
Molecular Targeted Therapies in Metastatic Prostate Cancer: Recent Advances and Future Challenges


Abstract
:Simple Summary
Abstract
1. Introduction
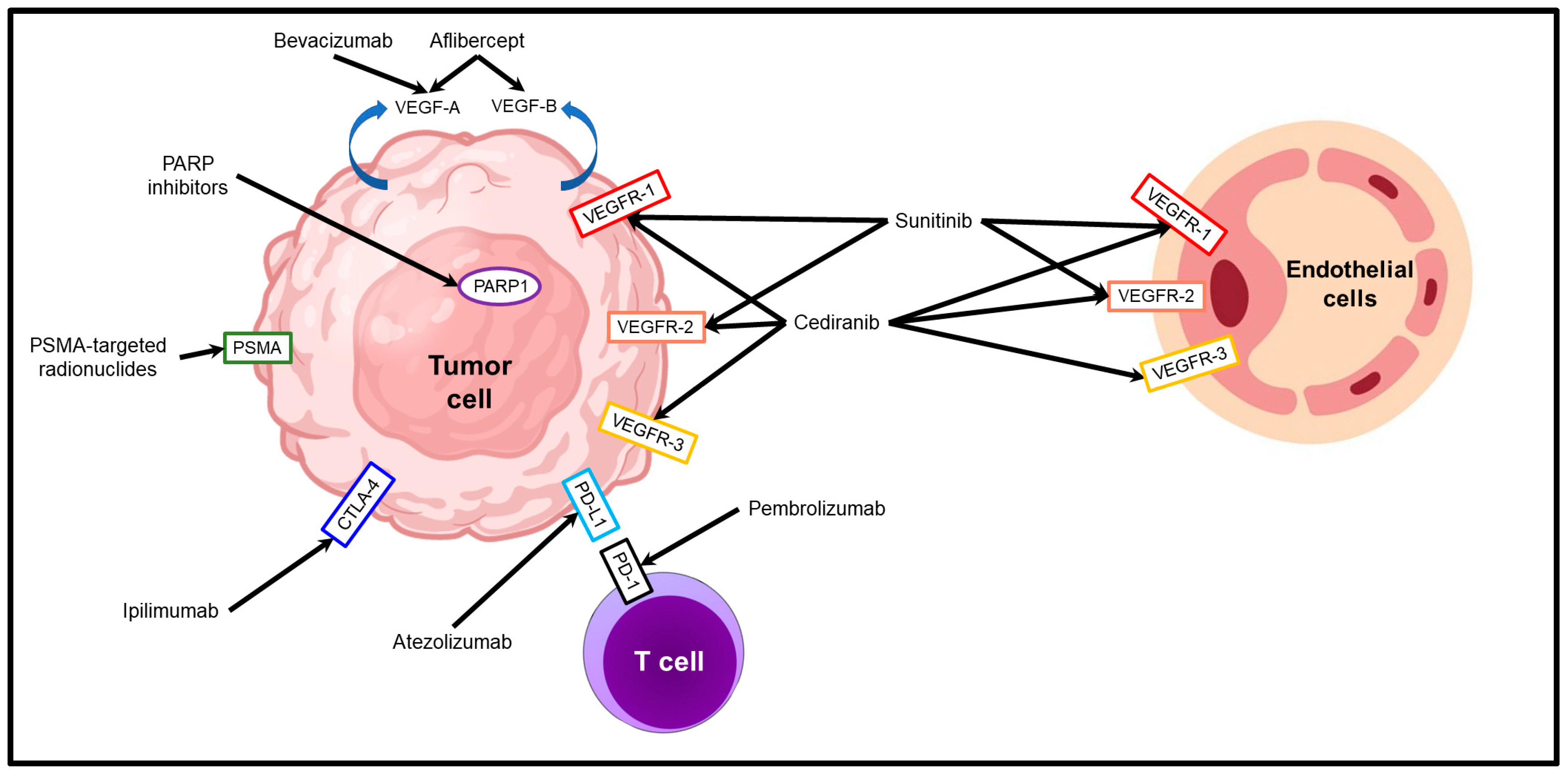
2. Molecular Targeted Therapies for the Treatment of Metastatic Prostate Cancer
2.1. PSMA-Targeted Radionuclide Therapies
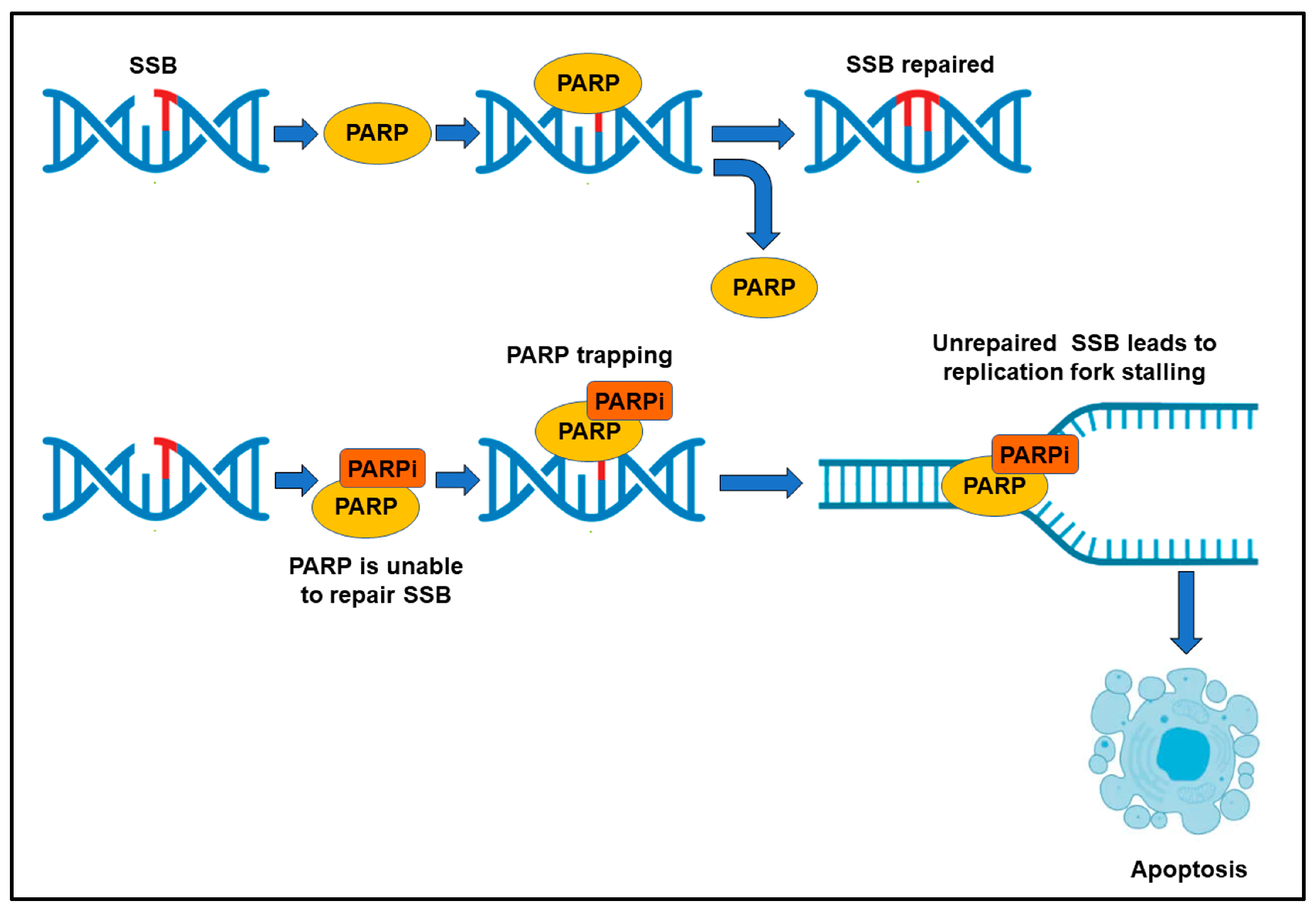
2.2. DNA Repair Inhibitors
2.3. Therapies Targeting Tumor Neovascularization
2.4. Immune Checkpoint Inhibitors
3. Future Challenges
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 19 May 2023).
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Survival Rates for Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer/detection-diagnosis-staging/survival-rates.html (accessed on 18 May 2023).
- Fitzpatrick, J.M. Management of localized prostate cancer in senior adults: The crucial role of comorbidity. BJU Int. 2008, 101 (Suppl. 2), 16–22. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Chen, K.; Jiang, K.; Tang, L.; Chen, X.; Hu, J.; Sun, F. Analysis of Clinical Trials on Therapies for Prostate Cancer in Mainland China and Globally from 2010 to 2020. Front. Oncol. 2021, 11, 647110. [Google Scholar] [CrossRef]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Porter, A.; McEwan, A.; Powe, J.; Reid, R.; McGowan, D.; Lukka, H.; Sathyanarayana, J.; Yakemchuk, V.; Thomas, G.; Erlich, L.; et al. Results of a randomized phase-III trial to evaluate the efficacy of strontium-89 adjuvant to local field external beam irradiation in the management of endocrine resistant metastatic prostate cancer. Int. J. Radiat. Oncol. 1993, 25, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Lepor, H. Management of clinically localized prostate cancer. Rev. Urol. 2004, 6, S3–S12. [Google Scholar]
- Chang, S.S. Overview of Prostate-Specific Membrane Antigen. Rev. Urol. 2004, 6 (Suppl. 10), S13–S18. [Google Scholar]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef]
- Hofman, M.S.; Emmett, L.; Sandhu, S.; Iravani, A.; Joshua, A.M.; Goh, J.C.; Pattison, D.A.; Tan, T.H.; Kirkwood, I.D.; Francis, R.J.; et al. TheraP: 177Lu-PSMA-617 (LuPSMA) versus cabazitaxel in metastatic castration-resistant prostate cancer (mCRPC) progressing after docetaxel—Overall survival after median follow-up of 3 years (ANZUP 1603). J. Clin. Oncol. 2022, 40, 5000. [Google Scholar] [CrossRef]
- El Fakiri, M.; Geis, N.M.; Ayada, N.; Eder, M.; Eder, A.-C. PSMA-Targeting Radiopharmaceuticals for Prostate Cancer Therapy: Recent Developments and Future Perspectives. Cancers 2021, 13, 3967. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Haberkorn, U.; Giesel, F.L. Radionuclide Therapy of Metastatic Prostate Cancer. Semin. Nucl. Med. 2019, 49, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Gafita, A.; Marcus, C.; Kostos, L.; Schuster, D.M.; Calais, J.; Hofman, M.S. Predictors and Real-World Use of Prostate-Specific Radioligand Therapy: PSMA and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 366–382. [Google Scholar] [CrossRef]
- Nauseef, J.T.; Osborne, J.; Gregos, P.; Thomas, C.; Bissassar, M.; Singh, S.; Patel, A.; Tan, A.; Naiz, M.O.; Zuloaga, J.M.; et al. Phase I/II study of 225Ac-J591 plus 177Lu-PSMA-I&T for progressive metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2022, 40, TPS5100. [Google Scholar] [CrossRef]
- Sena, L.A.; Wang, H.; ScM, S.J.L.; Rifkind, I.; Ngomba, N.; Isaacs, J.T.; Luo, J.; Pratz, C.; Sinibaldi, V.; Carducci, M.A.; et al. Bipolar androgen therapy sensitizes castration-resistant prostate cancer to subsequent androgen receptor ablative therapy. Eur. J. Cancer 2020, 144, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Featherstone, C.; Jackson, S.P. DNA double-strand break repair. Curr. Biol. 1999, 9, R759–R761. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Lin, S.-Y.; Brunicardi, F.C.; Goss, J.; Li, K. DNA Damage Response Pathways in Tumor Suppression and Cancer Treatment. World J. Surg. 2008, 33, 661–666. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Nombela, P.; Lozano, R.; Aytes, A.; Mateo, J.; Olmos, D.; Castro, E. BRCA2 and Other DDR Genes in Prostate Cancer. Cancers 2019, 11, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.D.; Ng, T.L.; Estrada-Bernal, A.; Le, A.T.; Ennever, P.R.; Camidge, D.R.; Doebele, R.C.; Aisner, D.L. Dramatic Response to Crizotinib in a Patient With Lung Cancer Positive for an HLA-DRB1-MET Gene Fusion. JCO Precis. Oncol. 2017, 1, PO.17.00117. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, P.; Lloyd, R.G. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 2002, 3, 859–870. [Google Scholar] [CrossRef]
- Dasovich, M.; Beckett, M.Q.; Bailey, S.; Ong, S.-E.; Greenberg, M.M.; Leung, A.K.L. Identifying Poly(ADP-ribose)-Binding Proteins with Photoaffinity-Based Proteomics. J. Am. Chem. Soc. 2021, 143, 3037–3042. [Google Scholar] [CrossRef]
- Krastev, D.B.; Pettitt, S.J.; Campbell, J.; Song, F.; Tanos, B.E.; Stoynov, S.S.; Ashworth, A.; Lord, C.J. Coupling bimolecular PARylation biosensors with genetic screens to identify PARylation targets. Nat. Commun. 2018, 9, 2016. [Google Scholar] [CrossRef] [Green Version]
- Teloni, F.; Altmeyer, M. Readers of poly(ADP-ribose): Designed to be fit for purpose. Nucleic Acids Res. 2015, 44, 993–1006. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Leung, D.K.W.; Chiu, P.K.F.; Ng, C.-F.; Teoh, J.Y.C. Novel Strategies for Treating Castration-Resistant Prostate Cancer. Biomedicines 2021, 9, 339. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Wang, H.; Teply, B.A.; Kelly, W.K.; Willms, J.; Sullivan, R.; King, S.; Marshall, C.H.; Lotan, T. Interim results from a phase 2 study of olaparib (without ADT) in men with biochemically-recurrent prostate cancer after prostatectomy, with integrated biomarker analysis. J. Clin. Oncol. 2019, 37, 5045. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Sautois, B.; Shapiro, J.; Vogelzang, N.; Bryce, A.; McDermott, R.; Ricci, F.; Rowe, J.; et al. Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)-deficient metastatic castration-resistant prostate cancer (mCRPC): Updated analyses. Ann. Oncol. 2019, 30, v327–v328. [Google Scholar] [CrossRef]
- Markowski, M.C.; Wang, H.; Sullivan, R.; Haffner, M.; De Marzo, A.M.; Lotan, T.L.; Antonarakis, E.S. Phase II trial of rucaparib (Without ADT) in patients with metastatic hormone-sensitive prostate cancer harboring germline DNA repair gene mutations (TRIUMPH). J. Clin. Oncol. 2018, 36, TPS5095. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; O’connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.-S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor–induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [Green Version]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual Roles of PARP-1 Promote Cancer Growth and Progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Rathkopf, D.E.; Smith, M.R.; Efstathiou, E.; Attard, G.; Olmos, D.; Lee, J.Y.; Small, E.J.; Gomes, A.J.; Roubaud, G.; et al. Phase 3 MAGNITUDE study: First results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J. Clin. Oncol. 2022, 40, 12. [Google Scholar] [CrossRef]
- Hussain, M.; Daignault, S.; Twardowski, P.; Albany, C.; Stein, M.N.; Kunju, L.P.; Robinson, D.R.; Cooney, K.A.; Montgomery, R.B.; Antonarakis, E.S.; et al. Abiraterone + prednisone (Abi) +/− veliparib (Vel) for patients (pts) with metastatic castration-resistant prostate cancer (CRPC): NCI 9012 updated clinical and genomics data. J. Clin. Oncol. 2017, 35, 5001. [Google Scholar] [CrossRef]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Wong, S.Y.; Haack, H.; Crowley, D.; Barry, M.; Bronson, R.T.; Hynes, R.O. Tumor-Secreted Vascular Endothelial Growth Factor-C Is Necessary for Prostate Cancer Lymphangiogenesis, but Lymphangiogenesis Is Unnecessary for Lymph Node Metastasis. Cancer Res 2005, 65, 9789–9798. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Bjartell, A.; Ekberg, J.; Gadaleanu, V.; Brunhoff, C.; Persson, J.L. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene 2005, 24, 6385–6393. [Google Scholar] [CrossRef] [Green Version]
- Green, M.M.; Hiley, C.T.; Shanks, J.H.; Bottomley, I.C.; West, C.M.; Cowan, R.A.; Stratford, I.J. Expression of vascular endothelial growth factor (VEGF) in locally invasive prostate cancer is prognostic for radiotherapy outcome. Int. J. Radiat. Oncol. 2007, 67, 84–90. [Google Scholar] [CrossRef]
- Duque, J.L.F.; Loughlin, K.R.; Adam, R.M.; Kantoff, P.W.; Zurakowski, D.; Freeman, M.R. Plasma levels of vascular endothelial growth factor are increased in patients with metastatic prostate cancer. Urology 1999, 54, 523–527. [Google Scholar] [CrossRef]
- Hrouda, D.; Nicol, D.; Gardiner, R. The role of angiogenesis in prostate development and the pathogenesis of prostate cancer. Urol. Res. 2003, 30, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Kelly, W.K.; Halabi, S.; Carducci, M.; George, D.; Mahoney, J.F.; Stadler, W.M.; Morris, M.; Kantoff, P.; Monk, J.P.; Kaplan, E.; et al. Randomized, Double-Blind, Placebo-Controlled Phase III Trial Comparing Docetaxel and Prednisone with or without Bevacizumab in Men with Metastatic Castration-Resistant Prostate Cancer: CALGB 90401. J. Clin. Oncol. 2012, 30, 1534–1540. [Google Scholar] [CrossRef]
- Tannock, I.F.; Fizazi, K.; Ivanov, S.; Karlsson, C.T.; Fléchon, A.; Skoneczna, I.; Orlandi, F.; Gravis, G.; Matveev, V.; Bavbek, S.; et al. Aflibercept versus placebo in combination with docetaxel and prednisone for treatment of men with metastatic castration-resistant prostate cancer (VENICE): A phase 3, double-blind randomised trial. Lancet Oncol. 2013, 14, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Michaelson, M.D.; Oudard, S.; Ou, Y.-C.; Sengeløv, L.; Saad, F.; Houede, N.; Ostler, P.; Stenzl, A.; Daugaard, G.; Jones, R.; et al. Randomized, Placebo-Controlled, Phase III Trial of Sunitinib Plus Prednisone Versus Prednisone Alone in Progressive, Metastatic, Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2014, 32, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; McKay, R.R.; Radke, M.R.; Zhao, S.; Taplin, M.-E.; Davis, N.B.; Monk, P.; Appleman, L.J.; Lara, P.N.; Vaishampayan, U.N.; et al. Randomized Trial of Olaparib With or Without Cediranib for Metastatic Castration-Resistant Prostate Cancer: The Results From National Cancer Institute 9984. J. Clin. Oncol. 2023, 41, 871–880. [Google Scholar] [CrossRef]
- Madan, R.A.; Karzai, F.H.; Ning, Y.-M.; Adesunloye, B.A.; Huang, X.; Harold, N.; Couvillon, A.; Chun, G.; Cordes, L.; Sissung, T.; et al. Phase II trial of docetaxel, bevacizumab, lenalidomide and prednisone in patients with metastatic castration-resistant prostate cancer. BJU Int. 2016, 118, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Tapia, J.C.; Niechi, I. Endothelin-converting enzyme-1 in cancer aggressiveness. Cancer Lett. 2019, 452, 152–157. [Google Scholar] [CrossRef]
- Zhang, X. Interactions between cancer cells and bone microenvironment promote bone metastasis in prostate cancer. Cancer Commun. 2019, 39, 76. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, T.; Nguyen, H.; Corey, E.; Nevedomskaya, E.; Politz, O.; Mumberg, D.; Haendler, B. Abstract 651: Combination of the androgen receptor inhibitor darolutamide and the PI3K inhibitor copanlisib leads to improved anti-tumor efficacy and apoptosis in prostate cancer models. Cancer Res. 2022, 82, 651. [Google Scholar] [CrossRef]
- Small, E.J.; Tchekmedyian, N.S.; Rini, B.I.; Fong, L.; Lowy, I.; Allison, J.P. A Pilot Trial of CTLA-4 Blockade with Human Anti-CTLA-4 in Patients with Hormone-Refractory Prostate Cancer. Clin. Cancer Res. 2007, 13, 1810–1815. [Google Scholar] [CrossRef] [Green Version]
- McNeel, D.G.; Smith, H.A.; Eickhoff, J.C.; Lang, J.M.; Staab, M.J.; Wilding, G.; Liu, G. Phase I trial of tremelimumab in combination with short-term androgen deprivation in patients with PSA-recurrent prostate cancer. Cancer Immunol. Immunother. 2011, 61, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Slovin, S.; Higano, C.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.; Scher, H.; Chin, K.; Gagnier, P.; McHenry, M.; et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase I/II study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Caruso, C. Anti–PD-1–CTLA4 Combo Hits Prostate Cancer. Cancer Discov. 2019, 9, 569–570. [Google Scholar]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, C.J.; Gillessen, S.; Rathkopf, D.; Matsubara, N.; Drake, C.; Fizazi, K.; Piulats, J.M.; Wysocki, P.J.; Buchschacher, G.L.; Doss, J.; et al. Abstract CT014: IMbassador250: A phase III trial comparing atezolizumab with enzalutamide vs enzalutamide alone in patients with metastatic castration-resistant prostate cancer (mCRPC). Cancer Res. 2020, 80, CT014. Available online: http://cancerres.aacrjournals.org/content/80/16_Supplement/CT014.abstract (accessed on 18 May 2023). [CrossRef]
- Gao, J.; Ward, J.F.; Pettaway, A.C.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Fléchon, A.; Gravis, G.; Galsky, M.D.; Mahammedi, H.; Patnaik, A.; Subudhi, S.K.; Ciprotti, M.; et al. Nivolumab Plus Ipilimumab for Metastatic Castration-Resistant Prostate Cancer: Preliminary Analysis of Patients in the CheckMate 650 Trial. Cancer Cell 2020, 38, 489–499.e3. [Google Scholar] [CrossRef]
- Gamat-Huber, M.; McNeel, D.G. Androgen deprivation and immunotherapy for the treatment of prostate cancer. Endocr.-Relat. Cancer 2017, 24, T297–T310. [Google Scholar] [CrossRef]
- Stultz, J.; Fong, L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 697–717. [Google Scholar] [CrossRef]
- Di Carlo, E.; D’Antuono, T.; Pompa, P.; Giuliani, R.; Rosini, S.; Stuppia, L.; Musiani, P.; Sorrentino, C. The Lack of Epithelial Interleukin-7 and BAFF/BLyS Gene Expression in Prostate Cancer as a Possible Mechanism of Tumor Escape from Immunosurveillance. Clin. Cancer Res. 2009, 15, 2979–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Yin, Z.; Ciummo, S.; Lanuti, P.; Lu, L.-F.; Marchisio, M.; Bellone, M.; Di Carlo, E. Targeting Interleukin(IL)-30/IL-27p28 signaling in cancer stem-like cells and host environment synergistically inhibits prostate cancer growth and improves survival. J. Immunother. Cancer 2019, 7, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-F.; Selli, C.; Zhou, H.-L.; Cao, J.; Wu, S.; Ma, R.-Y.; Lu, Y.; Zhang, C.-B.; Xun, B.; Lam, A.D.; et al. Macrophages promote anti-androgen resistance in prostate cancer bone disease. J. Exp. Med. 2023, 220, e20221007. [Google Scholar] [CrossRef] [PubMed]
- Martori, C.; Sanchez-Moral, L.; Paul, T.; Pardo, J.C.; Font, A.; de Porras, V.R.; Sarrias, M.-R. Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance? Cancers 2022, 14, 440. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168.e5. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Tang, J.; Peng, L.; Nie, H.; Zhang, Y.; Liu, P. Cancer-associated fibroblasts promote malignant phenotypes of prostate cancer cells via autophagy. Apoptosis 2023, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, M.; Zamagni, A.; Galasso, G.; Di Zazzo, E.; Giovannelli, P.; Barone, M.V.; Zanoni, M.; Gunelli, R.; Costantini, M.; Auricchio, F.; et al. The androgen receptor/filamin A complex as a target in prostate cancer microenvironment. Cell Death Dis. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Shah, K.; Mallik, S.B.; Gupta, P.; Iyer, A. Targeting Tumour-Associated Fibroblasts in Cancers. Front. Oncol. 2022, 12, 2863. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, B.; Yu, Q. Cancer-associated fibroblasts as accomplices to confer therapeutic resistance in cancer. Cancer Drug Resist. 2022, 5, 889–901. [Google Scholar] [CrossRef]
- Bedeschi, M.; Marino, N.; Cavassi, E.; Piccinini, F.; Tesei, A. Cancer-Associated Fibroblast: Role in Prostate Cancer Progression to Metastatic Disease and Therapeutic Resistance. Cells 2023, 12, 802. [Google Scholar] [CrossRef]
- Di Donato, M.; Cernera, G.; Auricchio, F.; Migliaccio, A.; Castoria, G. Cross-talk between androgen receptor and nerve growth factor receptor in prostate cancer cells: Implications for a new therapeutic approach. Cell Death Discov. 2018, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Di Donato, M.; Cernera, G.; Migliaccio, A.; Castoria, G. Nerve Growth Factor Induces Proliferation and Aggressiveness in Prostate Cancer Cells. Cancers 2019, 11, 784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Donato, M.; Giovannelli, P.; Migliaccio, A.; Castoria, G. The nerve growth factor-delivered signals in prostate cancer and its associated microenvironment: When the dialogue replaces the monologue. Cell Biosci. 2023, 13, 60. [Google Scholar] [CrossRef]
- Tolkach, Y.; Kristiansen, G. The Heterogeneity of Prostate Cancer: A Practical Approach. Pathobiology 2018, 85, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.S.; Stockert, J.A.; Hackert, V.; Yadav, K.K.; Tewari, A.K. Intratumor heterogeneity in prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Boyd, L.K.; Mao, X.; Lu, Y.-J. The complexity of prostate cancer: Genomic alterations and heterogeneity. Nat. Rev. Urol. 2012, 9, 652–664. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Colombino, M.; Cossu, A.; Marchetti, A.; Botti, G.; Ascierto, P.A. Genetic instability and increased mutational load: Which diagnostic tool best direct patients with cancer to immunotherapy? J. Transl. Med. 2017, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Ryan, F.J.; Hope, C.M.; Masavuli, M.G.; Lynn, M.A.; Mekonnen, Z.A.; Yeow, A.E.L.; Garcia-Valtanen, P.; Al-Delfi, Z.; Gummow, J.; Ferguson, C.; et al. Long-term perturbation of the peripheral immune system months after SARS-CoV-2 infection. BMC Med. 2022, 20, 26. [Google Scholar] [CrossRef]
- Di Lorenzo, G.; Buonerba, L.; Ingenito, C.; Crocetto, F.; Buonerba, C.; Libroia, A.; Sciarra, A.; Ragone, G.; Sanseverino, R.; Iaccarino, S.; et al. Clinical Characteristics of Metastatic Prostate Cancer Patients Infected with COVID-19 in South Italy. Oncology 2020, 98, 743–747. [Google Scholar] [CrossRef]
- Montecino-Rodriguez, E.; Berent-Maoz, B.; Dorshkind, K. Causes, consequences, and reversal of immune system aging. J. Clin. Investig. 2013, 123, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adelaiye-Ogala, R.; Gryder, B.E.; Nguyen, Y.T.M.; Alilin, A.N.; Grayson, A.R.; Bajwa, W.; Jansson, K.H.; Beshiri, M.L.; Agarwal, S.; Rodriguez-Nieves, J.A.; et al. Targeting the PI3K/AKT Pathway Overcomes Enzalutamide Resistance by Inhibiting Induction of the Glucocorticoid Receptor. Mol. Cancer Ther. 2020, 19, 1436–1447. [Google Scholar] [CrossRef]
- Kroon, J.; Puhr, M.; Buijs, J.T.; van der Horst, G.; Hemmer, D.M.; Marijt, A.K.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr.-Relat. Cancer 2015, 23, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasumizu, Y.; Miyajima, A.; Kosaka, T.; Miyazaki, Y.; Kikuchi, E.; Oya, M. Dual PI3K/mTOR Inhibitor NVP-BEZ235 Sensitizes Docetaxel in Castration Resistant Prostate Cancer. J. Urol. 2014, 191, 227–234. [Google Scholar] [CrossRef]
- Barreto-Andrade, J.C.; Efimova, E.V.; Mauceri, H.J.; Beckett, M.A.; Sutton, H.G.; Darga, T.E.; Vokes, E.E.; Posner, M.C.; Kron, S.J.; Weichselbaum, R.R. Response of Human Prostate Cancer Cells and Tumors to Combining PARP Inhibition with Ionizing Radiation. Mol. Cancer Ther. 2011, 10, 1185–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.; Choudhary, G.S.; Sharma, A.; Singh, K.; Heston, W.D.; Ciezki, J.; Klein, E.A.; Almasan, A. PARP Inhibition Sensitizes to Low Dose-Rate Radiation TMPRSS2-ERG Fusion Gene-Expressing and PTEN-Deficient Prostate Cancer Cells. PLoS ONE 2013, 8, e60408. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Xia, H.; Bai, S.; Zhao, J.; Edwards, H.; Li, X.; Yang, Y.; Lyu, J.; Wang, G.; Zhan, Y.; et al. CUDC-907, a novel dual PI3K and HDAC inhibitor, in prostate cancer: Antitumour activity and molecular mechanism of action. J. Cell. Mol. Med. 2020, 24, 7239–7253. [Google Scholar] [CrossRef]
- Gioeli, D.; Wunderlich, W.; Sebolt-Leopold, J.; Bekiranov, S.; Wulfkuhle, J.D.; Petricoin, E.F.; Conaway, M.; Weber, M.J. Compensatory Pathways Induced by MEK Inhibition Are Effective Drug Targets for Combination Therapy against Castration-Resistant Prostate Cancer. Mol. Cancer Ther. 2011, 10, 1581–1590. [Google Scholar] [CrossRef] [Green Version]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. JCO Oncol. Pract. 2021, 18, 45–55. [Google Scholar] [CrossRef]
- Ullah, A.; Aziz, T.; Ullah, N.; Nawaz, T. Molecular mechanisms of Sanguinarine in cancer prevention and treatment. Anti-Cancer Agents Med. Chem. 2022, 22, 765–778. [Google Scholar] [CrossRef]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.-C.; Sridhara, R.; Pazdur, R. FDA Drug Approval Summary: Azacitidine (5-azacytidine, Vidaza™) for Injectable Suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Tang, S.; Lian, X.; Meng, H.; Gu, X.; Jiang, J.; Li, X. The Differential Antitumor Activity of 5-Aza-2’-deoxycytidine in Prostate Cancer DU145, 22RV1, and LNCaP Cells. J. Cancer 2021, 12, 5593–5604. [Google Scholar] [CrossRef]
- Marrocco, D.L.; Tilley, W.D.; Bianco-Miotto, T.; Evdokiou, A.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; Butler, L.M. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol. Cancer Ther. 2007, 6, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Morel, K.L.; Sheahan, A.V.; Burkhart, D.L.; Baca, S.C.; Boufaied, N.; Liu, Y.; Qiu, X.; Cañadas, I.; Roehle, K.; Heckler, M.; et al. EZH2 inhibition activates a dsRNA–STING–interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat. Cancer 2021, 2, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, H.; Ren, L.; Gu, J.; Zhang, Y.; Zhang, Y. Demethylation of the miR-146a promoter by 5-Aza-2’-deoxycytidine correlates with delayed progression of castration-resistant prostate cancer. BMC Cancer 2014, 14, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fialova, B.; Luzna, P.; Gursky, J.; Langova, K.; Kolar, Z.; Trtkova, K.S. Epigenetic modulation of AR gene expression in prostate cancer DU145 cells with the combination of sodium butyrate and 5′-Aza-2′-deoxycytidine. Oncol. Rep. 2016, 36, 2365–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, S.; Li, Y.; Zhao, L.; Hou, P.; Shangguan, C.; Yao, R.; Zhang, W.; Zhang, Y.; Tan, J.; Huang, B.; et al. TSA-induced JMJD2B downregulation is associated with cyclin B1-dependent survivin degradation and apoptosis in LNCap cells. J. Cell. Biochem. 2012, 113, 2375–2382. [Google Scholar] [CrossRef]
- Chiam, K.; Centenera, M.M.; Butler, L.M.; Tilley, W.; Bianco-Miotto, T. GSTP1 DNA Methylation and Expression Status Is Indicative of 5-aza-2′-Deoxycytidine Efficacy in Human Prostate Cancer Cells. PLoS ONE 2011, 6, e25634. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, Y.; Lu, X.; Song, B.; Fong, K.-W.; Cao, Q.; Licht, J.D.; Zhao, J.C.; Yu, J. Polycomb- and Methylation-Independent Roles of EZH2 as a Transcription Activator. Cell Rep. 2018, 25, 2808–2820.e4. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Weston, A.; Bearrs, J.; Thode, T.; Neiss, A.; Soldi, R.; Sharma, S. Reversible lysine-specific demethylase 1 antagonist HCI-2509 inhibits growth and decreases c-MYC in castration- and docetaxel-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 2016, 19, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Graham, P.H.; Hao, J.; Chang, L.; Ni, J.; Power, C.A.; Dong, Q.; Kearsley, J.H.; Li, Y. Combination Therapy with the Histone Deacetylase Inhibitor LBH589 and Radiation Is an Effective Regimen for Prostate Cancer Cells. PLoS ONE 2013, 8, e74253. [Google Scholar] [CrossRef] [Green Version]
- Zucconi, B.E.; Makofske, J.L.; Meyers, D.J.; Hwang, Y.; Wu, M.; Kuroda, M.I.; Cole, P.A. Combination Targeting of the Bromodomain and Acetyltransferase Active Site of p300/CBP. Biochemistry 2019, 58, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, C.; Wang, W.; Li, C.; Mu, X.; Hu, K. Current progress of nanomedicine for prostate cancer diagnosis and treatment. Biomed. Pharmacother. 2022, 155, 113714. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Fukushima, S.; Kanayama, N.; Aoyagi, K.; Nishiyama, N.; Koyama, H.; Kataoka, K. Cyclic RGD Peptide-Conjugated Polyplex Micelles as a Targetable Gene Delivery System Directed to Cells Possessing αvβ3 and αvβ5 Integrins. Bioconj. Chem. 2007, 18, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Dirisala, A.; Osada, K.; Chen, Q.; Tockary, T.A.; Machitani, K.; Osawa, S.; Liu, X.; Ishii, T.; Miyata, K.; Oba, M.; et al. Optimized rod length of polyplex micelles for maximizing transfection efficiency and their performance in systemic gene therapy against stroma-rich pancreatic tumors. Biomaterials 2014, 35, 5359–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Chen, Q.; Osada, K.; Liu, X.; Tockary, T.A.; Uchida, S.; Dirisala, A.; Ishii, T.; Nomoto, T.; Toh, K.; et al. Targeted gene delivery by polyplex micelles with crowded PEG palisade and cRGD moiety for systemic treatment of pancreatic tumors. Biomaterials 2014, 35, 3416–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Osada, K.; Ge, Z.; Uchida, S.; Tockary, T.A.; Dirisala, A.; Matsui, A.; Toh, K.; Takeda, K.M.; Liu, X.; et al. Polyplex micelle installing intracellular self-processing functionalities without free catiomers for safe and efficient systemic gene therapy through tumor vasculature targeting. Biomaterials 2017, 113, 253–265. [Google Scholar] [CrossRef] [Green Version]
- Christie, R.J.; Matsumoto, Y.; Miyata, K.; Nomoto, T.; Fukushima, S.; Osada, K.; Halnaut, J.; Pittella, F.; Kim, H.J.; Nishiyama, N.; et al. Targeted Polymeric Micelles for siRNA Treatment of Experimental Cancer by Intravenous Injection. ACS Nano 2012, 6, 5174–5189. [Google Scholar] [CrossRef] [PubMed]
- Buonerba, C.; Ferro, M.; Dolce, P.; Crocetto, F.; Verde, A.; Lucarelli, G.; Scafuri, L.; Facchini, S.; Vaia, A.; Marinelli, A.; et al. Predictors of efficacy of androgen-receptor-axis-targeted therapies in patients with metastatic castration-sensitive prostate cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. 2020, 151, 102992. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.; Zhang, J.; Sun, J.; Bakht, S. Recent Development and Future Prospects of Molecular Targeted Therapy in Prostate Cancer. Curr. Mol. Pharmacol. 2021, 15, 159–169. [Google Scholar] [CrossRef]
- Gao, P.; Li, T.; Zhang, K.; Luo, G. Recent advances in the molecular targeted drugs for prostate cancer. Int. Urol. Nephrol. 2023, 55, 777–789. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapeutic Agent | Mechanism of Action |
|---|---|
| PSMA-targeted radionuclides | |
| Beta-emitting radioisotope Lutetium-177 conjugated with the PSMA ligand PSMA-617 |
| Lutetium-177 conjugated, via the chelating agent dodecanetetraacetic acid (DOTA), with rosopatamab, a humanized monoclonal antibody against PSMA |
| Alpha-emitting radioisotope Actinium-225 conjugated with PSMA-617 |
| Actinium-225 conjugated with the PSMA ligand PSMA-I&T |
| DNA repair inhibitors | |
| Inhibitor of PARP1 and PARP2 |
| Inhibitor of PARP1, PARP2 and PARP3 |
| Inhibitor of PARP1 and PARP2 |
| Therapies targeting tumor neovascularization | |
| Monoclonal antibody specific for VEGF-A |
| Inhibitor of VEGF-A and -B |
| Inhibitor of VEGFR-1, -2 and -3 |
| Inhibitor of VEGFR-1, -2, -3; PDGFR-A and -B |
| Immune checkpoint inhibitors | |
| Monoclonal antibody specific for PD-L1 |
| Monoclonal antibody specific for CTLA-4 |
| Monoclonal antibody specific for PD-1 |
| Therapeutic Agent | Clinical Trial N. (Acronym) | Patients Eligibility Criteria | Outcomes |
|---|---|---|---|
| 177Lu-PSMA-617 | NCT03511664 (VISION) |
| Improvement in overall survival |
| 177Lu-DOTA-rosopatamab | NCT04876651 (PROSTACT) |
| Ongoing. Data not yet available |
| 225Ac-PSMA-617 | NCT04597411 (AcTION) |
| Ongoing. Data not yet available |
| 225Ac-PSMA-I&T | NCT05219500 (TATCIST) |
| Ongoing. Data not yet available |
| Olaparib | NCT02987543 (PROFOUND) |
| Improvement in overall survival |
| Rucaparib | NCT02952534 (TRITON2) |
| Improvement in PSA response rate |
| NCT03413995 (TRIUMPH) |
| Ongoing. Data not yet available | |
| NCT03533946 (ROAR) |
| Ongoing. Data not yet available | |
| Talazoparib | NCT03148795 (TALAPRO-1) |
| Improvement in radiolgical response rate |
| Ipilimumab | NCT01057810 |
| No improvement in overall survival |
| NCT00861614 |
| No improvement in overall survival | |
| Combination Tested | Clinical Trial N. (Acronym) | Patients Eligibility Criteria | Outcomes |
|---|---|---|---|
| 177Lu-PSMA-617 + androgen deprivation therapy | NCT04720157 (PSMAddition) |
| Ongoing. Data not yet available |
| 177Lu-PSMA-617 + enzalutamide | NCT04419402 (ENZA-p) |
| Ongoing. Data not yet available |
| 177Lu-PSMA-617 + olaparib | NCT03874884 (LuPARP) |
| Ongoing. Data not yet available |
| 177Lu-PSMA-617 + pembrolizumab | NCT03658447 (PRINCE) |
| Ongoing. Data not yet available |
| 177Lu-PSMA-617 + ipilimumab and nivolumab | NCT05150236 (EVOLUTION) |
| Ongoing. Data not yet available |
| 177Lu-PSMA-I&T + 225Ac-J591 * | NCT04886986 |
| Ongoing. Data not yet available |
| 223Radium + bipolar androgen therapy (BAT) | NCT04704505 (BAT-RAD) |
| Ongoing. Data not yet available |
| Niraparib + abiraterone acetate + prednisone | NCT03748641 (MAGNITUDE) |
| Improvement in progression-free survival in patients with alterations in HR genes |
| Olaparib + abiraterone acetate | NCT01972217 (PROpel) |
| Reduction of radiographic progression of 34% |
| Olaparib + cediranib | NCT02893917 |
| Improvement in progression-free survival compared with olaparib alone |
| Veliparib + abiraterone acetate + prednisone | NCT01576172 |
| Improvement in PFS |
| Atezolizumab + enzalutamide | NCT03016312 |
| No improvement in overall survival |
| Bevacizumab + docetaxel + prednisone | NCT00110214 |
| No improvement in overall survival |
| Sunitinib + prednisone | NCT00676650 (SUN 1120) |
| No improvement in overall survival |
| Aflibercept + docetaxel + prednisone | NCT00519285 (VENICE) |
| No improvement in overall survival |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorrentino, C.; Di Carlo, E. Molecular Targeted Therapies in Metastatic Prostate Cancer: Recent Advances and Future Challenges. Cancers 2023, 15, 2885. https://doi.org/10.3390/cancers15112885
Sorrentino C, Di Carlo E. Molecular Targeted Therapies in Metastatic Prostate Cancer: Recent Advances and Future Challenges. Cancers. 2023; 15(11):2885. https://doi.org/10.3390/cancers15112885
Chicago/Turabian StyleSorrentino, Carlo, and Emma Di Carlo. 2023. "Molecular Targeted Therapies in Metastatic Prostate Cancer: Recent Advances and Future Challenges" Cancers 15, no. 11: 2885. https://doi.org/10.3390/cancers15112885
APA StyleSorrentino, C., & Di Carlo, E. (2023). Molecular Targeted Therapies in Metastatic Prostate Cancer: Recent Advances and Future Challenges. Cancers, 15(11), 2885. https://doi.org/10.3390/cancers15112885