
Unraveling the Role of Epithelial–Mesenchymal Transition in Adenoid Cystic Carcinoma of the Salivary Glands: A Comprehensive Review




Abstract
:Simple Summary
Abstract
1. Introduction
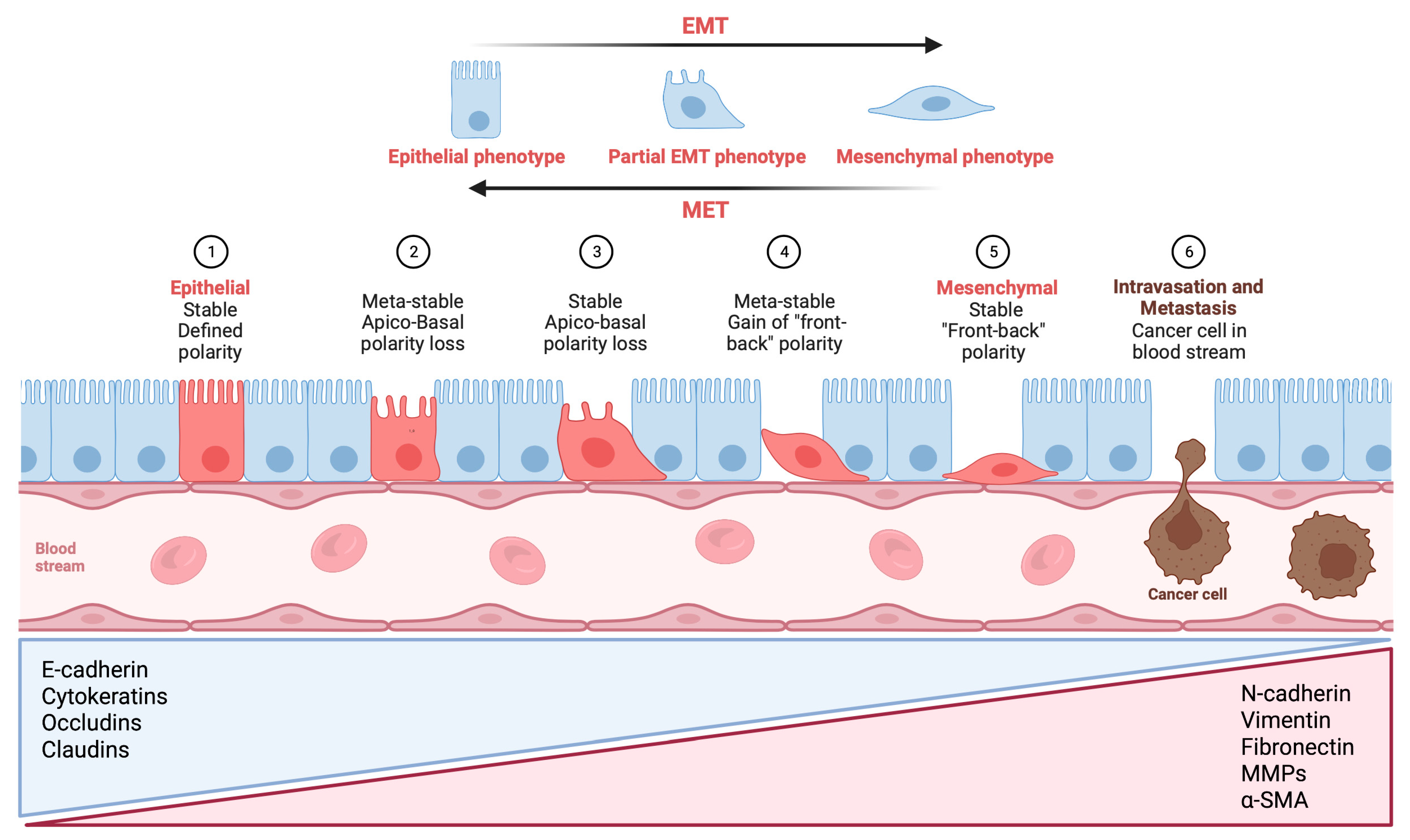
2. Multiple Molecular Signals Driving EMT in Cancer Progression and Metastasis
3. The Role of EMT-TFs in Cancer Invasion and Metastasis in SACC
3.1. TWIST Expression in SACC and Its Significance in Pathogenesis and Prognosis
3.2. SNAIL and SLUG: Key Transcription Factors Driving EMT and Metastasis in SACC
3.3. ZEB1/ZEB2: A Key Regulator of EMT in SACC
4. Expression of Major EMT Markers in SACC
4.1. Expression of CK in SACC
4.2. The Role of E-Cadherin in SACC: Implications for Tumor Invasion, Metastasis, and EMT
4.3. Vimentin: A Key Intermediate Filament in Mesenchymal Cells and Its Role in SACC Metastasis
4.4. The Complex Role of Fibronectin in SACC: Promoting PNI, Inhibiting Invasion, and Potential Prognostic Marker
4.5. N-Cadherin Expression in SACC: Association with PNI and EMT
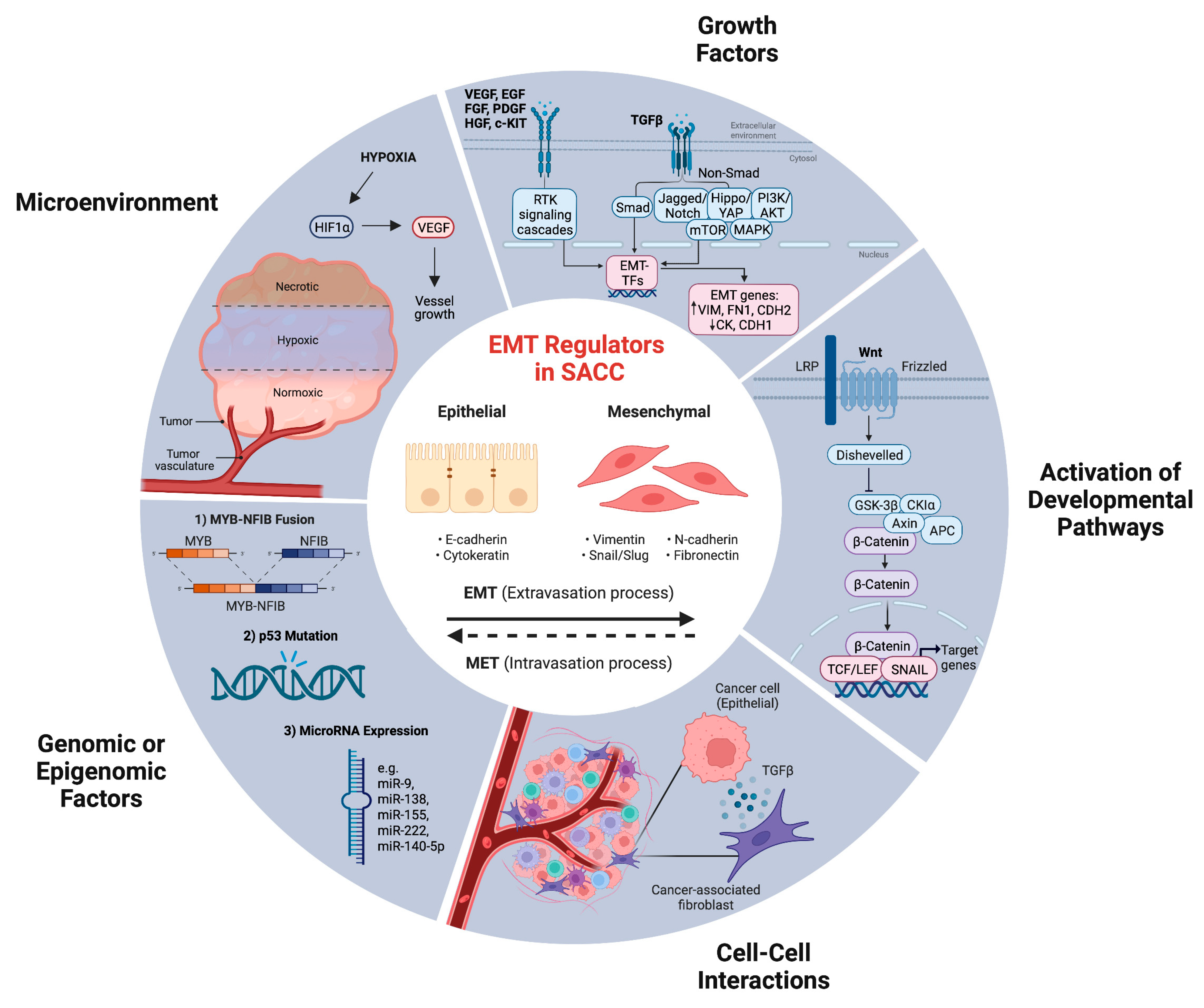
5. TGF-ß-Mediated EMT in SACC
6. MYB Promotes SACC Metastasis by Regulating EMT
7. Hypoxia as an Essential Factor Related to EMT in SACC
8. The Role of MicroRNAs in the Regulation of EMT Progression in SACC
9. The Role of c-Kit in EMT Progression of SACC
10. p53 as a Potential Therapeutic Target for Inhibiting PNI in SACC
11. Clinical Trials Evaluating Therapies Influencing the EMT-Axis in SACC
12. Future Directions and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Boukheris, H.; Curtis, R.E.; Land, C.E.; Dores, G.M. Incidence of Carcinoma of the Major Salivary Glands According to the WHO Classification, 1992 to 2006: A Population-Based Study in the United States. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2899–2906. [Google Scholar] [CrossRef]
- Amit, M.; Binenbaum, Y.; Sharma, K.; Ramer, N.; Ramer, I.; Agbetoba, A.; Miles, B.; Yang, X.; Lei, D.; Bjørndal, K.; et al. Analysis of failure in patients with adenoid cystic carcinoma of the head and neck. An international collaborative study. Head Neck 2014, 36, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Cantù, G. Adenoid cystic carcinoma. An indolent but aggressive tumour. Part A: From aetiopathogenesis to diagnosis. Acta Otorhinolaryngol. Ital. 2021, 41, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Coca-Pelaz, A.; Rodrigo, J.P.; Bradley, P.J.; Poorten, V.V.; Triantafyllou, A.; Hunt, J.L.; Strojan, P.; Rinaldo, A.; Haigentz, M.; Takes, R.P.; et al. Adenoid cystic carcinoma of the head and neck–An update. Oral Oncol. 2015, 51, 652–661. [Google Scholar] [CrossRef] [PubMed]
- Dillon, P.M.; Chakraborty, S.; Moskaluk, C.A.; Joshi, P.J.; Thomas, C.Y. Adenoid cystic carcinoma: A review of recent advances, molecular targets, and clinical trials. Head Neck 2016, 38, 620–627. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chung, S.Y.; Davis, A.A.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Kalyan, A.; Giles, F.J. Adenoid cystic carcinoma: Current therapy and potential therapeutic advances based on genomic profiling. Oncotarget 2015, 6, 37117–37134. [Google Scholar] [CrossRef]
- Moskaluk, C.A. Adenoid Cystic Carcinoma: Clinical and Molecular Features. Head Neck Pathol. 2013, 7, 17–22. [Google Scholar] [CrossRef]
- Chang, C.-F.; Hsieh, M.-Y.; Chen, M.-K.; Chou, M.-C. Adenoid cystic carcinoma of head and neck: A retrospective clinical analysis of a single institution. Auris Nasus Larynx 2018, 45, 831–837. [Google Scholar] [CrossRef]
- Shingaki, S.; Kanemaru, S.; Oda, Y.; Niimi, K.; Mikami, T.; Funayama, A.; Saito, C. Distant metastasis and survival of adenoid cystic carcinoma after definitive treatment. J. Oral Maxillofac. Surg. Med. Pathol. 2014, 26, 312–316. [Google Scholar] [CrossRef]
- van der Wal, J.E.; Becking, A.G.; Snow, G.B.; van der Waal, I. Distant metastases of adenoid cystic carcinoma of the salivary glands and the value of diagnostic examinations during follow-up. Head Neck 2002, 24, 779–783. [Google Scholar] [CrossRef]
- Drier, Y.; Cotton, M.J.; Williamson, K.E.; Gillespie, S.M.; Ryan, R.J.H.; Kluk, M.J.; Carey, C.D.; Rodig, S.J.; Sholl, L.M.; Afrogheh, A.H.; et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat. Genet. 2016, 48, 265–272. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Mader, S.; Pantel, K. Epithelial-mesenchymal plasticity in circulating tumor cells. J. Mol. Med. 2017, 95, 133–142. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schäfer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef]
- Bommi, P.V.; Ravindran, S.; Raychaudhuri, P.; Bagchi, S. DDB2 regulates Epithelial-to-Mesenchymal Transition (EMT) in Oral/Head and Neck Squamous Cell Carcinoma. Oncotarget 2018, 9, 34708–34718. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Yang, W.-W.; Yang, L.-Q.; Zhao, F.; Chen, C.-W.; Xu, L.-H.; Fu, J.; Li, S.-L.; Ge, X.-Y. Epiregulin Promotes Lung Metastasis of Salivary Adenoid Cystic Carcinoma. Theranostics 2017, 7, 3700–3714. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Hong, W.; Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef] [PubMed]
- Diepenbruck, M.; Christofori, G. Epithelial–mesenchymal transition (EMT) and metastasis: Yes, no, maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Casanova, J. A common framework for EMT and collective cell migration. Development 2016, 143, 4291–4300. [Google Scholar] [CrossRef]
- Thompson, L.; Chang, B.; Barsky, S.H. Monoclonal Origins of Malignant Mixed Tumors (Carcinosarcomas). Am. J. Surg. Pathol. 1996, 20, 277–285. [Google Scholar] [CrossRef]
- Karacosta, L.G.; Anchang, B.; Ignatiadis, N.; Kimmey, S.C.; Benson, J.A.; Shrager, J.B.; Tibshirani, R.; Bendall, S.C.; Plevritis, S.K. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat. Commun. 2019, 10, 5587. [Google Scholar] [CrossRef]
- Shinde, A.; Paez, J.S.; Libring, S.; Hopkins, K.; Solorio, L.; Wendt, M.K. Transglutaminase-2 facilitates extracellular vesicle-mediated establishment of the metastatic niche. Oncogenesis 2020, 9, 16. [Google Scholar] [CrossRef]
- Tsai, J.H.; Donaher, J.L.; Murphy, D.A.; Chau, S.; Yang, J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition Is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell 2012, 22, 725–736. [Google Scholar] [CrossRef]
- Stylianou, N.; Lehman, M.L.; Wang, C.; Fard, A.T.; Rockstroh, A.; Fazli, L.; Jovanovic, L.; Ward, M.; Sadowski, M.C.; Kashyap, A.S.; et al. A molecular portrait of epithelial–mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogene 2019, 38, 913–934. [Google Scholar] [CrossRef]
- Tsarfaty, I.; Rong, S.; Resau, J.H.; Rulong, S.; da Silva, P.P.; Woude, G.F.V. The Met Proto-Oncogene Mesenchymal to Epithelial Cell Conversion. Science 1994, 263, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Vincan, E.; Darcy, P.K.; A Farrelly, C.; Faux, M.C.; Brabletz, T.; Ramsay, R.G. Frizzled-7 dictates three-dimensional organization of colorectal cancer cell carcinoids. Oncogene 2007, 26, 2340–2352. [Google Scholar] [CrossRef]
- Darmon, M.; Nicolas, J.; Lamblin, D. 5-Azacytidine is able to induce the conversion of teratocarcinoma-derived mesenchymal cells into epithelia cells. EMBO J. 1984, 3, 961–967. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, K.; Qian, C.-N.; Leach, R. DNA methylation is associated with transcription of Snail and Slug genes. Biochem. Biophys. Res. Commun. 2013, 430, 1083–1090. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Wang, S.M.; Coljee, V.W.; Pignolo, R.J.; Rotenberg, M.O.; Cristofalo, V.J.; Sierra, F. Cloning of the human twist gene: Its expression is retained in adult mesodermally-derived tissues. Gene 1997, 187, 83–92. [Google Scholar] [CrossRef]
- Belulescu, I.C.; Mărgăritescu, C.; Dumitrescu, C.I.; Munteanu, M.C.; Dăguci, L.; Mărgăritescu, O.C.; Matei, M. The immunophenotype of epithelial to mesenchymal transition inducing transcription factors in salivary gland adenoid cystic carcinomas. Rom. J. Morphol. Embryol. 2020, 61, 769–782. [Google Scholar] [CrossRef]
- Shen, M.; Wen, Y.; Hua, C.; Xiao, J. The expression of Twist in salivary adenoid cystic carcinoma and its clinicopathological significance. Chin. -Ger. J. Clin. Oncol. 2010, 9, 187–192. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, J.; Tang, Y.; Zhu, G.; Zheng, M.; Jiang, J.; Yang, J.; Liang, X. Coexpression of hypoxia-inducible factor-2α, TWIST2, and SIP1 may correlate with invasion and metastasis of salivary adenoid cystic carcinoma. J. Oral Pathol. Med. 2012, 41, 424–431. [Google Scholar] [CrossRef]
- Pardis, S.; Zare, R.; Jaafari-Ashkavandi, Z.; Ashraf, M.J.; Khademi, B. Twist expression in pleomorphic adenoma, adenoid cystic carcinoma and mucoepidermoid carcinoma of salivary glands. Turk. J. Pathol. 2016, 32, 15–21. [Google Scholar] [CrossRef]
- Kerche, L.E.; de Sousa, E.A.; Squarize, C.H.; Oliveira, K.K.; Marchi, F.A.; Bettim, B.B.; Kowalski, L.P.; Soares, F.A.; Lourenço, S.V.; Coutinho-Camillo, C.M. EMT in salivary gland tumors: The expression of microRNAs miR-155 and miR-200c is associated with clinical-pathological parameters. Mol. Biol. Rep. 2022, 49, 2157–2167. [Google Scholar] [CrossRef]
- Silva, B.S.D.F.; Silva, F.P.Y.; Pontes, H.A.R.; Junior, D.D.S.P. E-cadherin downregulation and Twist overexpression since early stages of oral carcinogenesis. J. Oral Pathol. Med. 2014, 43, 125–131. [Google Scholar] [CrossRef]
- Yuen, H.-F.; Chua, C.-W.; Chan, Y.-P.; Wong, Y.-C.; Wang, X.; Chan, K.-W. Significance of TWIST and E-cadherin expression in the metastatic progression of prostatic cancer. Histopathology 2007, 50, 648–658. [Google Scholar] [CrossRef]
- Zhang, M.; Zheng, M.; Dai, L.; Zhang, W.; Fan, H.; Yu, X.; Pang, X.; Liao, P.; Chen, B.; Wang, S.; et al. CXCL12/CXCR4 facilitates perineural invasion via induction of the Twist/S100A4 axis in salivary adenoid cystic carcinoma. J. Cell. Mol. Med. 2021, 25, 7901–7912. [Google Scholar] [CrossRef]
- Isenmann, S.; Arthur, A.; Zannettino, A.C.; Turner, J.L.; Shi, S.; Glackin, C.A.; Gronthos, S. TWIST Family of Basic Helix-Loop-Helix Transcription Factors Mediate Human Mesenchymal Stem Cell Growth and Commitment. Stem Cells 2009, 27, 2457–2468. [Google Scholar] [CrossRef]
- Yokoyama, K.; Kamata, N.; Hayashi, E.; Hoteiya, T.; Ueda, N.; Fujimoto, R.; Nagayama, M. Reverse correlation of E-cadherin and snail expression in oral squamous cell carcinoma cells in vitro. Oral Oncol. 2001, 37, 65–71. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor Snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, Y.; Zhu, G.; Zheng, M.; Yang, J.; Liang, X. Correlation between transcription factor Snail1 expression and prognosis in adenoid cystic carcinoma of salivary gland. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2010, 110, 764–769. [Google Scholar] [CrossRef]
- Alberga, A.; Boulay, J.-L.; Kempe, E.; Dennefeld, C.; Haenlin, M. The snail gene required for mesoderm formation in Drosophila is expressed dynamically in derivatives of all three germ layers. Development 1991, 111, 983–992. [Google Scholar] [CrossRef]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Wu, Y.; Zhou, B.P. Snail. Cell Adhes. Migr. 2010, 4, 199–203. [Google Scholar] [CrossRef]
- Zhao, D.; Yang, K.; Tang, X.-F.; Lin, N.-N.; Liu, J.-Y. Expression of integrin-linked kinase in adenoid cystic carcinoma of salivary glands correlates with epithelial–mesenchymal transition markers and tumor progression. Med. Oncol. 2013, 30, 619. [Google Scholar] [CrossRef]
- Chen, W.; Ren, X.; Wu, J.; Gao, X.; Cen, X.; Wang, S.; Sheng, S.; Chen, Q.; Tang, Y.; Liang, X. HSP 27 associates with epithelial–mesenchymal transition, stemness and radioresistance of salivary adenoid cystic carcinoma. J. Cell. Mol. Med. 2018, 22, 2283–2298. [Google Scholar] [CrossRef]
- Nieto, M.A.; Sargent, M.G.; Wilkinson, D.G.; Cooke, J. Control of Cell Behavior During Vertebrate Development by Slug, a Zinc Finger Gene. Science 1994, 264, 835–839. [Google Scholar] [CrossRef]
- Du, Y.; Lv, D.; Cui, B.; Li, X.; Chen, H.; Kang, Y.; Chen, Q.; Feng, Y.; Zhang, P.; Chen, J.; et al. Protein kinase D1 induced epithelial–mesenchymal transition and invasion in salivary adenoid cystic carcinoma via E-cadherin/Snail regulation. Oral Dis. 2022, 28, 1539–1554. [Google Scholar] [CrossRef]
- Tang, Y.; Liang, X.; Zhu, G.; Zheng, M.; Yang, J.; Chen, Y. Expression and importance of zinc-finger transcription factor Slug in adenoid cystic carcinoma of salivary gland. J. Oral Pathol. Med. 2010, 39, 775–780. [Google Scholar] [CrossRef]
- Vandewalle, C.; van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef]
- He, Q.; Zhou, X.; Li, S.; Jin, Y.; Chen, Z.; Chen, D.; Cai, Y.; Liu, Z.; Zhao, T.; Wang, A. MicroRNA-181a suppresses salivary adenoid cystic carcinoma metastasis by targeting MAPK–Snai2 pathway. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 5258–5266. [Google Scholar] [CrossRef]
- Wu, B.; Wei, J.; Hu, Z.; Shan, C.; Wang, L.; Zhang, C.; Yang, X.; Yang, X.; Lei, D. Slug silencing inhibited perineural invasion through regulation of EMMPRIN expression in human salivary adenoid cystic carcinoma. Tumor Biol. 2016, 37, 2161–2169. [Google Scholar] [CrossRef]
- Yang, L.; Wang, T.; Zhang, J.; Wang, X. BTBD7 silencing inhibited epithelial- mesenchymal transition (EMT) via regulating Slug expression in human salivary adenoid cystic carcinoma. Cancer Biomark. 2017, 20, 461–468. [Google Scholar] [CrossRef]
- Yang, L.; Wang, T.; Zhang, J.; Liu, Z.; Wang, X. Expression of BTBD7 in primary salivary adenoid cystic carcinoma and correlation with Slug and prognosis. Cancer Biomark. 2016, 17, 179–185. [Google Scholar] [CrossRef]
- Liu, S.; Ye, D.; Xu, D.; Liao, Y.; Zhang, L.; Liu, L.; Yu, W.; Wang, Y.; He, Y.; Hu, J.; et al. Autocrine epiregulin activates EGFR pathway for lung metastasis via EMT in salivary adenoid cystic carcinoma. Oncotarget 2016, 7, 25251–25263. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, J.; Wang, Y.; Ye, W.; Zhang, X.; Ju, H.; Xu, D.; Liu, L.; Ye, D.; Zhang, L.; et al. EGFR activation induced Snail-dependent EMT and myc-dependent PD-L1 in human salivary adenoid cystic carcinoma cells. Cell Cycle 2018, 17, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ge, X.-Y.; Liu, S.-M.; Zheng, L.; Huang, M.-W.; Shi, Y.; Fu, J.; Zhang, J.-G.; Li, S.-L. Nimotuzumab suppresses epithelial–mesenchymal transition and enhances apoptosis in low-dose UV-C treated salivary adenoid cystic carcinoma cell lines in vitro. Anti-Cancer Drugs 2014, 25, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Li, B.-B.; Zhou, C.-X. Bmi-1 expression predicts prognosis in salivary adenoid cystic carcinoma and correlates with epithelial-mesenchymal transition–related factors. Ann. Diagn. Pathol. 2016, 22, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.M.; Liu, W.; Cao, Z.-H.; Liu, M.-X. Effects of ZEB1 on regulating osteosarcoma cells via NF-κB/iNOS. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1184–1190. [Google Scholar]
- Gu, K.; Ming-Ming, L.; Jing, S.; Fang, L.; Jing-Yan, C.; Shi, J.; Yan, Y. Interleukin-17-induced EMT promotes lung cancer cell migration and invasion via NF-κB/ZEB1 signal pathway. Am. J. Cancer Res. 2015, 5, 1169–1179. [Google Scholar]
- Yao, X.; Wang, Y.; Duan, Y.; Zhang, Q.; Li, P.; Jin, R.; Tao, Y.; Zhang, W.; Wang, X.; Jing, C.; et al. IGFBP2 promotes salivary adenoid cystic carcinoma metastasis by activating the NF-κB/ZEB1 signaling pathway. Cancer Lett. 2018, 432, 38–46. [Google Scholar] [CrossRef]
- Peng, J.; Wang, H.-C.; Liu, Y.; Jiang, J.-H.; Lv, W.-Q.; Yang, Y.; Li, C.-Y.; Qiu, X.-Y. Involvement of non-B cell-derived immunoglobulin G in the metastasis and prognosis of salivary adenoid cystic carcinoma. Oncol. Lett. 2017, 14, 4491–4498. [Google Scholar] [CrossRef]
- Slack, J.M.; Tosh, D. Transdifferentiation and metaplasia—switching cell types. Curr. Opin. Genet. Dev. 2001, 11, 581–586. [Google Scholar] [CrossRef]
- Matsumiya-Matsumoto, Y.; Morita, Y.; Uzawa, N. Pleomorphic Adenoma of the Salivary Glands and Epithelial–Mesenchymal Transition. J. Clin. Med. 2022, 11, 4210. [Google Scholar] [CrossRef]
- Kuburich, N.A.; Hollander, P.D.; Pietz, J.T.; Mani, S.A. Vimentin and cytokeratin: Good alone, bad together. Semin. Cancer Biol. 2022, 86 Pt 3, 816–826. [Google Scholar] [CrossRef]
- Belulescu, I.C.; Mărgăritescu, C.; Dumitrescu, C.I.; Munteanu, M.C.; Mărgăritescu, O.C. Immunophenotypical alterations with impact on the epithelial–mesenchymal transition (EMT) process in salivary gland adenoid cystic carcinomas. Rom. J. Morphol. Embryol. 2020, 61, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Meer, S.; Altini, M. CK7+/CK20? immunoexpression profile is typical of salivary gland neoplasia. Histopathology 2007, 51, 26–32. [Google Scholar] [CrossRef]
- Lee, J.-H.; Lee, J.H.; Kim, A.; Kim, I.; Chae, Y.-S. Unique expression of MUC3, MUC5AC and cytokeratins in salivary gland carcinomas. Pathol. Int. 2005, 55, 386–390. [Google Scholar] [CrossRef]
- Ben Salha, I.; Bhide, S.; Mourtzoukou, D.; Fisher, C.; Thway, K. Solid Variant of Adenoid Cystic Carcinoma: Difficulties in Diagnostic Recognition. Int. J. Surg. Pathol. 2016, 24, 419–424. [Google Scholar] [CrossRef]
- Gao, X.-L.; Wu, J.-S.; Cao, M.-X.; Gao, S.-Y.; Cen, X.; Jiang, Y.-P.; Wang, S.-S.; Tang, Y.-J.; Chen, Q.-M.; Liang, X.-H.; et al. Cytokeratin-14 contributes to collective invasion of salivary adenoid cystic carcinoma. PLoS ONE 2017, 12, e0171341. [Google Scholar] [CrossRef]
- Ge, M.-H.; Ling, Z.-Q.; Tan, Z.; Chen, C.; Xu, J.-J.; Yu, J.-L. Expression and significance of E-cadherin in adenoid cystic carcinoma of salivary glands. Zhonghua Yi Xue Za Zhi 2012, 92, 106–109. [Google Scholar]
- Lai, F.-Y.; Zhang, Q.; Wu, Q.-L.; Qing, J.; Cao, Y. Expression and significance of E-cadherin in adenoid cystic carcinoma of the salivary glands. Ai Zheng 2007, 26, 1025–1028. [Google Scholar]
- Maruya, S.-I.; Kurotaki, H.; Wada, R.; Saku, T.; Shinkawa, H.; Yagihashi, S. Promoter methylation and protein expression of the E-cadherin gene in the clinicopathologic assessment of adenoid cystic carcinoma. Mod. Pathol. 2004, 17, 637–645. [Google Scholar] [CrossRef]
- Prabhu, S.; Kaveri, H.; Rekha, K. Benign; malignant salivary gland tumors: Comparison of immunohistochemical expression of e-cadherin. Oral Oncol. 2009, 45, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, Z.; Wang, H.; Wang, S.; Yu, X.; Wu, J.; Pang, X.; Wu, J.; Yang, X.; Tang, Y.; et al. MIF promotes perineural invasion through EMT in salivary adenoid cystic carcinoma. Mol. Carcinog. 2019, 58, 898–912. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Feng, Y.; Lin, T.; Huang, X.-Y.; Gan, R.-H.; Zhao, Y.; Su, B.-H.; Ding, L.-C.; She, L.; Chen, J.; et al. CDH4 suppresses the progression of salivary adenoid cystic carcinoma via E-cadherin co-expression. Oncotarget 2016, 7, 82961–82971. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Wang, W.; Hu, Z.; Shan, C.; Wang, L.; Wu, B.; Yang, Z.; Yang, X.; Lei, D. BDNF mediated TrkB activation contributes to the EMT progression and the poor prognosis in human salivary adenoid cystic carcinoma. Oral Oncol. 2015, 51, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, R.B.; Nonaka, C.F.W.; Rabenhorst, S.H.B.; Miguel, M.C.D.C.; Pinto, L.P.; de Souza, L.B. Pleomorphic adenoma and adenoid cystic carcinoma of salivary glands: E-cadherin immunoexpression and analysis of the CDH1 -160C/A polymorphism. Arch. Oral Biol. 2017, 73, 48–54. [Google Scholar] [CrossRef]
- Phattarataratip, E.; Kositkittiwanit, N.; Kajornkiatkul, P.; Yeunyong, P.; Ratanapitak, R. P120 catenin expression and its correlation with E-cadherin in salivary gland neoplasms. J. Oral Biol. Craniofacial Res. 2019, 9, 57–62. [Google Scholar] [CrossRef]
- Van Der Wal, J.E.; Sgaramella, N.; Spaak, L.N.; Zborayova, K.; Nylander, K. High podoplanin and low E-cadherin levels correlate with better prognosis in adenoid cystic carcinoma. Clin. Exp. Dent. Res. 2019, 5, 350–355. [Google Scholar] [CrossRef]
- Wu, J.; Li, Z.; Wang, H.; Yu, X.; Pang, X.; Wu, J.; Wang, S.; Zhang, M.; Yang, X.; Cao, M.; et al. Cathepsin B defines leader cells during the collective invasion of salivary adenoid cystic carcinoma. Int. J. Oncol. 2019, 54, 1233–1244. [Google Scholar] [CrossRef]
- Westcott, J.M.; Prechtl, A.M.; Maine, E.A.; Dang, T.; Esparza, M.; Sun, H.; Zhou, Y.; Xie, Y.; Pearson, G.W. An epigenetically distinct breast cancer cell subpopulation promotes collective invasion. J. Clin. Investig. 2015, 125, 1927–1943. [Google Scholar] [CrossRef]
- Konen, J.; Summerbell, E.; Dwivedi, B.; Galior, K.; Hou, Y.; Rusnak, L.; Chen, A.; Saltz, J.; Zhou, W.; Boise, L.H.; et al. Image-guided genomics of phenotypically heterogeneous populations reveals vascular signalling during symbiotic collective cancer invasion. Nat. Commun. 2017, 8, 15078. [Google Scholar] [CrossRef]
- Krakhmal, N.V.; Zavyalova, M.; Denisov, E.V.; Vtorushin, S.V.; Perelmuter, V. Cancer Invasion: Patterns and Mechanisms. Acta Nat. 2015, 7, 17–28. [Google Scholar] [CrossRef]
- Krebs, M.G.; Metcalf, R.L.; Carter, L.; Brady, G.; Blackhall, F.H.; Dive, C. Molecular analysis of circulating tumour cells—Biology and biomarkers. Nat. Rev. Clin. Oncol. 2014, 11, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska-Podhorodecka, Z.; McCulloch, C.A. Vimentin regulates the assembly and function of matrix adhesions. Wound Repair Regen. 2021, 29, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Caselitz, J.; Becker, J.; Seifert, G.; Weber, K.; Osborn, M. Coexpression of keratin and vimentin filaments in adenoid cystic carcinomas of salivary glands. Virchows Arch. 1984, 403, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Chomette, G.; Auriol, M.; Vaillant, J.M.; Kasai, T.; Okada, Y.; Mori, M. Heterogeneity and co-expression of intermediate filament proteins in adenoid cystic carcinoma of salivary glands. Pathol. Biol. 1991, 39, 110–116. [Google Scholar] [PubMed]
- Patten, J.; Wang, K. Fibronectin in development and wound healing. Adv. Drug Deliv. Rev. 2021, 170, 353–368. [Google Scholar] [CrossRef]
- D’Ardenne, A.J.; Kirkpatrick, P.; Wells, C.A.; Davies, J.D. Laminin and fibronectin in adenoid cystic carcinoma. J. Clin. Pathol. 1986, 39, 138–144. [Google Scholar] [CrossRef]
- Dong, F.; Wang, X.; Zhang, P. Study of electron microscopy histochemistry and immunohistochemistry of extracellular matrix in adenoid cystic carcinoma. Hua Xi Kou Qiang Yi Xue Za Zhi 1997, 15, 306–307. [Google Scholar]
- Wegner, A.; Waśniewska, E.; Golusiński, W.; Golusiński, P. Assessment of extracellular matrix proteins (laminin and fibronectin) in adenoid cystic carcinoma of salivary gland using morphometric method. Rep. Pract. Oncol. Radiother. 2007, 12, 339–343. [Google Scholar] [CrossRef]
- Liu, Y.; Song, J.; Zhang, J.; Yang, L.; Liu, Z.; Wang, X. BTB/POZ domain-containing protein 7 is inversely associated with fibronectin expression in salivary adenoid cystic carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 125, 468–477. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, Y.; Zhang, W. Immunohistochemical study of basement membrane proteins expression in adenoid cystic carcinoma of salivary glands. Hua Xi Kou Qiang Yi Xue Za Zhi 1999, 17, 236–238,253. [Google Scholar]
- Cao, Z.-Q.; Wang, Z.; Leng, P. Aberrant N-cadherin expression in cancer. Biomed. Pharmacother. 2019, 118, 109320. [Google Scholar] [CrossRef]
- Jiang, Y.; Feng, X.; Zheng, L.; Li, S.-L.; Ge, X.-Y.; Zhang, J.-G. Thioredoxin 1 mediates TGF-β-induced epithelial-mesenchymal transition in salivary adenoid cystic carcinoma. Oncotarget 2015, 6, 25506–25519. [Google Scholar] [CrossRef]
- Nawshad, A.; LaGamba, D.; Polad, A.; Hay, E.D. Transforming Growth Factor-β Signaling during Epithelial-Mesenchymal Transformation: Implications for Embryogenesis and Tumor Metastasis. Cells Tissues Organs 2005, 179, 11–23. [Google Scholar] [CrossRef]
- Dong, L.; Wang, Y.; Li, S.; Yu, G.; Gan, Y.; Li, D.; Wang, C. TGF-β1 Promotes Migration and Invasion of Salivary Adenoid Cystic Carcinoma. J. Dent. Res. 2011, 90, 804–809. [Google Scholar] [CrossRef]
- Dong, L.; Ge, X.-Y.; Wang, Y.; Yang, L.-Q.; Li, S.-L.; Yu, G.-Y.; Gao, Y.; Fu, J. Transforming growth factor-β and epithelial–mesenchymal transition are associated with pulmonary metastasis in adenoid cystic carcinoma. Oral Oncol. 2013, 49, 1051–1058. [Google Scholar] [CrossRef]
- Knust, E.; Bossinger, O. Composition and Formation of Intercellular Junctions in Epithelial Cells. Science 2002, 298, 1955–1959. [Google Scholar] [CrossRef]
- Birchmeier, W.; Hülsken, J.; Behrens, J. E-Cadherin as an Invasion Suppressor. Ciba Found. Symp. 2007, 189, 124–141. [Google Scholar] [CrossRef]
- Furuse, C.; Cury, P.R.; Altemani, A.; Júnior, D.D.S.P.; de Araújo, N.S.; Araújo, V. β-Catenin and E-Cadherin Expression in Salivary Gland Tumors. Int. J. Surg. Pathol. 2006, 14, 212–217. [Google Scholar] [CrossRef]
- Strillacci, A.; Valerii, M.C.; Sansone, P.; Caggiano, C.; Sgromo, A.; Vittori, L.; Fiorentino, M.; Poggioli, G.; Rizzello, F.; Campieri, M.; et al. Loss of miR-101 expression promotes Wnt/β-catenin signalling pathway activation and malignancy in colon cancer cells. J. Pathol. 2013, 229, 379–389. [Google Scholar] [CrossRef]
- Ma, S.-R.; Mao, L.; Deng, W.-W.; Li, Y.-C.; Bu, L.-L.; Yu, G.-T.; Zhang, W.-F.; Sun, Z.-J. AGR2 promotes the proliferation, migration and regulates epithelial-mesenchymal transition in salivary adenoid cystic carcinoma. Am. J. Transl. Res. 2017, 9, 507–519. [Google Scholar]
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef]
- West, R.B.; Kong, C.; Clarke, N.; Gilks, T.; Lipsick, J.S.; Cao, H.; Kwok, S.; Montgomery, K.D.; Varma, S.; Le, Q.-T. MYB Expression and Translocation in Adenoid Cystic Carcinomas and Other Salivary Gland Tumors With Clinicopathologic Correlation. Am. J. Surg. Pathol. 2011, 35, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.G.; Gonda, T.J. MYB function in normal and cancer cells. Nat. Rev. Cancer 2008, 8, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y. Myb proteins: Angels and demons in normal and transformed cells. Front. Biosci. 2011, 16, 1109–1131. [Google Scholar] [CrossRef] [PubMed]
- George, O.L.; Ness, S.A. Situational Awareness: Regulation of the Myb Transcription Factor in Differentiation, the Cell Cycle and Oncogenesis. Cancers 2014, 6, 2049–2071. [Google Scholar] [CrossRef]
- Xu, L.-H.; Zhao, F.; Yang, W.-W.; Chen, C.-W.; Du, Z.-H.; Fu, M.; Ge, X.-Y.; Li, S.-L. MYB promotes the growth and metastasis of salivary adenoid cystic carcinoma. Int. J. Oncol. 2019, 54, 1579–1590. [Google Scholar] [CrossRef]
- Li, Y.; Jin, K.; van Pelt, G.W.; van Dam, H.; Yu, X.; Mesker, W.E.; Dijke, P.T.; Zhou, F.; Zhang, L. c-Myb Enhances Breast Cancer Invasion and Metastasis through the Wnt/β-Catenin/Axin2 Pathway. Cancer Res. 2016, 76, 3364–3375. [Google Scholar] [CrossRef]
- Sala, A. c-MYB and TGFβ: EMT’s dynamic duo in breast cancer. Cell Cycle 2012, 11, 17. [Google Scholar] [CrossRef]
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Yang, M.-H.; Wu, K.-J. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle 2008, 7, 2090–2096. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, Y.-L.; Liang, X.-H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia in cancer cell metabolism and pH regulation. Essays Biochem. 2007, 43, 165–178. [Google Scholar] [CrossRef]
- Wang, H.; Wang, S.; Zheng, M.; Dai, L.; Wang, K.; Gao, X.; Cao, M.; Yu, X.; Pang, X.; Zhang, M.; et al. Hypoxia promotes vasculogenic mimicry formation by vascular endothelial growth factor A mediating epithelial-mesenchymal transition in salivary adenoid cystic carcinoma. Cell Prolif. 2019, 52, e12600. [Google Scholar] [CrossRef]
- Andreasen, S. Molecular features of adenoid cystic carcinoma with an emphasis on microRNA expression. Apmis 2018, 126 (Suppl. S140), 7–57. [Google Scholar] [CrossRef]
- Warzecha, C.C.; Carstens, R.P. Complex changes in alternative pre-mRNA splicing play a central role in the epithelial-to-mesenchymal transition (EMT). Semin. Cancer Biol. 2012, 22, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Subramanyam, D.; Blelloch, R.; Derynck, R. Regulation of epithelial–mesenchymal and mesenchymal–epithelial transitions by microRNAs. Curr. Opin. Cell Biol. 2013, 25, 200–207. [Google Scholar] [CrossRef]
- Sun, D.-K.; Wang, J.-M.; Zhang, P.; Wang, Y.-Q. MicroRNA-138 Regulates Metastatic Potential of Bladder Cancer Through ZEB2. Cell. Physiol. Biochem. 2015, 37, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.R.B.; Coutinho-Camillo, C.M.; Soares, F.A.; Freitas, V.; Vilas-Bôas, D.S.; Xavier, F.; Rocha, C.A.G.; de Araújo, I.B.; dos Santos, J.N. MicroRNAs expression pattern related to mast cell activation and angiogenesis in paraffin-embedded salivary gland tumors. Pathol. Res. Pr. 2017, 213, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hu, Y.; Fu, J.; Yang, X.; Zhang, Z. MicroRNA155 in the growth and invasion of salivary adenoid cystic carcinoma. J. Oral Pathol. Med. 2013, 42, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhou, L.; Jiang, F.; Zeng, B.; Wei, C.; Zhao, W.; Yu, D. Downregulation of miR-222 Induces Apoptosis and Cellular Migration in Adenoid Cystic Carcinoma Cells. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2017, 25, 207–214. [Google Scholar] [CrossRef]
- Denaro, M.; Navari, E.; Ugolini, C.; Seccia, V.; Donati, V.; Casani, A.P.; Basolo, F. A microRNA signature for the differential diagnosis of salivary gland tumors. PLoS ONE 2019, 14, e0210968. [Google Scholar] [CrossRef]
- Qiao, Z.; Zou, Y.; Zhao, H. MicroRNA-140-5p inhibits salivary adenoid cystic carcinoma progression and metastasis via targeting survivin. Cancer Cell Int. 2019, 19, 301. [Google Scholar] [CrossRef]
- Cheng, M.; Qin, G. Progenitor Cell Mobilization and Recruitment: SDF-1, CXCR4, α4-integrin, and c-kit. Prog. Mol. Biol. Transl. Sci. 2012, 111, 243–264. [Google Scholar] [CrossRef]
- Liang, J.; Wu, Y.-L.; Chen, B.-J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-Kit Receptor-Mediated Signal Transduction and Tumor-Related Diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef]
- Tang, Y.-L.; Fan, Y.-L.; Jiang, J.; Li, K.-D.; Zheng, M.; Chen, W.; Ma, X.-R.; Geng, N.; Chen, Q.-M.; Chen, Y.; et al. C-kit induces epithelial-mesenchymal transition and contributes to salivary adenoid cystic cancer progression. Oncotarget 2014, 5, 1491–1501. [Google Scholar] [CrossRef]
- Ghazi, A.; Salehinejad, J.; Mohtasham, N.; Bagherpour, A.; Abbaszadeh-Bidokhty, H. Evaluation of c-kit protein (CD117) expression in common salivary gland neoplasms. J. Oral Maxillofac. Pathol. 2014, 18, 177–182. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Fordice, J.; Kershaw, C.; El-Naggar, A.; Goepfert, H. Adenoid Cystic Carcinoma of the Head and Neck. Arch. Otolaryngol. Neck Surg. 1999, 125, 149–152. [Google Scholar] [CrossRef]
- Yang, X.; Jing, D.; Liu, L.; Shen, Z.; Ju, J.; Ma, C.; Sun, M. Downregulation of p53 promotes in vitro perineural invasive activity of human salivary adenoid cystic carcinoma cells through epithelial-mesenchymal transition-like changes. Oncol. Rep. 2015, 33, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Laurie, S.A.; Ho, A.L.; Fury, M.G.; Sherman, E.; Pfister, D.G. Systemic therapy in the management of metastatic or locally recurrent adenoid cystic carcinoma of the salivary glands: A systematic review. Lancet Oncol. 2011, 12, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Locati, L.; Bossi, P.; Perrone, F.; Potepan, P.; Crippa, F.; Mariani, L.; Casieri, P.; Orsenigo, M.; Losa, M.; Bergamini, C.; et al. Cetuximab in recurrent and/or metastatic salivary gland carcinomas: A phase II study. Oral Oncol. 2009, 45, 574–578. [Google Scholar] [CrossRef]
- Agulnik, M.; Cohen, E.W.; Cohen, R.B.; Chen, E.X.; Vokes, E.E.; Hotte, S.J.; Winquist, E.; Laurie, S.; Hayes, D.N.; Dancey, J.E.; et al. Phase II Study of Lapatinib in Recurrent or Metastatic Epidermal Growth Factor Receptor and/or erbB2 Expressing Adenoid Cystic Carcinoma and Non–Adenoid Cystic Carcinoma Malignant Tumors of the Salivary Glands. J. Clin. Oncol. 2007, 25, 3978–3984. [Google Scholar] [CrossRef]
- Hotte, S.J.; Winquist, E.W.; Lamont, E.; MacKenzie, M.; Vokes, E.; Chen, E.X.; Brown, S.; Pond, G.R.; Murgo, A.; Siu, L.L. Imatinib Mesylate in Patients With Adenoid Cystic Cancers of the Salivary Glands Expressing c-kit: A Princess Margaret Hospital Phase II Consortium Study. J. Clin. Oncol. 2005, 23, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Locati, L.D.; Galbiati, D.; Calareso, G.; Alfieri, S.; Singer, S.; Cavalieri, S.; Bergamini, C.; Bossi, P.; Orlandi, E.; Resteghini, C.; et al. Patients with adenoid cystic carcinomas of the salivary glands treated with lenvatinib: Activity and quality of life. Cancer 2020, 126, 1888–1894. [Google Scholar] [CrossRef]
- Gao, M.; Hao, Y.; Huang, M.; Ma, D.; Luo, H.; Gao, Y.; Peng, X.; Yu, G. Clinicopathological study of distant metastases of salivary adenoid cystic carcinoma. Int. J. Oral Maxillofac. Surg. 2013, 42, 923–928. [Google Scholar] [CrossRef]
- Zavadil, J.; Haley, J.; Kalluri, R.; Muthuswamy, S.K.; Thompson, E. Epithelial-Mesenchymal Transition. Cancer Res 2008, 68, 9574–9577. [Google Scholar] [CrossRef]
- Revenu, C.; Gilmour, D. EMT 2.0: Shaping epithelia through collective migration. Curr. Opin. Genet. Dev. 2009, 19, 338–342. [Google Scholar] [CrossRef]
- Souid, S.; Elsayed, H.E.; Ebrahim, H.Y.; Mohyeldin, M.M.; Siddique, A.B.; Karoui, H.; El Sayed, K.A.; Essafi-Benkhadir, K. 13 1 -Oxophorbine protopheophorbide A from Ziziphus lotus as a novel mesenchymal-epithelial transition factor receptor inhibitory lead for the control of breast tumor growth in vitro and in vivo. Mol. Carcinog. 2018, 57, 1507–1524. [Google Scholar] [CrossRef]
- Hamilton, G.; Hochmair, M.; Rath, B.; Klameth, L.; Zeillinger, R. Small cell lung cancer: Circulating tumor cells of extended stage patients express a mesenchymal-epithelial transition phenotype. Cell Adhes. Migr. 2016, 10, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gong, W.; Ma, X.; Sun, X.; Jiang, H.; Chen, T. Smad7 maintains epithelial phenotype of ovarian cancer stem-like cells and supports tumor colonization by mesenchymal-epithelial transition. Mol. Med. Rep. 2015, 11, 309–316. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| EMT-TF | Original Function | Role of EMT in SACC | References |
|---|---|---|---|
| TWIST1/TWIST2 |
|
| [13,36,37,38,39,40,41,42,43,44,45] |
| SNAIL |
|
| [37,46,47,48,49,50,51] |
| SLUG |
|
| [37,52,53,54] |
| ZEB1/ZEB2 |
|
| [37,55,56,57] |
| Intervention | Targets | EMT-Axis | Phase | Status | References |
|---|---|---|---|---|---|
| Lenvatinib | VEGFR, FGFR, PDGFR, RET, KIT | Pi3K/AKT/GSK3β/Snail | II | Completed | NCT02860936 |
| Dovitinib | VEGFR, FGFR, PDGFR, RET, KIT | Pi3K/AKT/GSK3β/Snail | II | Completed | NCT01678105 |
| Sunitinib | VEGFR, PDGFR, KIT | Pi3K/AKT/GSK3β/Snail | II | Completed | NCT00886132 |
| Amivantamab | EGFR, MET | Pi3K/AKT/GSK3β/Snail, Src/ERK/Slug | II | Recruiting | NCT05074940 |
| Dasatinib | BCR-ABL, SRC | Pi3K/AKT/GSK3β/Snail, Src/ERK/Slug | II | Completed | NCT00859937 |
| Lapatinib | HER2, EGFR | Pi3K/AKT/GSK3β/Snail, Src/ERK/Slug | II | Completed | NCT00095563 |
| 9-ING-41 Plus Carboplatin | GSK3β | WNT/β-catenin/Snail | II | Recruiting | NCT05010629 |
| Akt Inhibitor MK2206 | Akt | Pi3K/AKT/GSK3β/Snail | II | Completed | NCT01604772 |
| Imatinib | BCR-ABL, KIT | Pi3K/AKT/GSK3β/Snail | II | Completed | NCT00045669 |
| Trastuzumab | HER2 | Pi3K/AKT/GSK3β/Snail, Src/ERK/Slug | II | Completed | NCT00004163 |
| Gefitinib | EGFR | Pi3K/AKT/GSK3β/Snail, Src/ERK/Slug | I | Completed | NCT00068497 |
| Erlotinib and Cetuximab with or without Bevacizumab | EGFR, VEGFR | Pi3K/AKT/GSK3β/Snail, Src/ERK/Slug | I | Completed | NCT00101348 |
| Cetuximab and Everolimus | EGFR, mTOR, HIF | Pi3K/AKT/GSK3β/Snail, Src/ERK/Slug, HIF-1α | I | Completed | NCT01637194 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hoch, C.C.; Stögbauer, F.; Wollenberg, B. Unraveling the Role of Epithelial–Mesenchymal Transition in Adenoid Cystic Carcinoma of the Salivary Glands: A Comprehensive Review. Cancers 2023, 15, 2886. https://doi.org/10.3390/cancers15112886
Hoch CC, Stögbauer F, Wollenberg B. Unraveling the Role of Epithelial–Mesenchymal Transition in Adenoid Cystic Carcinoma of the Salivary Glands: A Comprehensive Review. Cancers. 2023; 15(11):2886. https://doi.org/10.3390/cancers15112886
Chicago/Turabian StyleHoch, Cosima C., Fabian Stögbauer, and Barbara Wollenberg. 2023. "Unraveling the Role of Epithelial–Mesenchymal Transition in Adenoid Cystic Carcinoma of the Salivary Glands: A Comprehensive Review" Cancers 15, no. 11: 2886. https://doi.org/10.3390/cancers15112886
APA StyleHoch, C. C., Stögbauer, F., & Wollenberg, B. (2023). Unraveling the Role of Epithelial–Mesenchymal Transition in Adenoid Cystic Carcinoma of the Salivary Glands: A Comprehensive Review. Cancers, 15(11), 2886. https://doi.org/10.3390/cancers15112886