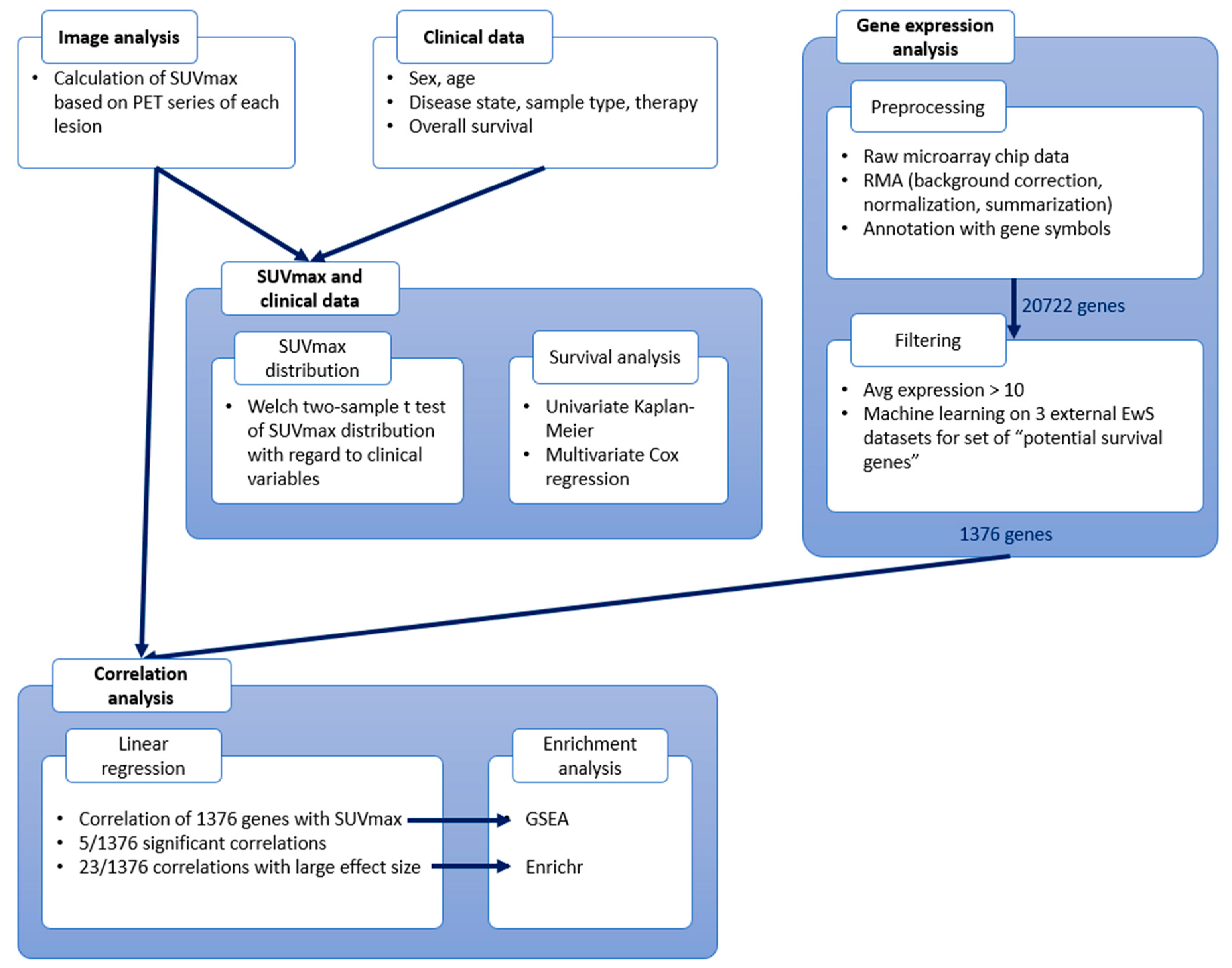
2. Materials and Methods
2.1. Dataset: The Munich Cohort (n = 19)
2.2. Gene Expression Data
2.2.1. Tissue
2.2.2. Preprocessing
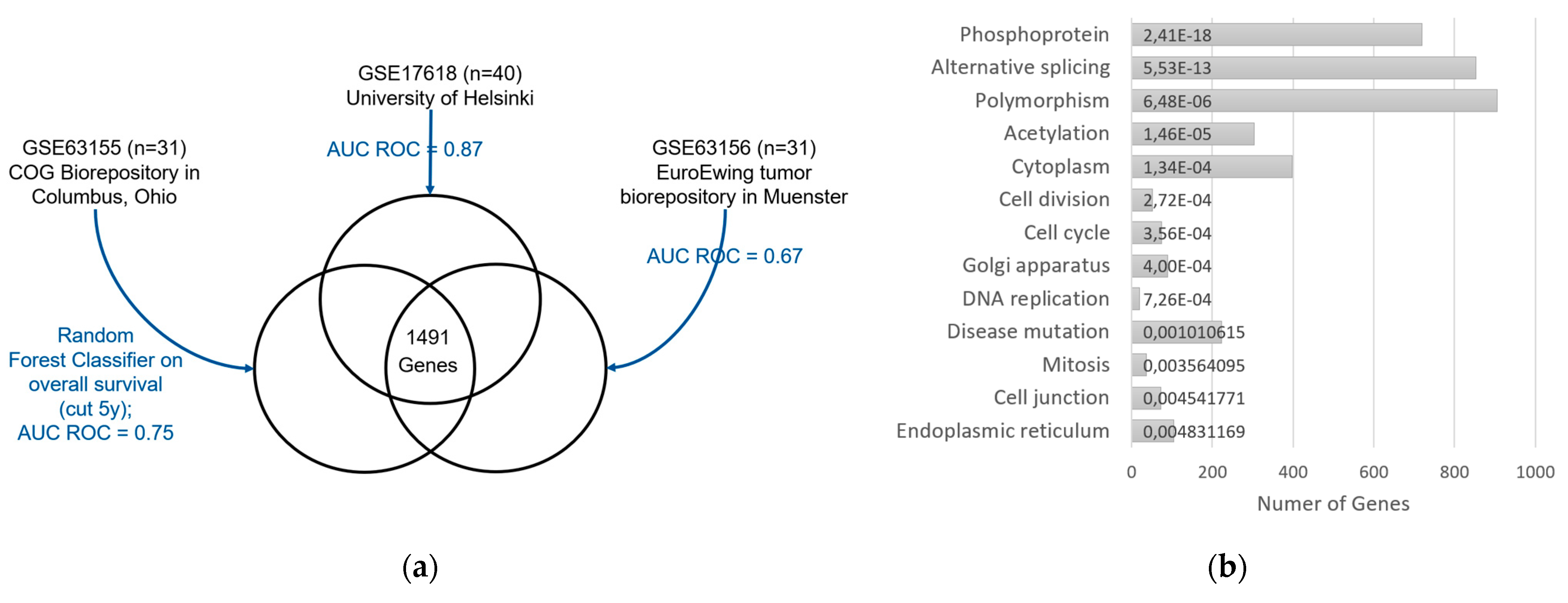
2.2.3. Filtering
2.3. Imaging Data
2.4. Statistical Analysis of SUVmax and Clinical Variables
2.4.1. Distribution of SUVmax Regarding Clinical Variables
2.4.2. Survival Analysis
2.5. Correlation Analysis
2.5.1. Linear Regression
2.5.2. Enrichment Analyses
3. Results
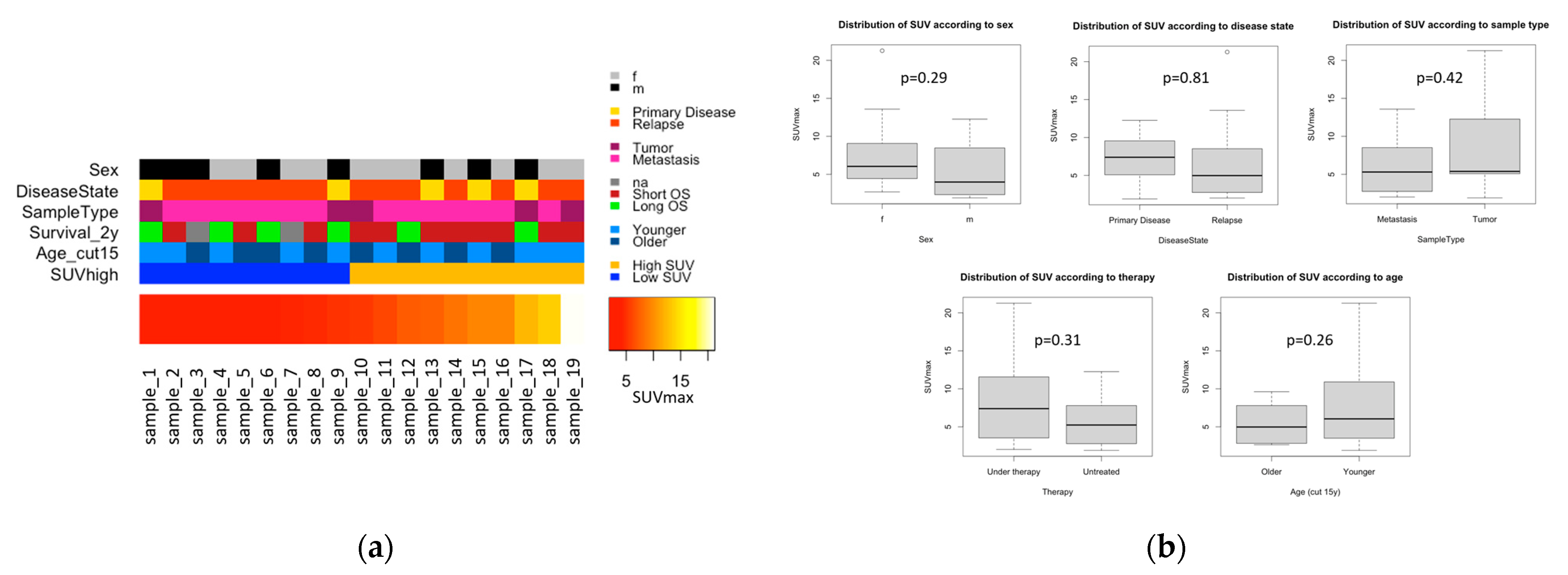
3.1. The Munich Cohort (n = 19)
3.2. Statistical Analysis of SUVmax and Clinical Variables
3.2.1. SUVmax Distribution with Regard to Clinical Variables
3.2.2. Survival Analysis
3.3. Gene Expression Analysis
3.4. Correlation Analysis of SUVmax and Gene Expression
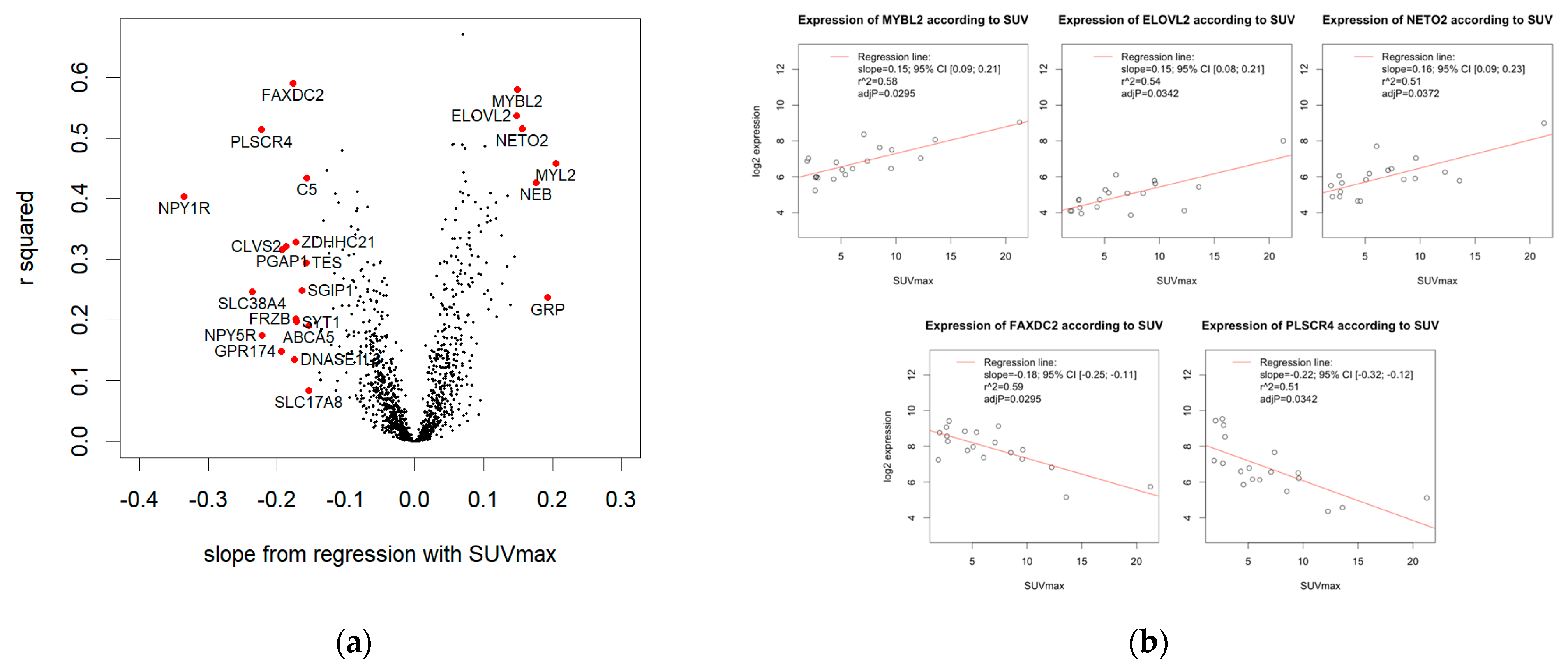
3.4.1. Linear Regression
3.4.2. Significant Association of SUVmax and Five Genes
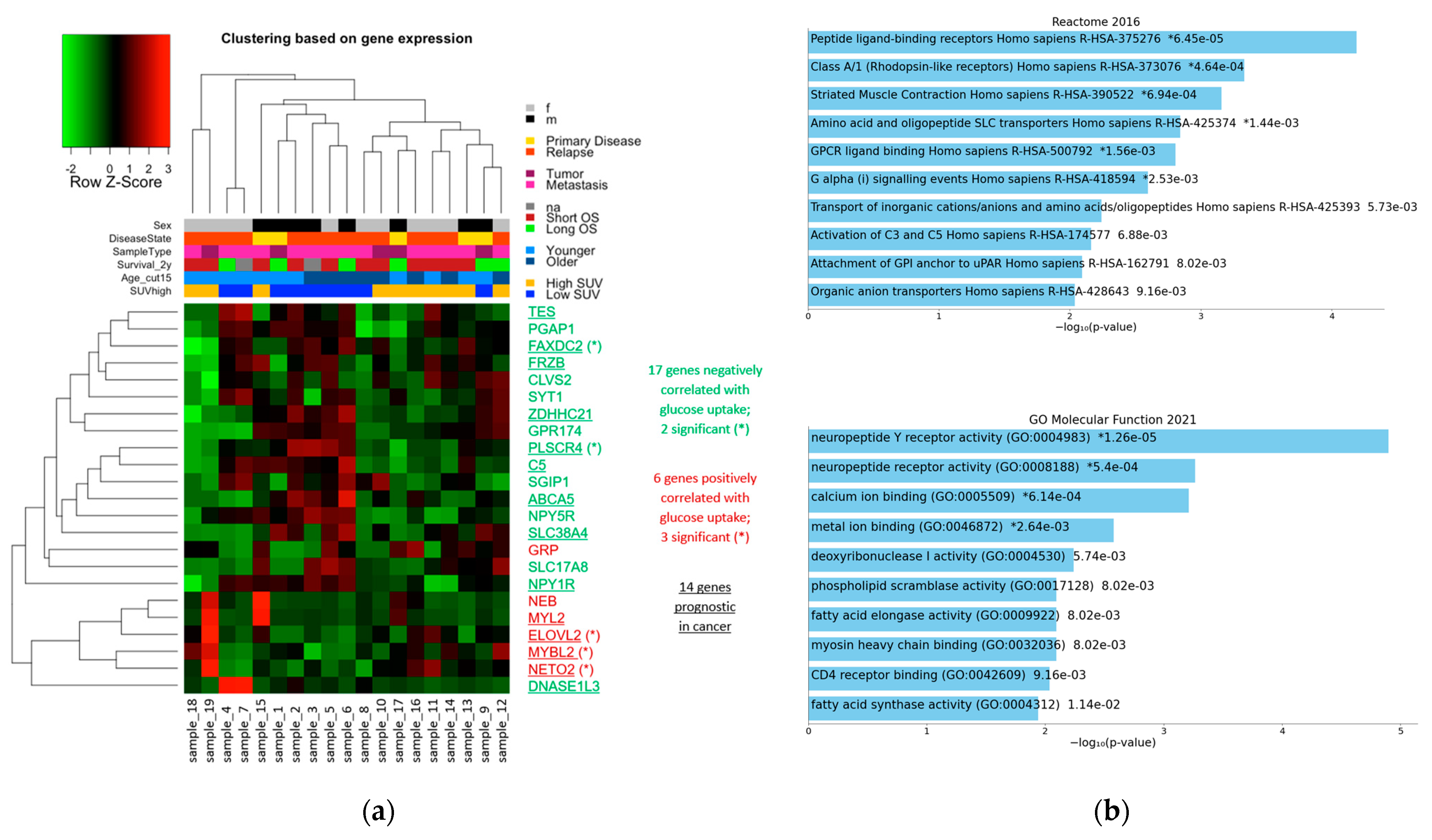
3.4.3. 23 Genes with High Effect Size
3.5. Enrichment Analyses
3.5.1. Enrichments among the 23 Genes with High Effect Size
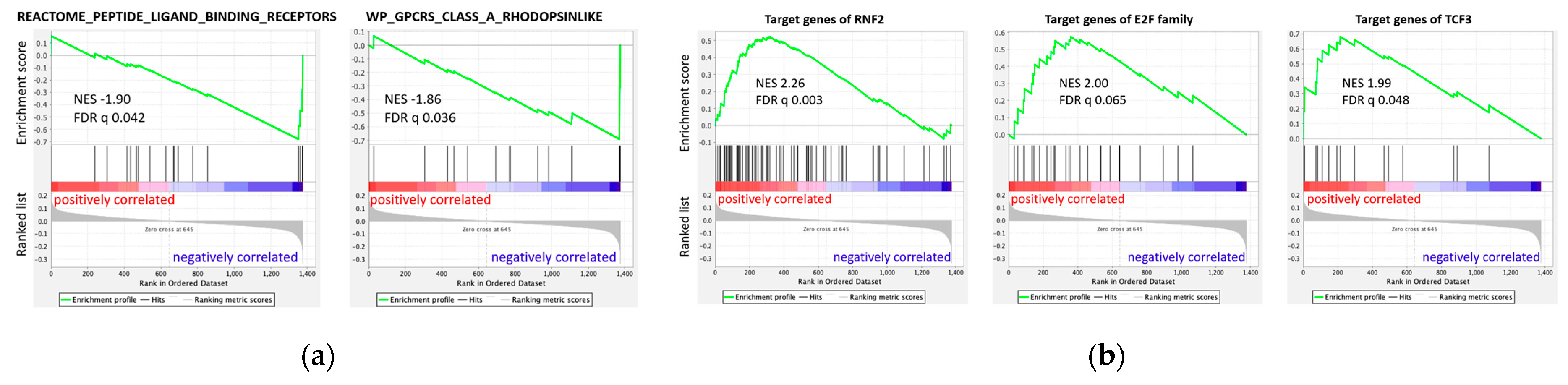
3.5.2. GSEA Based on Regression Results of All 1376 Genes
4. Discussion
4.1. Limitations
4.2. Negative Correlation of NPY Axis and SUVmax in Enrichment Analysis
4.3. Spectrum of Stemness to Differentiation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [CrossRef]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetête-Lalami, S.; Rusch, M.; et al. Genomic Landscape of Ewing Sarcoma Defines an Aggressive Subtype with Co-Association of STAG2 and TP53 Mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Schmidkonz, C.; Krumbholz, M.; Atzinger, A.; Cordes, M.; Goetz, T.I.; Prante, O.; Ritt, P.; Schaefer, C.; Agaimy, A.; Hartmann, W.; et al. Assessment of treatment responses in children and adolescents with Ewing sarcoma with metabolic tumor parameters derived from 18F-FDG-PET/CT and circulating tumor DNA. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1564–1575. [Google Scholar] [CrossRef]
- Burdach, S.; Jürgens, H.; Peters, C.; Nürnberger, W.; Mauz-Körholz, C.; Körholz, D.; Paulussen, M.; Pape, H.; Dilloo, D.; Koscielniak, E. Myeloablative radiochemotherapy and hematopoietic stem-cell rescue in poor-prognosis Ewing’s sarcoma. J. Clin. Oncol. 1993, 11, 1482–1488. [Google Scholar] [CrossRef]
- Koch, R.; Gelderblom, H.; Haveman, L.; Brichard, B.; Jürgens, H.; Cyprova, S.; Berg, H.V.D.; Hassenpflug, W.; Raciborska, A.; Ek, T.; et al. High-Dose Treosulfan and Melphalan as Consolidation Therapy Versus Standard Therapy for High-Risk (Metastatic) Ewing Sarcoma. J. Clin. Oncol. 2022, 40, 2307–2320. [Google Scholar] [CrossRef]
- Haveman, L.M.; van Ewijk, R.; van Dalen, E.C.; Breunis, W.B.; Kremer, L.C.; Berg, H.V.D.; Dirksen, U.; Merks, J.H. High-dose chemotherapy followed by autologous haematopoietic cell transplantation for children, adolescents, and young adults with primary metastatic Ewing sarcoma. Cochrane Database Syst. Rev. 2021, 9, CD011405. [Google Scholar] [CrossRef] [PubMed]
- Burdach, S.E.G.; Westhoff, M.-A.; Steinhauser, M.F.; Debatin, K.-M. Precision medicine in pediatric oncology. Mol. Cell. Pediatr. 2018, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Burdach, S. Molecular Precision Chemotherapy: Overcoming Resistance to Targeted Therapies? Clin. Cancer Res. 2014, 20, 1064–1066. [Google Scholar] [CrossRef] [Green Version]
- Sand, L.G.L.; Szuhai, K.; Hogendoorn, P.C.W. Sequencing Overview of Ewing Sarcoma: A Journey across Genomic, Epigenomic and Transcriptomic Landscapes. Int. J. Mol. Sci. 2015, 16, 16176–16215. [Google Scholar] [CrossRef] [Green Version]
- Agelopoulos, K.; Richter, G.H.; Schmidt, E.; Dirksen, U.; von Heyking, K.; Moser, B.; Klein, H.-U.; Kontny, U.; Dugas, M.; Poos, K.; et al. Deep Sequencing in Conjunction with Expression and Functional Analyses Reveals Activation of FGFR1 in Ewing Sarcoma. Clin. Cancer Res. 2015, 21, 4935–4946. [Google Scholar] [CrossRef]
- Stahl, D.; Gentles, A.J.; Thiele, R.; Gütgemann, I. Prognostic profiling of the immune cell microenvironment in Ewing’s Sarcoma Family of Tumors. OncoImmunology 2019, 8, e1674113. [Google Scholar] [CrossRef] [Green Version]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rössler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Löwel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef] [Green Version]
- Ewing, J. Diffuse endothelioma of bone. CA Cancer J. Clin. 1972, 22, 95–98. [Google Scholar] [CrossRef]
- Staege, M.S.; Hutter, C.; Neumann, I.; Foja, S.; Hattenhorst, U.E.; Hansen, G.; Afar, D.; Burdach, S.E.G. DNA Microarrays Reveal Relationship of Ewing Family Tumors to Both Endothelial and Fetal Neural Crest-Derived Cells and Define Novel Targets. Cancer Res. 2004, 64, 8213–8221. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D.; Harms, D.; Burdach, S. Malignant peripheral neuroectodermal tumours of childhood and adolescence. Virchows Arch. 1985, 406, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The Genomic Landscape of Pediatric Ewing Sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; De Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Burchill, S.A. Molecular abnormalities in Ewing’s sarcoma. Expert Rev. Anticancer Ther. 2008, 8, 1675–1687. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Pierron, G.; Klughammer, J.; Datlinger, P.; Schönegger, A.; Schuster, M.; Hadler, J.; Surdez, D.; Guillemot, D.; Lapouble, E.; et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat. Med. 2017, 23, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Singh, I.; Bikas, A.; Garcia, C.A.; Desale, S.; Wartofsky, L.; Burman, K.D. 18F-FDG-PET Suv as a Prognostic Marker of Increasing Size in Thyroid Cancer Tumors. Endocr. Pract. Off. J. Am. Coll. Endocrinol. Am. Assoc. Clin. Endocrinol. 2017, 23, 182–189. [Google Scholar] [CrossRef]
- Floberg, J.; Zhang, J.; DeWees, T.; Markovina, S.; Grigsby, P.; Schwarz, J. Pre-Treatment [F-18]FDG-PET SUVmax as a Prognostic and Radiogenomic Marker in Cervical Cancer. Int. J. Radiat. Oncol. 2018, 102, S82–S83. [Google Scholar] [CrossRef] [Green Version]
- Diao, W.; Tian, F.; Jia, Z. The prognostic value of SUVmax measuring on primary lesion and ALN by 18F-FDG PET or PET/CT in patients with breast cancer. Eur. J. Radiol. 2018, 105, 1–7. [Google Scholar] [CrossRef]
- Zhang, Q.; Gao, X.; Wei, G.; Qiu, C.; Qu, H.; Zhou, X. Prognostic Value of MTV, SUVmax and the T/N Ratio of PET/CT in Patients with Glioma: A Systematic Review and Meta-Analysis. J. Cancer 2019, 10, 1707–1716. [Google Scholar] [CrossRef] [Green Version]
- Perrone, A.M.; Dondi, G.; Lima, G.M.; Castellucci, P.; Tesei, M.; Coluccelli, S.; Gasparre, G.; Porcelli, A.M.; Nanni, C.; Fanti, S.; et al. Potential Prognostic Role of 18F-FDG PET/CT in Invasive Epithelial Ovarian Cancer Relapse. A Preliminary Study. Cancers 2019, 11, 713. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-Y.; Jeong, S.Y.; Chong, G.O.; Son, S.H.; Jung, J.-H.; Kim, D.-H.; Lee, S.-W.; Ahn, B.-C.; Lee, J. Quantitative metabolic parameters measured on F-18 FDG PET/CT predict survival after relapse in patients with relapsed epithelial ovarian cancer. Gynecol. Oncol. 2015, 136, 498–504. [Google Scholar] [CrossRef]
- Torizuka, T.; Tanizaki, Y.; Kanno, T.; Futatsubashi, M.; Naitou, K.; Ueda, Y.; Ouchi, Y. Prognostic Value of 18F-FDG PET in Patients with Head and Neck Squamous Cell Cancer. Am. J. Roentgenol. 2009, 192, W156–W160. [Google Scholar] [CrossRef]
- Pankowska, V.; Malkowski, B.; Wedrowski, M.; Wedrowska, E.; Roszkowski, K. FDG PET/CT as a survival prognostic factor in patients with advanced renal cell carcinoma. Clin. Exp. Med. 2019, 19, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Berghmans, T.; Dusart, M.; Paesmans, M.; Hossein-Foucher, C.; Buvat, I.; Castaigne, C.; Scherpereel, A.; Mascaux, C.; Moreau, M.; Roelandts, M.; et al. Primary Tumor Standardized Uptake Value (SUVmax) Measured on Fluorodeoxyglucose Positron Emission Tomography (FDG-PET) is of Prognostic Value for Survival in Non-small Cell Lung Cancer (NSCLC): A Systematic Review and Meta-Analysis (MA) by the European Lung Cancer Working Party for the IASLC Lung Cancer Staging Project. J. Thorac. Oncol. 2008, 3, 6–12. [Google Scholar] [CrossRef]
- Cerfolio, R.J.; Bryant, A.S. Maximum Standardized Uptake Values on Positron Emission Tomography of Esophageal Cancer Predicts Stage, Tumor Biology, and Survival. Ann. Thorac. Surg. 2006, 82, 391–394; discussion 394–395. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Gu, P.; Huang, G.; Xue, H.; Wu, S. Prognostic significance of SUV on PET/CT in patients with esophageal cancer: A systematic review and meta-analysis. Eur. J. Gastroenterol. Hepatol. 2009, 21, 1008–1015. [Google Scholar] [CrossRef]
- Lee, W.; Oh, M.; Kim, J.S.; Park, Y.; Kwon, J.W.; Jun, E.; Song, K.B.; Lee, J.H.; Hwang, D.W.; Yoo, C.; et al. Metabolic activity by FDG-PET/CT after neoadjuvant chemotherapy in borderline resectable and locally advanced pancreatic cancer and association with survival. Br. J. Surg. 2021, 109, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, D.; Gröschel, A.; Graeter, T.P.; Hellwig, A.P.; Nestle, U.; Schäfers, H.-J.; Sybrecht, G.W.; Kirsch, C.-M. Diagnostic performance and prognostic impact of FDG-PET in suspected recurrence of surgically treated non-small cell lung cancer. Eur. J. Nucl. Med. 2006, 33, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Hou, G.; Wu, F.; Zhu, Z.; Zhang, W.; Cheng, W. The maximum standardized uptake value and extent of peritoneal involvement may predict the prognosis of patients with recurrent ovarian cancer after primary treatment: A retrospective clinical study. Medicine 2020, 99, e19228. [Google Scholar] [CrossRef] [PubMed]
- Sala, E.; Kataoka, M.; Pandit-Taskar, N.; Ishill, N.; Mironov, S.; Moskowitz, C.S.; Mironov, O.; Collins, M.A.; Chi, D.S.; Larson, S.; et al. Recurrent Ovarian Cancer: Use of Contrast-enhanced CT and PET/CT to Accurately Localize Tumor Recurrence and to Predict Patients’ Survival. Radiology 2010, 257, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzius, C.; Schulte, M.; Hillmann, A.; Winkelmann, W.; Jürgens, H.; Bockisch, A.; Schober, O. Clinical value of positron emission tomography (PET) in the diagnosis of bone and soft tissue tumors. 3rd Interdisciplinary Consensus Conference “PET in Oncology”: Results of the Bone and Soft Tissue Study Group. Chirurgie 2001, 72, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.P.; Lim, I.; Kong, C.-B.; Jeon, D.G.; Byun, B.H.; Kim, B.I.; Choi, C.W.; Lim, S.M. Prognostic Value of SUVmax Measured by Pretreatment Fluorine-18 Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography in Patients with Ewing Sarcoma. PLoS ONE 2016, 11, e0153281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamet, B.; Carlier, T.; Campion, L.; Bompas, E.; Girault, S.; Borrely, F.; Ferrer, L.; Rousseau, M.; Venel, Y.; Kraeber-Bodéré, F.; et al. Initial FDG-PET/CT predicts survival in adults Ewing sarcoma family of tumors. Oncotarget 2017, 8, 77050–77060. [Google Scholar] [CrossRef]
- Raciborska, A.; Bilska, K.; Drabko, K.; Michalak, E.; Chaber, R.; Pogorzala, M.; Polczynska, K.; Sobol, G.; Wieczorek, M.; Muszynskaroslan, K.; et al. Response to chemotherapy estimates by FDG PET is an important prognostic factor in patients with Ewing sarcoma. Clin. Transl. Oncol. 2016, 18, 189–195. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Schuetze, S.M.; Butrynski, J.E.; Rajendran, J.G.; Vernon, C.B.; Conrad, E.U.; Eary, J.F. [18F]Fluorodeoxyglucose Positron Emission Tomography Predicts Outcome for Ewing Sarcoma Family of Tumors. J. Clin. Oncol. 2005, 23, 8828–8834. [Google Scholar] [CrossRef]
- Salem, U.; Amini, B.; Chuang, H.H.; Daw, N.C.; Wei, W.; Haygood, T.M.; Madewell, J.E.; Costelloe, C.M. 18F-FDG PET/CT as an Indicator of Survival in Ewing Sarcoma of Bone. J. Cancer 2017, 8, 2892–2898. [Google Scholar] [CrossRef]
- Annovazzi, A.; Ferraresi, V.; Anelli, V.; Covello, R.; Vari, S.; Zoccali, C.; Biagini, R.; Sciuto, R. [18F]FDG PET/CT quantitative parameters for the prediction of histological response to induction chemotherapy and clinical outcome in patients with localised bone and soft-tissue Ewing sarcoma. Eur. Radiol. 2021, 31, 7012–7021. [Google Scholar] [CrossRef]
- Macpherson, R.E.; Pratap, S.; Tyrrell, H.; Khonsari, M.; Wilson, S.; Gibbons, M.; Whitwell, D.; Giele, H.; Critchley, P.; Cogswell, L.; et al. Retrospective audit of 957 consecutive 18F-FDG PET–CT scans compared to CT and MRI in 493 patients with different histological subtypes of bone and soft tissue sarcoma. Clin. Sarcoma Res. 2018, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, M.; Wang, P.; Boyd, A.D.; Kostov, G.; Athey, B.; Jones, E.G.; Bunney, W.E.; Myers, R.M.; Speed, T.P.; Akil, H.; et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Zhang, X. The effect of GeneChip gene definitions on the microarray study of cancers. BioEssays 2006, 28, 739–746. [Google Scholar] [CrossRef]
- Sandberg, R.; Larsson, O. Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinform. 2007, 8, 48. [Google Scholar] [CrossRef] [Green Version]
- Draghici, S.; Khatri, P.; Eklund, A.C.; Szallasi, Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006, 22, 101–109. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volchenboum, S.L.; Andrade, J.; Huang, L.; Barkauskas, D.A.; Krailo, M.; Womer, R.B.; Ranft, A.; Potratz, J.; Dirksen, U.; Triche, T.J.; et al. Gene expression profiling of Ewing sarcoma tumours reveals the prognostic importance of tumour-stromal interactions: A report from the Children’s Oncology Group. J. Pathol. Clin. Res. 2015, 1, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savola, S.; Klami, A.; Myllykangas, S.; Manara, C.; Scotlandi, K.; Picci, P.; Knuutila, S.; Vakkila, J. High Expression of Complement Component 5 (C5) at Tumor Site Associates with Superior Survival in Ewing’s Sarcoma Family of Tumour Patients. ISRN Oncol. 2011, 2011, 168712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Caret: Classification and Regression Training, R package version 6.0-90. Available online: https://CRAN.R-project.org/package=caret:2021 (accessed on 1 November 2021).
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Rosset, A.; Spadola, L.; Ratib, O. OsiriX: An Open-Source Software for Navigating in Multidimensional DICOM Images. J. Digit. Imaging 2004, 17, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Zwanenburg, A.; Vallières, M.; Abdalah, M.A.; Aerts, H.J.W.L.; Andrearczyk, V.; Apte, A.; Ashrafinia, S.; Bakas, S.; Beukinga, R.J.; Boellaard, R.; et al. The Image Biomarker Standardization Initiative: Standardized Quantitative Radiomics for High-Throughput Image-based Phenotyping. Radiology 2020, 295, 328–338. [Google Scholar] [CrossRef] [Green Version]
- van Griethuysen, J.J.M.; Fedorov, A.; Parmar, C.; Hosny, A.; Aucoin, N.; Narayan, V.; Beets-Tan, R.G.H.; Fillion-Robin, J.-C.; Pieper, S.; Aerts, H.J.W.L. Computational Radiomics System to Decode the Radiographic Phenotype. Cancer Res. 2017, 77, e104–e107. [Google Scholar] [CrossRef] [Green Version]
- Therneau, T.M. A Package for Survival Analysis in R, R package version 3.2-13. Available online: https://CRAN.R-project.org/package=survival:2021 (accessed on 1 September 2021).
- Therneau, T.; Grambsch, P. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000; ISBN 0-387-98784-3. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawing Survival Curves using ‘ggplot2′, R package version 0.4.9. 2021. Available online: https://CRAN.R-project.org/package=survminer (accessed on 17 October 2022).
- Ritchie, M.E.; Belinda, P.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Heatmap.3. Available online: https://raw.githubusercontent.com/obigriffith/biostar-tutorials/master/Heatmaps/heatmap.3.R (accessed on 1 January 2018).
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 1 July 2020).
- Human Protein Atlas. Available online: www.proteinatlas.org (accessed on 7 December 2021).
- Burdach, S.E.G.; Combs, S.E.; Dürr, H.R.; Eisenhart-Rothe, R.v.; Lindner, L.H. Tumorzentrum München an den Medizinischen Fakultäten der Ludwig-Maximilians-Universität und der Technischen Universität München. Klinik und multidisziplinäre Therapie der Ewing-Sarkome, MANUAL Knochentumoren und Weichteilsarkome. Zuckschwerdt Verlag: München, Germany, 2017. [Google Scholar]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, N.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A.; et al. Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Sipol, A.; Hameister, E.; Xue, B.; Hofstetter, J.; Barenboim, M.; Öllinger, R.; Jain, G.; Prexler, C.; Rubio, R.A.; Baldauf, M.C.; et al. MondoA Drives B-ALL Malignancy through Enhanced Adaptation to Metabolic Stress. Blood 2022, 139, 1184–1197. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Ranft, A.; Paulussen, M.; Bölling, T.; Vieth, V.; Bielack, S.; Görtitz, I.; Braun-Munzinger, G.; Hardes, J.; Jürgens, H.; et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr. Blood Cancer 2011, 57, 549–553. [Google Scholar] [CrossRef]
- Groneberg, D.A.; Folkerts, G.; Peiser, C.; Chung, K.F.; Fischer, A. Neuropeptide Y (NPY). Pulm. Pharmacol. Ther. 2004, 17, 173–180. [Google Scholar] [CrossRef]
- Wheway, J.; Herzog, H.; Mackay, F. NPY and Receptors in Immune and Inflammatory Diseases. Curr. Top. Med. Chem. 2007, 7, 1743–1752. [Google Scholar] [CrossRef]
- Li, J.; Tian, Y.; Wu, A. Neuropeptide Y receptors: A promising target for cancer imaging and therapy. Regen. Biomater. 2015, 2, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Wormser, L.; Fritz, V.; Seitz, T.; De Maria, M.; Schambony, A.; Kremer, A.E.; Günther, C.; Itzel, T.; Thasler, W.E.; et al. Molecular crosstalk between Y5 receptor and neuropeptide Y drives liver cancer. J. Clin. Investig. 2020, 130, 2509–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, X.; Zhao, F.; Huo, X.; Tang, W.; Hu, B.; Gong, X.; Yang, J.; Shen, Q.; Qin, W. Neuropeptide Y1 receptor inhibits cell growth through inactivating mitogen-activated protein kinase signal pathway in human hepatocellular carcinoma. Med. Oncol. 2016, 33, 70. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Sun, J.; Chen, Y.; Li, J.; Huang, J.; Du, H.; Gan, L.; Qiu, Z.; Li, H.; et al. The Novel Methylation Biomarker NPY5R Sensitizes Breast Cancer Cells to Chemotherapy. Front. Cell Dev. Biol. 2022, 9, 798221. [Google Scholar] [CrossRef] [PubMed]
- Körner, M.; Waser, B.; Reubi, J.C. High Expression of Neuropeptide Y1 Receptors in Ewing Sarcoma Tumors. Clin. Cancer Res. 2008, 14, 5043–5049. [Google Scholar] [CrossRef] [Green Version]
- van Valen, F.; Keck, E.; Jurgens, H. Neuropeptide Y inhibits vasoactive intestinal peptide- and dopamine-induced cyclic AMP formation in human Ewing’s sarcoma WE-68 cells. FEBS Lett. 1989, 249, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Kitlinska, J. Neuropeptide Y in neural crest-derived tumors: Effect on growth and vascularization. Cancer Lett. 2007, 245, 293–302. [Google Scholar] [CrossRef]
- Tilan, J.U.; Lu, C.; Galli, S.; Izycka-Swieszewska, E.; Earnest, J.P.; Shabbir, A.; Everhart, L.M.; Wang, S.; Martin, S.; Horton, M.; et al. Hypoxia shifts activity of neuropeptide Y in Ewing sarcoma from growth-inhibitory to growth-promoting effects. Oncotarget 2013, 4, 2487–2501. [Google Scholar] [CrossRef] [Green Version]
- Tilan, J.; Kitlinska, J. Neuropeptide Y (NPY) in tumor growth and progression: Lessons learned from pediatric oncology. Neuropeptides 2016, 55, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Mahajan, A.; Hong, S.-H.; Galli, S.; Zhu, S.; Tilan, J.U.; Abualsaud, N.; Adnani, M.; Chung, S.; Elmansy, N.; et al. Hypoxia-activated neuropeptide Y/Y5 receptor/RhoA pathway triggers chromosomal instability and bone metastasis in Ewing sarcoma. Nat. Commun. 2022, 13, 2323. [Google Scholar] [CrossRef]
- Fedorova, M.S.; Snezhkina, A.V.; Lipatova, A.V.; Pavlov, V.S.; Kobelyatskaya, A.A.; Guvatova, Z.G.; Pudova, E.A.; Savvateeva, M.V.; Ishina, I.A.; Demidova, T.B.; et al. NETO2 Is Deregulated in Breast, Prostate, and Colorectal Cancer and Participates in Cellular Signaling. Front. Genet. 2020, 11, 594933. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bian, Z.; Hou, C.; Li, M.; Jiang, W.; Zhu, L. Neuropilin and tolloid-like 2 regulates the progression of osteosarcoma. Gene 2021, 768, 145292. [Google Scholar] [CrossRef]
- Gimple, R.C.; Kidwell, R.L.; Kim, L.J.Y.; Sun, T.; Gromovsky, A.D.; Wu, Q.; Wolf, M.; Lv, D.; Bhargava, S.; Jiang, L.; et al. Glioma Stem Cell–Specific Superenhancer Promotes Polyunsaturated Fatty-Acid Synthesis to Support EGFR Signaling. Cancer Discov. 2019, 9, 1248–1267. [Google Scholar] [CrossRef] [PubMed]
- Musa, J.; Aynaud, M.-M.; Mirabeau, O.; Delattre, O.; Grünewald, T.G. MYBL2 (B-Myb): A central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Cell Death Dis. 2017, 8, e2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvageau, M.; Sauvageau, G. Polycomb Group Genes: Keeping Stem Cell Activity in Balance. PLOS Biol. 2008, 6, e113. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Wu, C.-I.; Hoffman, J.; Shy, B.R.; Ford, E.M.; Fuchs, E.; Nguyen, H.; Merrill, B.J. Function of Wnt/β-catenin in counteracting Tcf3 repression through the Tcf3–β-catenin interaction. Development 2012, 139, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.-Y.; Wang, W.; Li, M.; Li, Y.; Guo, Y.-M. Predictive significance of standardized uptake value parameters of FDG-PET in patients with non-small cell lung carcinoma. Braz. J. Med. Biol. Res. 2015, 48, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Gomez, N.C.; Adam, R.C.; Nikolova, M.; Yang, H.; Verma, A.; Lu, C.P.-J.; Polak, L.; Yuan, S.; Elemento, O.; et al. Stem Cell Lineage Infidelity Drives Wound Repair and Cancer. Cell 2017, 169, 636–650.e614. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Fraction | ||
|---|---|---|---|
| Total | 19 | 1 | |
| Sex | Female | 11 | 0.58 |
| Male | 8 | 0.42 | |
| Disease state | Primary disease | 5 | 0.26 |
| Relapse | 14 | 0.74 | |
| Sample type | Tumor | 5 | 0.26 |
| Metastasis | 14 | 0.74 | |
| Therapy | Untreated | 12 | 0.63 |
| Under therapy | 7 | 0.37 | |
| Age at PET (all) | Number | 19 | 1 |
| Range | 3–31 | ||
| Median | 14 | ||
| Mean | 14.8 | ||
| Age at PET (≤15 years) | Number | 11 | 0.58 |
| Range | 3–15 | ||
| Median | 10 | ||
| Mean | 9.2 | ||
| Age at PET (>15 years) | Number | 8 | 0.42 |
| Range | 17–31 | ||
| Median | 21 | ||
| Mean | 22.5 | ||
| Imaging modality | PET-CT | 15 | 0.79 |
| PET-MR | 4 | 0.21 | |
| SUVmax (all) | Number | 19 | 1 |
| Range | 1.898–21.269 | ||
| Median | 5.387 | ||
| Mean | 6.819 | ||
| SUVmax low | Number | 9 | 0.47 |
| Range | 1.898–5.084 | ||
| Median | 2.756 | ||
| Mean | 3.207 | ||
| SUVmax high | Number | 10 | 0.53 |
| Range | 5.387–21.269 | ||
| Median | 9.035 | ||
| Mean | 10.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prexler, C.; Knape, M.S.; Erlewein-Schweizer, J.; Roll, W.; Specht, K.; Woertler, K.; Weichert, W.; von Luettichau, I.; Rossig, C.; Hauer, J.; et al. Correlation of Transcriptomics and FDG-PET SUVmax Indicates Reciprocal Expression of Stemness-Related Transcription Factor and Neuropeptide Signaling Pathways in Glucose Metabolism of Ewing Sarcoma. Cancers 2022, 14, 5999. https://doi.org/10.3390/cancers14235999
Prexler C, Knape MS, Erlewein-Schweizer J, Roll W, Specht K, Woertler K, Weichert W, von Luettichau I, Rossig C, Hauer J, et al. Correlation of Transcriptomics and FDG-PET SUVmax Indicates Reciprocal Expression of Stemness-Related Transcription Factor and Neuropeptide Signaling Pathways in Glucose Metabolism of Ewing Sarcoma. Cancers. 2022; 14(23):5999. https://doi.org/10.3390/cancers14235999
Chicago/Turabian StylePrexler, Carolin, Marie Sophie Knape, Janina Erlewein-Schweizer, Wolfgang Roll, Katja Specht, Klaus Woertler, Wilko Weichert, Irene von Luettichau, Claudia Rossig, Julia Hauer, and et al. 2022. "Correlation of Transcriptomics and FDG-PET SUVmax Indicates Reciprocal Expression of Stemness-Related Transcription Factor and Neuropeptide Signaling Pathways in Glucose Metabolism of Ewing Sarcoma" Cancers 14, no. 23: 5999. https://doi.org/10.3390/cancers14235999
APA StylePrexler, C., Knape, M. S., Erlewein-Schweizer, J., Roll, W., Specht, K., Woertler, K., Weichert, W., von Luettichau, I., Rossig, C., Hauer, J., Richter, G. H. S., Weber, W., & Burdach, S. (2022). Correlation of Transcriptomics and FDG-PET SUVmax Indicates Reciprocal Expression of Stemness-Related Transcription Factor and Neuropeptide Signaling Pathways in Glucose Metabolism of Ewing Sarcoma. Cancers, 14(23), 5999. https://doi.org/10.3390/cancers14235999