

Folic Acid Supplementation Promotes Hypomethylation in Both the Inflamed Colonic Mucosa and Colitis-Associated Dysplasia
 ,
,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
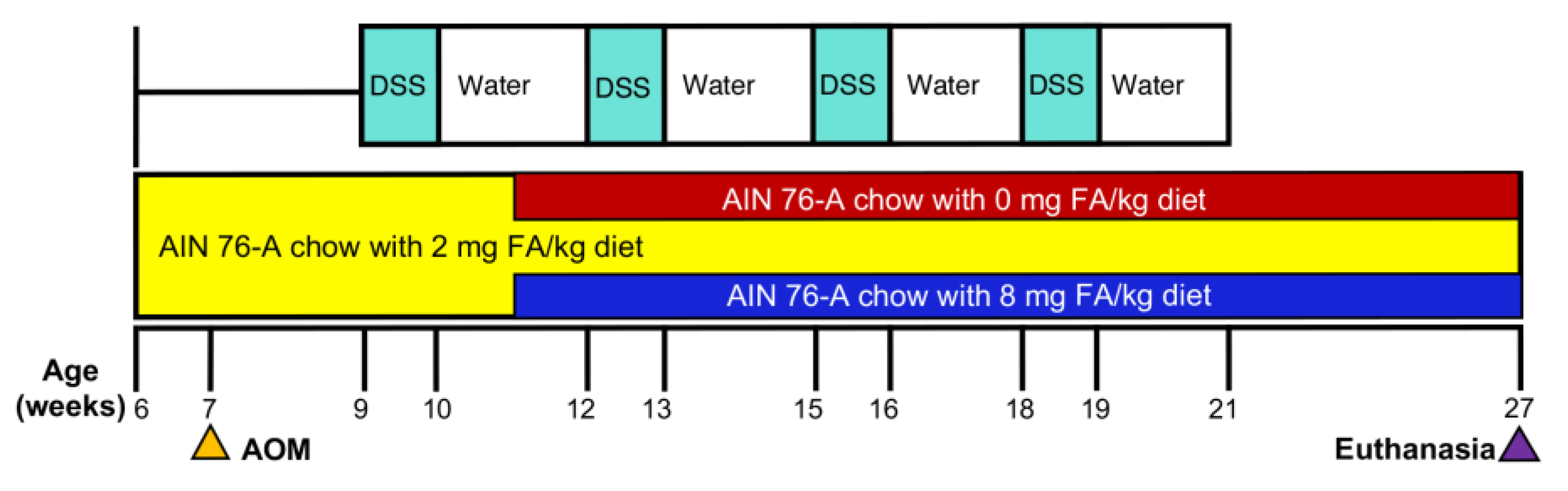
2.1. Diet and Animal Treatment
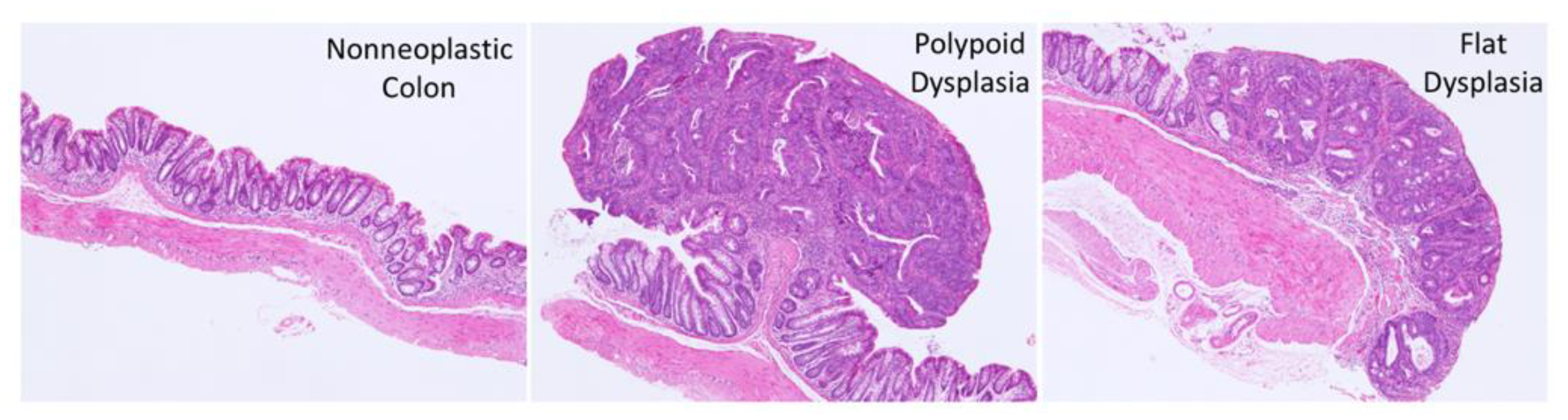
2.2. Histopathology
2.3. Isolation of DNA and RNA from Laser Microdissected (LMD) Colonic Epithelial Cells
2.4. Genome-Wide CpG island Methylation Analysis
2.5. RNA-Seq Analysis
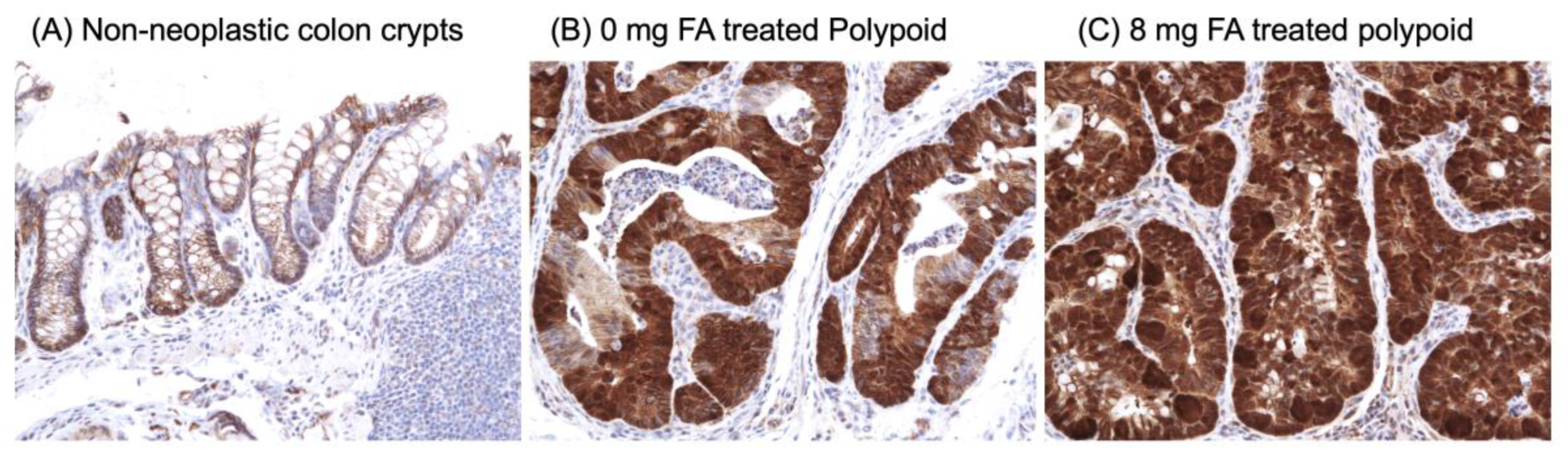
2.6. Immunohistochemistry (IHC) of β-Catenin
2.7. Real-Time Quantitative PCR (RT-qPCR)
2.8. Biostatistical Analyses
3. Results
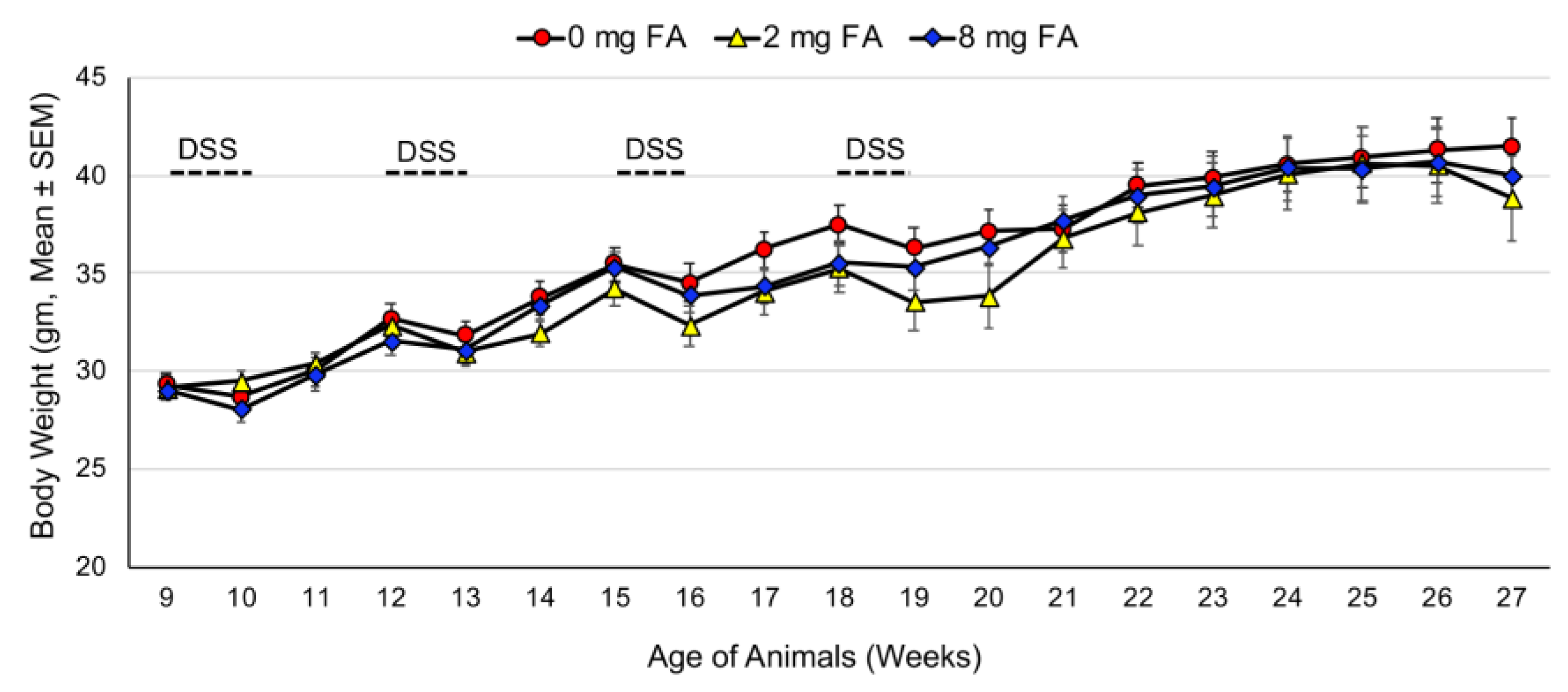
3.1. Treatment Tolerance
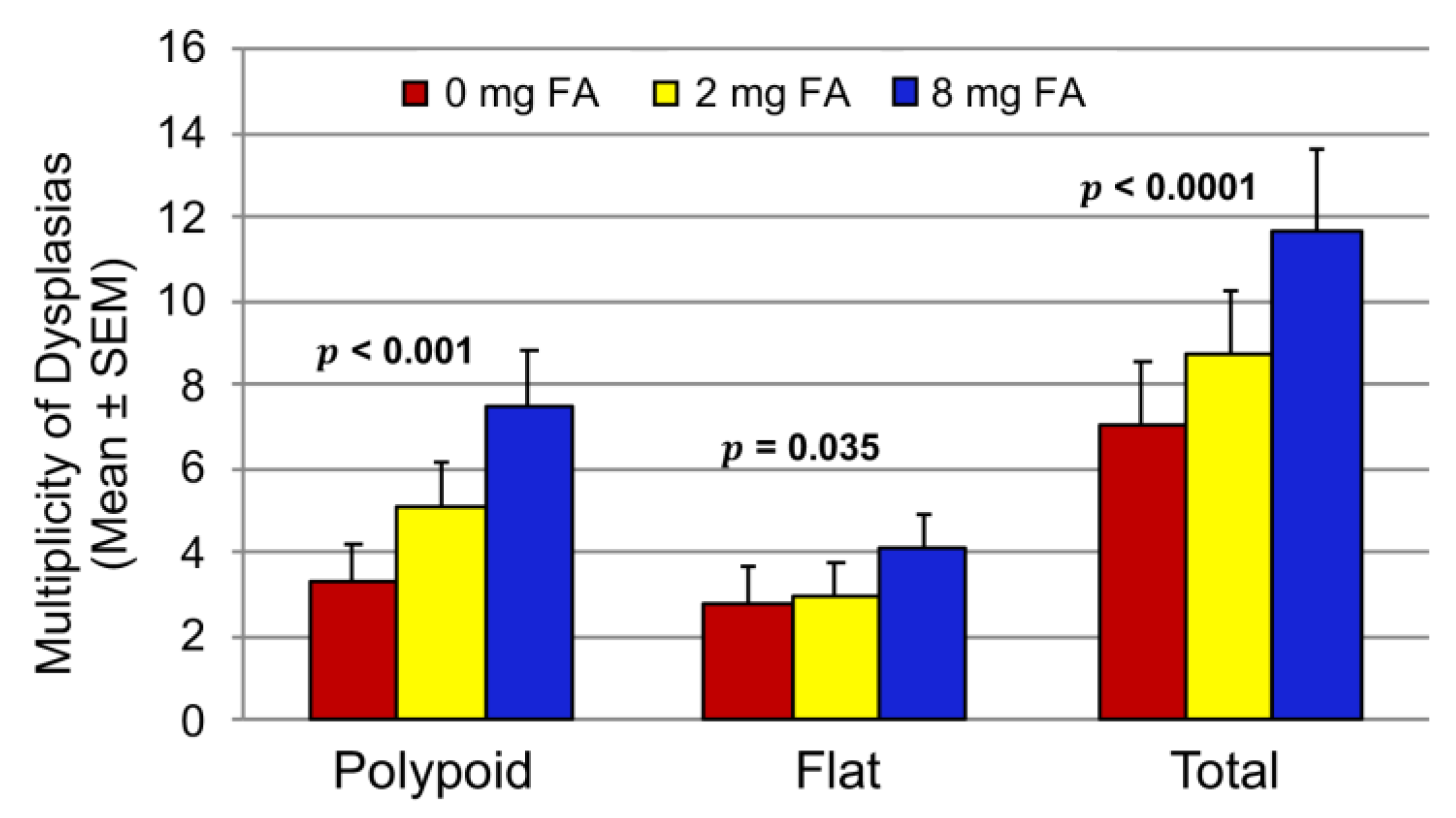
3.2. Effect of FA on the Development of Colitis-Associated Dysplasia and Cancer
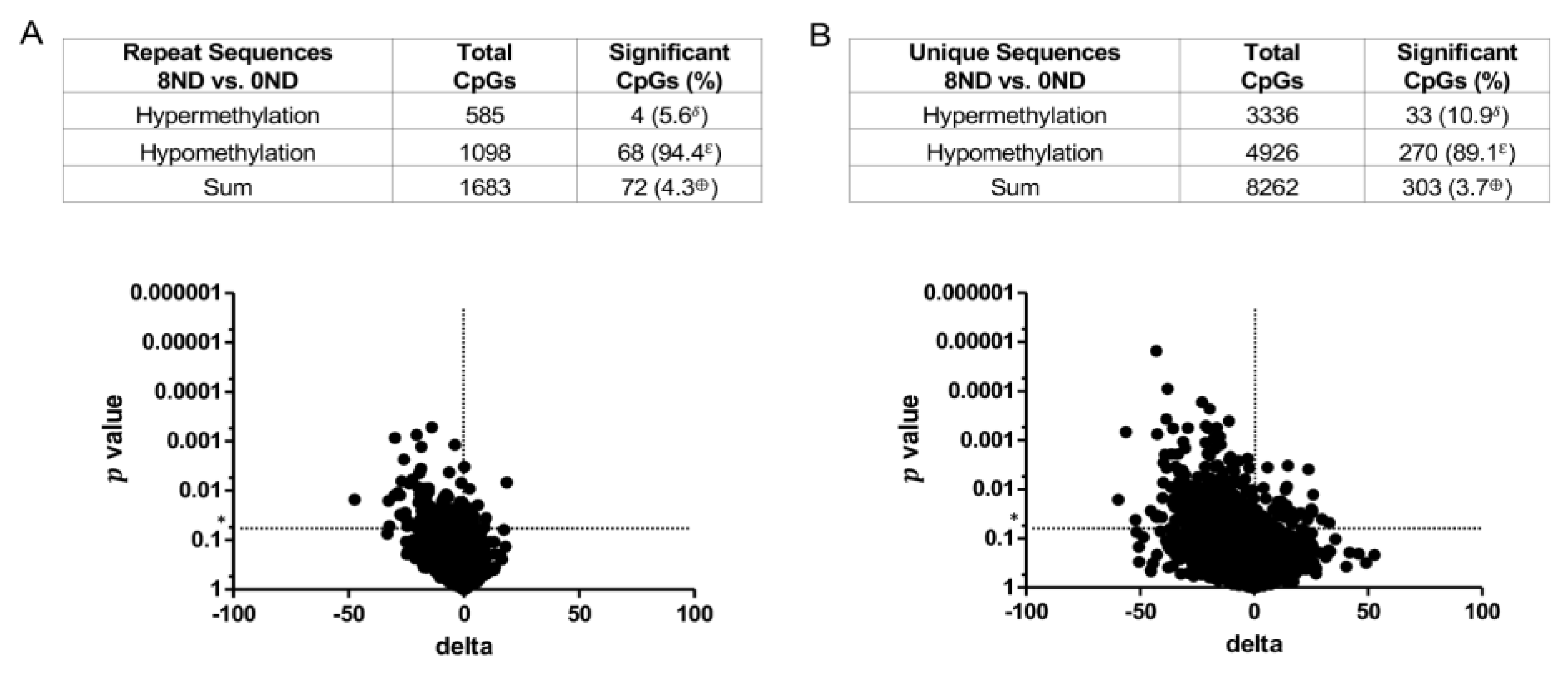
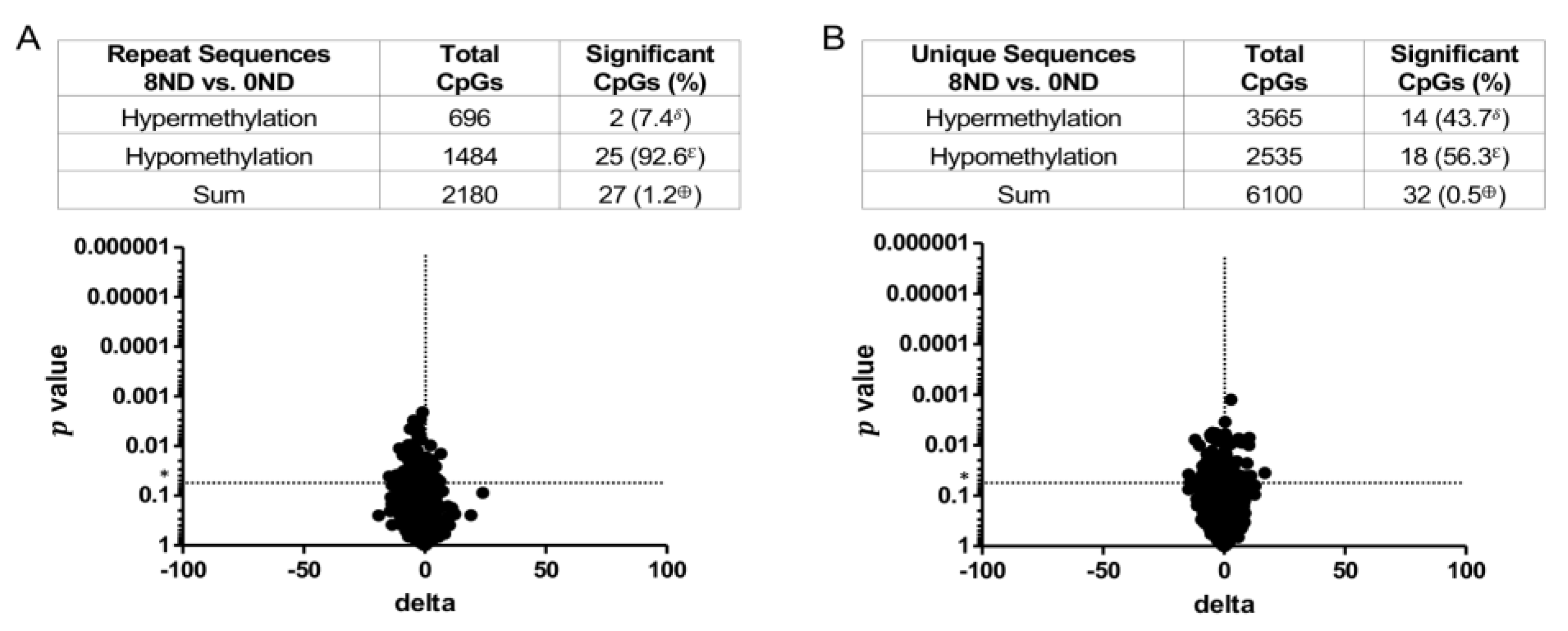
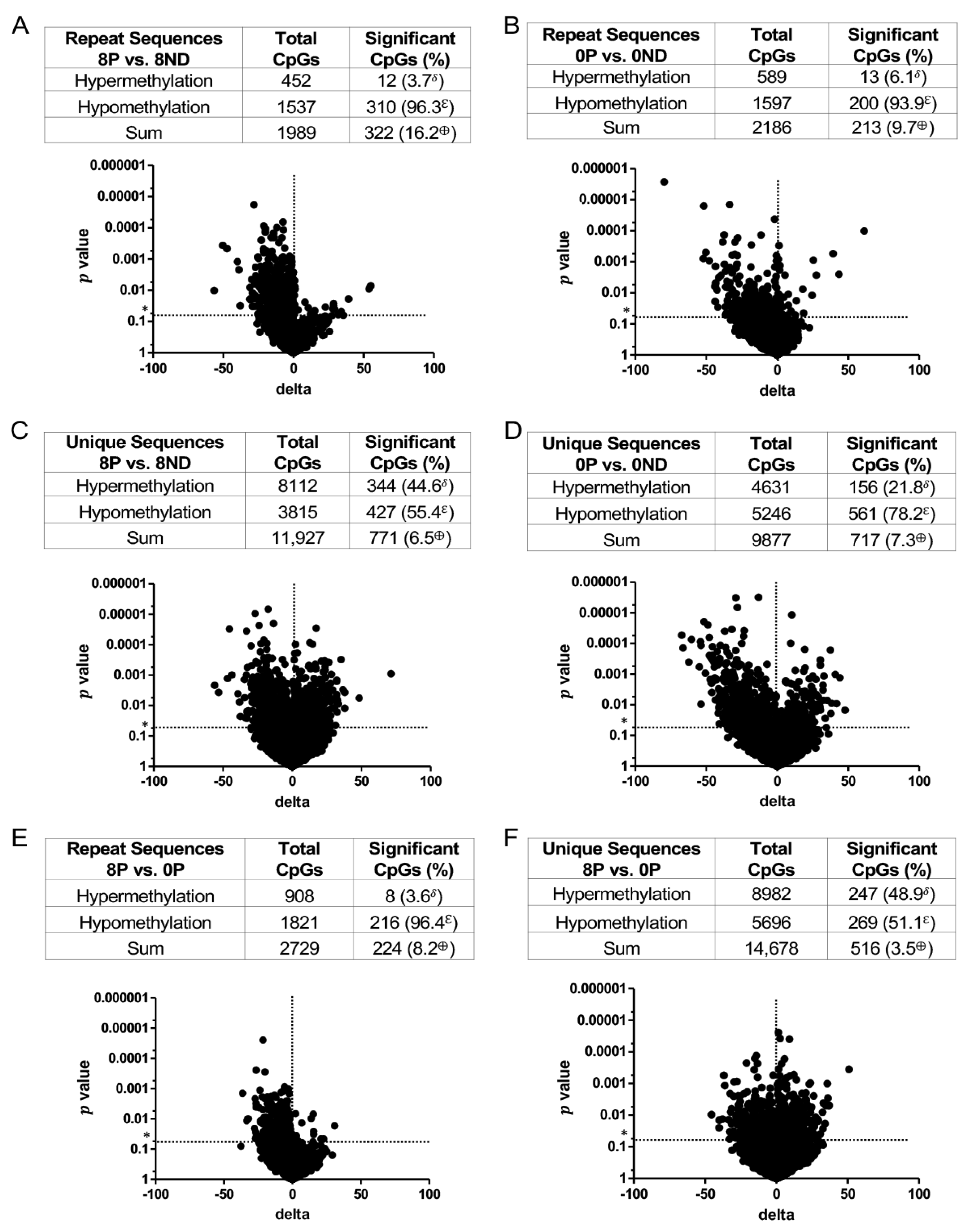
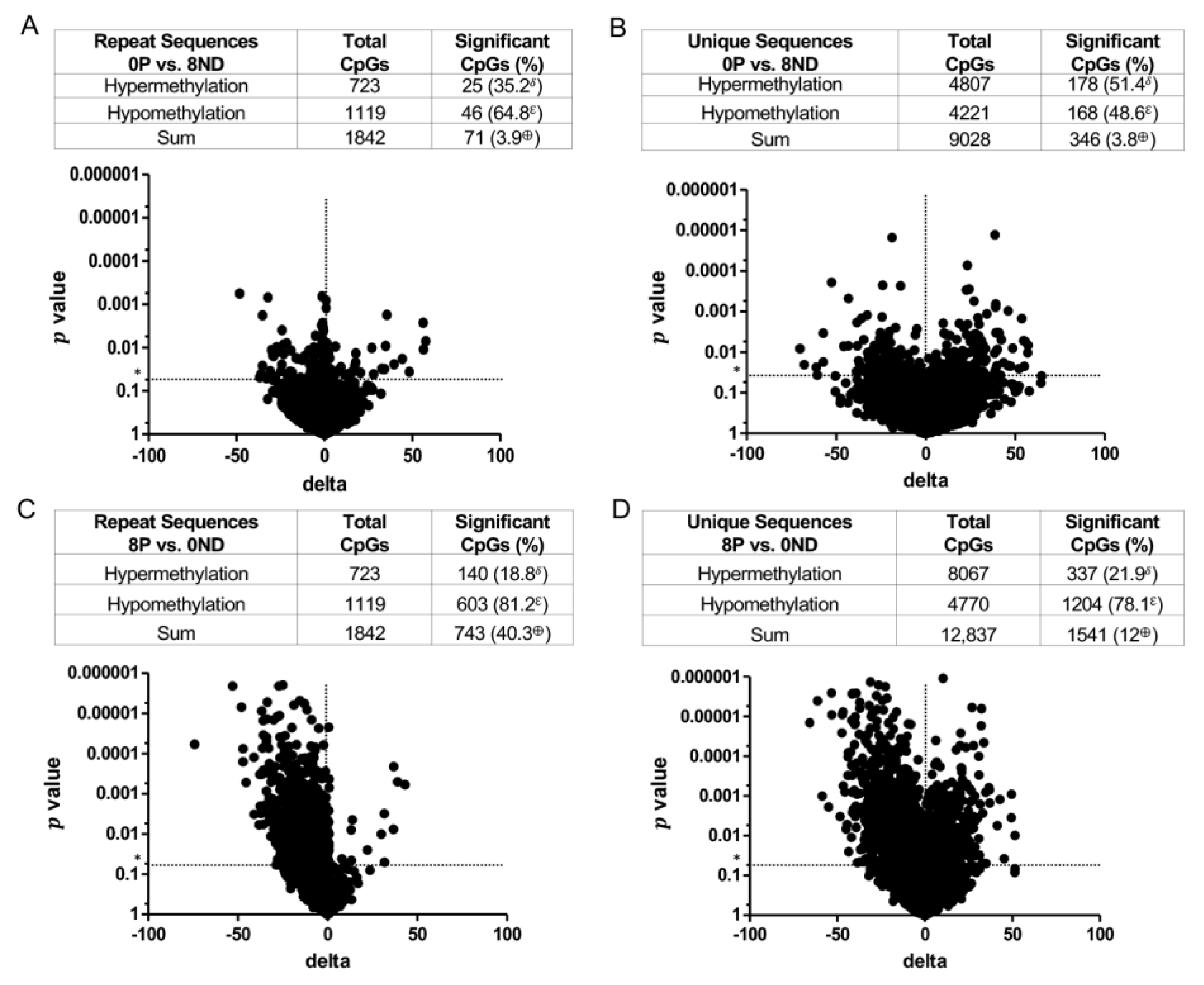
3.3. Effect of FA on Genome-Wide DNA Methylation at CCCGGG Sites
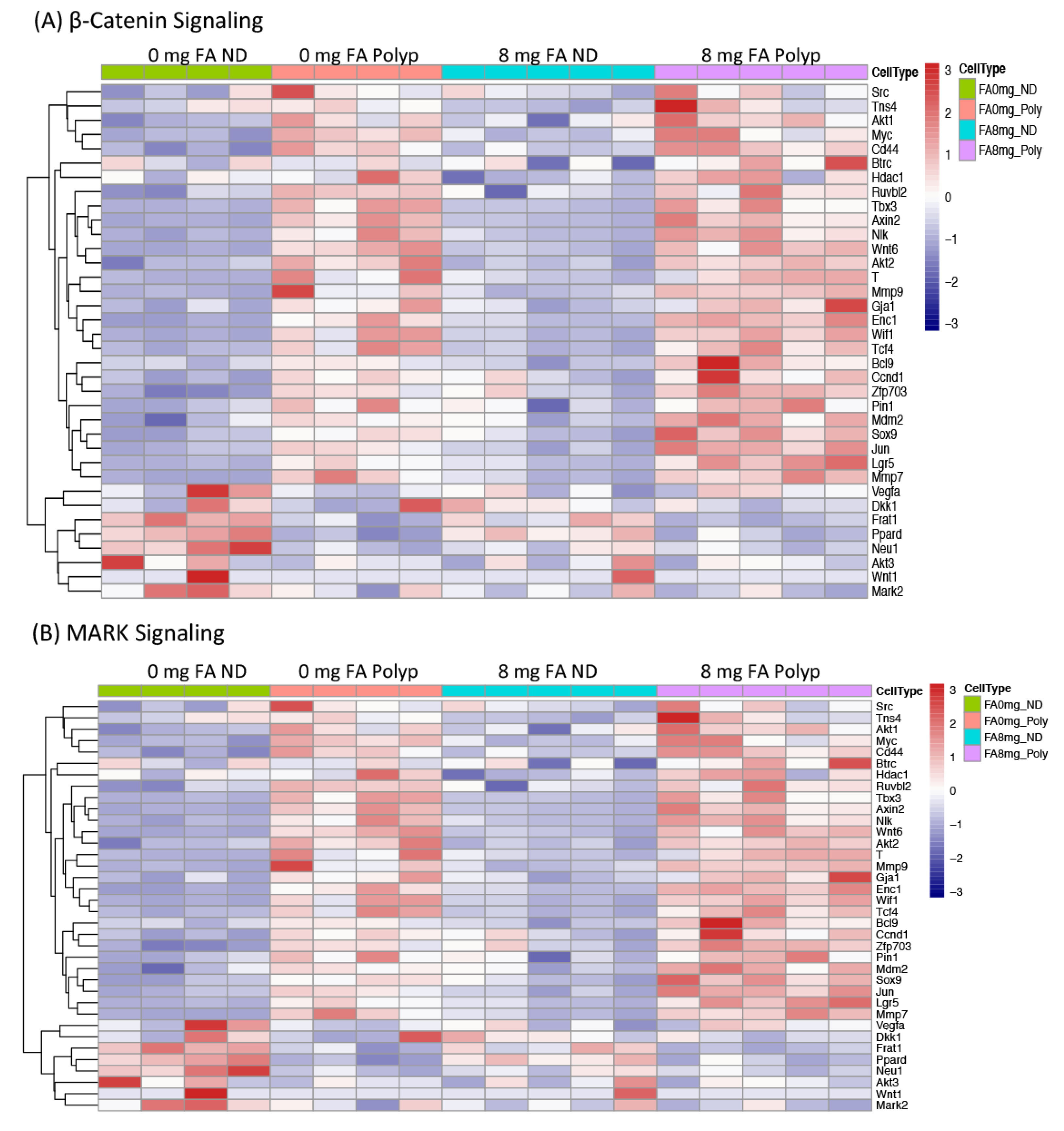
3.4. Expression of Genes whose Methylation Status was Modified by FA Treatment
3.5. Effect of FA on Inflammatory Markers
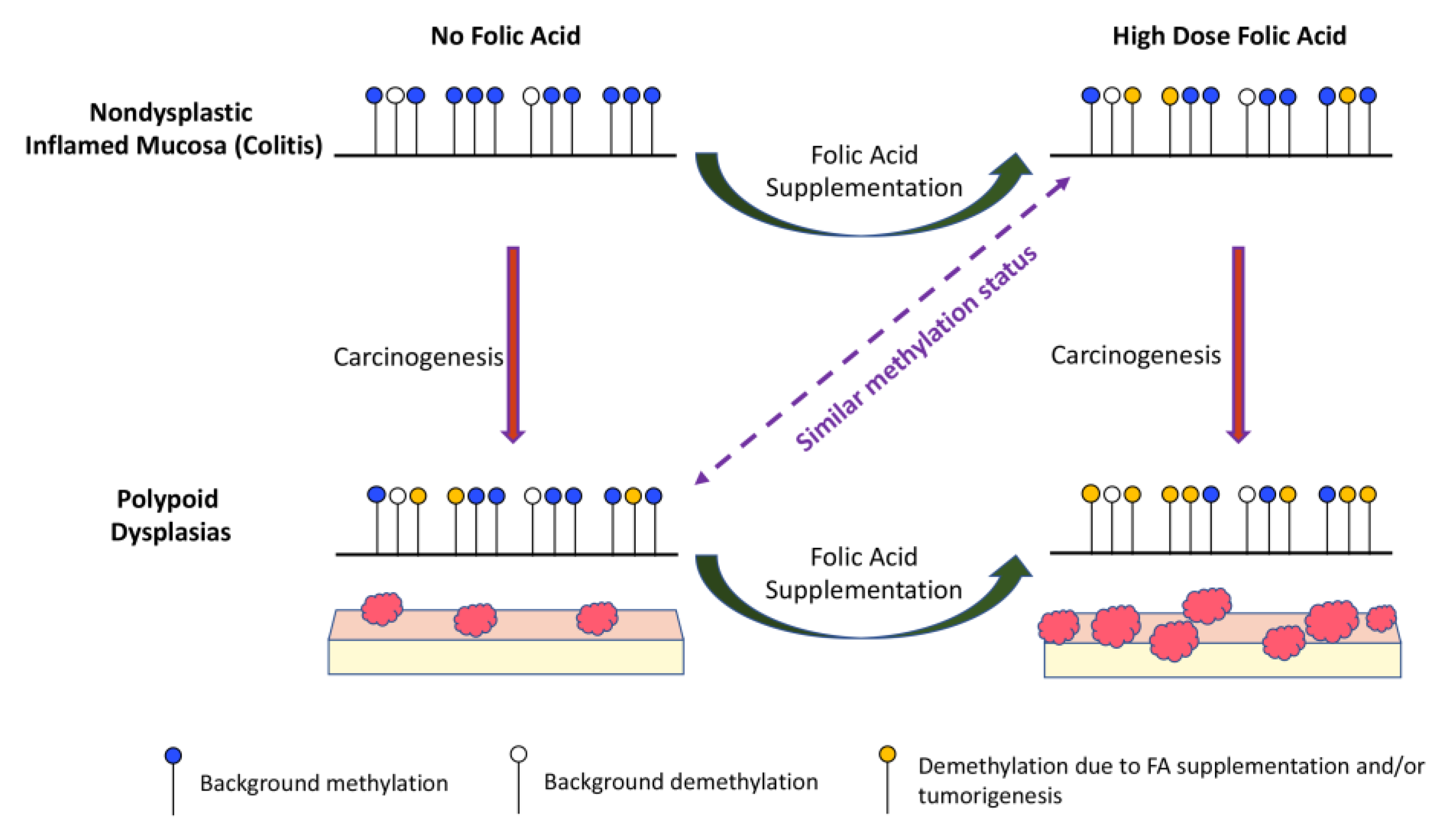
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Rutter, M.D.; Saunders, B.P.; Wilkinson, K.H.; Rumbles, S.; Schofield, G.; Kamm, M.A.; Williams, C.B.; Price, A.B.; Talbot, I.C.; Forbes, A. Thirty-year analysis of a colonoscopic surveillance program for neoplasia in ulcerative colitis. Gastroenterology 2006, 130, 1030–1038. [Google Scholar] [CrossRef]
- Hardy, R.G.; Meltzer, S.J.; Jankowski, J.A. ABC of colorectal cancer. Molecular basis for risk factors. BMJ 2000, 321, 886–889. [Google Scholar] [CrossRef]
- Hartnett, L.; Egan, L.J. Inflammation, DNA methylation and colitis-associated cancer. Carcinogenesis 2012, 33, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Akbulut, S.; Altiparmak, E.; Topal, F.; Ozaslan, E.; Kucukazman, M.; Yonem, O. Increased levels of homocysteine in patients with ulcerative colitis. World J. Gastroenterol. 2010, 16, 2411–2416. [Google Scholar] [CrossRef]
- Yakut, M.; Ustun, Y.; Kabacam, G.; Soykan, I. Serum vitamin B12 and folate status in patients with inflammatory bowel diseases. Eur. J. Intern. Med. 2010, 21, 320–323. [Google Scholar] [CrossRef]
- Mullin, G.E. Micronutrients and inflammatory bowel disease. Nutr. Clin. Pr. 2012, 27, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.B. Folate, cancer risk, and the Greek god, Proteus: A tale of two chameleons. Nutr. Rev. 2009, 67, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Smith-Warner, S.A.; Spiegelman, D.; Yaun, S.S.; Colditz, G.A.; Freudenheim, J.L.; Giovannucci, E.; Goldbohm, R.A.; Graham, S.; Harnack, L.; et al. Pooled analyses of 13 prospective cohort studies on folate intake and colon cancer. Cancer Causes Control 2010, 21, 1919–1930. [Google Scholar] [CrossRef]
- Cole, B.F.; Baron, J.A.; Sandler, R.S.; Haile, R.W.; Ahnen, D.J.; Bresalier, R.S.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.I.; Burke, C.A.; et al. Folic acid for the prevention of colorectal adenomas: A randomized clinical trial. JAMA 2007, 297, 2351–2359. [Google Scholar] [CrossRef]
- Figueiredo, J.C.; Levine, A.J.; Lee, W.H.; Conti, D.V.; Poynter, J.N.; Campbell, P.T.; Duggan, D.; Lewinger, J.P.; Martinez, M.E.; Ulrich, C.M.; et al. Genes involved with folate uptake and distribution and their association with colorectal cancer risk. Cancer Causes Control 2010, 21, 597–608. [Google Scholar] [CrossRef]
- Logan, R.F.; Grainge, M.J.; Shepherd, V.C.; Armitage, N.C.; Muir, K.R. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology 2008, 134, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Van Guelpen, B.; Hultdin, J.; Johansson, I.; Hallmans, G.; Stenling, R.; Riboli, E.; Winkvist, A.; Palmqvist, R. Low folate levels may protect against colorectal cancer. Gut 2006, 55, 1461–1466. [Google Scholar] [CrossRef]
- Kim, Y.I. Folate, colorectal carcinogenesis, and DNA methylation: Lessons from animal studies. Environ. Mol. Mutagen 2004, 44, 10–25. [Google Scholar] [CrossRef]
- Song, J.; Medline, A.; Mason, J.B.; Gallinger, S.; Kim, Y.I. Effects of dietary folate on intestinal tumorigenesis in the apcMin mouse. Cancer Res. 2000, 60, 5434–5440. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Sohn, K.J.; Medline, A.; Ash, C.; Gallinger, S.; Kim, Y.I. Chemopreventive effects of dietary folate on intestinal polyps in Apc+/-Msh2-/- mice. Cancer Res. 2000, 60, 3191–3199. [Google Scholar] [PubMed]
- Lindzon, G.M.; Medline, A.; Sohn, K.J.; Depeint, F.; Croxford, R.; Kim, Y.I. Effect of folic acid supplementation on the progression of colorectal aberrant crypt foci. Carcinogenesis 2009, 30, 1536–1543. [Google Scholar] [CrossRef]
- Lashner, B.A. Red blood cell folate is associated with the development of dysplasia and cancer in ulcerative colitis. J. Cancer Res. Clin. Oncol. 1993, 119, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Lashner, B.A.; Provencher, K.S.; Seidner, D.L.; Knesebeck, A.; Brzezinski, A. The effect of folic acid supplementation on the risk for cancer or dysplasia in ulcerative colitis. Gastroenterology 1997, 112, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Lashner, B.A.; Heidenreich, P.A.; Su, G.L.; Kane, S.V.; Hanauer, S.B. Effect of folate supplementation on the incidence of dysplasia and cancer in chronic ulcerative colitis. A case-control study. Gastroenterology 1989, 97, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Heyman, M.B.; Garnett, E.A.; Shaikh, N.; Huen, K.; Jose, F.A.; Harmatz, P.; Winter, H.S.; Baldassano, R.N.; Cohen, S.A.; Gold, B.D.; et al. Folate concentrations in pediatric patients with newly diagnosed inflammatory bowel disease. Am. J. Clin. Nutr. 2009, 89, 545–550. [Google Scholar] [CrossRef]
- Clapper, M.L.; Gary, M.A.; Coudry, R.A.; Litwin, S.; Chang, W.-C.L.; Devarajan, K.; Lubet, R.A.; Cooper, H.S. 5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm. Bowel Dis. 2008, 14, 1341–1347. [Google Scholar] [CrossRef]
- Riddell, R.H.; Goldman, H.; Ransohoff, D.F.; Appelman, H.D.; Fenoglio, C.M.; Haggitt, R.C.; Ahren, C.; Correa, P.; Hamilton, S.R.; Morson, B.C.; et al. Dysplasia in inflammatory bowel disease: Standardized classification with provisional clinical applications. Hum. Pathol. 1983, 14, 931–968. [Google Scholar] [CrossRef]
- Jelinek, J.; Liang, S.; Lu, Y.; He, R.; Ramagli, L.S.; Shpall, E.J.; Estecio, M.R.; Issa, J.P. Conserved DNA methylation patterns in healthy blood cells and extensive changes in leukemia measured by a new quantitative technique. Epigenetics 2012, 7, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Statist. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Arnold, S.J.; Stappert, J.; Bauer, A.; Kispert, A.; Herrmann, B.G.; Kemler, R. Brachyury is a target gene of the Wnt/beta-catenin signaling pathway. Mech. Dev. 2000, 91, 249–258. [Google Scholar] [CrossRef]
- Kilic, N.; Feldhaus, S.; Kilic, E.; Tennstedt, P.; Wicklein, D.; Wasielewski, R.; Viebahn, C.; Kreipe, H.; Schumacher, U. Brachyury expression predicts poor prognosis at early stages of colorectal cancer. Eur. J. Cancer 2011, 47, 1080–1085. [Google Scholar] [CrossRef]
- Kao, L.; Azimov, R.; Shao, X.M.; Abuladze, N.; Newman, D.; Zhekova, H.; Noskov, S.; Pushkin, A.; Kurtz, I. SLC4A11 function: Evidence for H(+)(OH(-)) and NH3-H(+) transport. Am. J. Physiol. Cell Physiol. 2020, 318, C392–C405. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Seet, L.F.; Koh, L.W.; Venkatraman, A.; Venkataraman, D.; Mohan, R.R.; Praetorius, J.; Bonanno, J.A.; Aung, T.; Vithana, E.N. Depletion of SLC4A11 causes cell death by apoptosis in an immortalized human corneal endothelial cell line. Invest. Ophthalmol. Vis. Sci. 2012, 53, 3270–3279. [Google Scholar] [CrossRef]
- Groschl, B.; Bettstetter, M.; Giedl, C.; Woenckhaus, M.; Edmonston, T.; Hofstadter, F.; Dietmaier, W. Expression of the MAP kinase phosphatase DUSP4 is associated with microsatellite instability in colorectal cancer (CRC) and causes increased cell proliferation. Int. J. Cancer 2013, 132, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Menyhart, O.; Kakisaka, T.; Pongor, L.S.; Uetake, H.; Goel, A.; Gyorffy, B. Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes. Cancers 2019, 11, 983. [Google Scholar] [CrossRef] [PubMed]
- Noah, T.K.; Kazanjian, A.; Whitsett, J.; Shroyer, N.F. SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Exp. Cell Res. 2010, 316, 452–465. [Google Scholar] [CrossRef]
- Wang, X.; Yu, Q.; Ghareeb, W.M.; Zhang, Y.; Lu, X.; Huang, Y.; Huang, S.; Sun, Y.; Lin, J.; Liu, J.; et al. Downregulated SPINK4 is associated with poor survival in colorectal cancer. BMC Cancer 2019, 19, 1258. [Google Scholar] [CrossRef]
- Sundaram, P.; Hultine, S.; Smith, L.M.; Dews, M.; Fox, J.L.; Biyashev, D.; Schelter, J.M.; Huang, Q.; Cleary, M.A.; Volpert, O.V.; et al. p53-responsive miR-194 inhibits thrombospondin-1 and promotes angiogenesis in colon cancers. Cancer Res. 2011, 71, 7490–7501. [Google Scholar] [CrossRef]
- Alkim, C.; Sakiz, D.; Alkim, H.; Livaoglu, A.; Kendir, T.; Demirsoy, H.; Erdem, L.; Akbayir, N.; Sokmen, M. Thrombospondin-1 and VEGF in inflammatory bowel disease. Libyan J. Med. 2012, 7, 8942. [Google Scholar] [CrossRef]
- Hamaoka, Y.; Negishi, M.; Katoh, H. EphA2 is a key effector of the MEK/ERK/RSK pathway regulating glioblastoma cell proliferation. Cell Signal. 2016, 28, 937–945. [Google Scholar] [CrossRef]
- Martini, G.; Cardone, C.; Vitiello, P.P.; Belli, V.; Napolitano, S.; Troiani, T.; Ciardiello, D.; Della Corte, C.M.; Morgillo, F.; Matrone, N.; et al. EPHA2 Is a Predictive Biomarker of Resistance and a Potential Therapeutic Target for Improving Antiepidermal Growth Factor Receptor Therapy in Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Chalamalasetty, R.B.; Kennedy, M.W.; Thomas, S.; Inala, S.N.; Garriock, R.J.; Yamaguchi, T.P. Zfp703 Is a Wnt/beta-Catenin Feedback Suppressor Targeting the beta-Catenin/Tcf1 Complex. Mol. Cell. Biol. 2016, 36, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Furukawa, Y.; Tsunoda, T.; Tanaka, T.; Ogawa, M.; Nakamura, Y. Up-regulation of the ectodermal-neural cortex 1 (ENC1) gene, a downstream target of the beta-catenin/T-cell factor complex, in colorectal carcinomas. Cancer Res. 2001, 61, 7722–7726. [Google Scholar] [PubMed]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Nagai, K.; Uemura, M.; Sekimoto, M.; Doki, Y.; Mori, M. ATP11A is a novel predictive marker for metachronous metastasis of colorectal cancer. Oncol. Rep. 2010, 23, 505–510. [Google Scholar]
- Zhou, W.; He, M.R.; Jiao, H.L.; He, L.Q.; Deng, D.L.; Cai, J.J.; Xiao, Z.Y.; Ye, Y.P.; Ding, Y.Q.; Liao, W.T.; et al. The tumor-suppressor gene LZTS1 suppresses colorectal cancer proliferation through inhibition of the AKT-mTOR signaling pathway. Cancer Lett. 2015, 360, 68–75. [Google Scholar] [CrossRef]
- Vecchione, A.; Croce, C.M.; Baldassarre, G. Fez1/Lzts1 a new mitotic regulator implicated in cancer development. Cell Div. 2007, 2, 24. [Google Scholar] [CrossRef]
- Liao, Y.C.; Chen, N.T.; Shih, Y.P.; Dong, Y.; Lo, S.H. Up-regulation of C-terminal tensin-like molecule promotes the tumorigenicity of colon cancer through beta-catenin. Cancer Res. 2009, 69, 4563–4566. [Google Scholar] [CrossRef]
- van de Vlekkert, D.; Demmers, J.; Nguyen, X.X.; Campos, Y.; Machado, E.; Annunziata, I.; Hu, H.; Gomero, E.; Qiu, X.; Bongiovanni, A.; et al. Excessive exosome release is the pathogenic pathway linking a lysosomal deficiency to generalized fibrosis. Sci. Adv. 2019, 5, eaav3270. [Google Scholar] [CrossRef]
- Uriu, Y.; Kiyonaka, S.; Miki, T.; Yagi, M.; Akiyama, S.; Mori, E.; Nakao, A.; Beedle, A.M.; Campbell, K.P.; Wakamori, M.; et al. Rab3-interacting molecule gamma isoforms lacking the Rab3-binding domain induce long lasting currents but block neurotransmitter vesicle anchoring in voltage-dependent P/Q-type Ca2+ channels. J. Biol. Chem. 2010, 285, 21750–21767. [Google Scholar] [CrossRef]
- Auslander, N.; Cunningham, C.E.; Toosi, B.M.; McEwen, E.J.; Yizhak, K.; Vizeacoumar, F.S.; Parameswaran, S.; Gonen, N.; Freywald, T.; Bhanumathy, K.K.; et al. An integrated computational and experimental study uncovers FUT9 as a metabolic driver of colorectal cancer. Mol. Syst. Biol. 2017, 13, 956. [Google Scholar] [CrossRef] [PubMed]
- Pufulete, M.; Al-Ghnaniem, R.; Leather, A.J.; Appleby, P.; Gout, S.; Terry, C.; Emery, P.W.; Sanders, T.A. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: A case control study. Gastroenterology 2003, 124, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Hoyt, L.; Crowell, J.; Kim, Y.I. Folate and DNA methylation. Antioxid. Redox Signal 2012, 17, 302–326. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A. Folate, colorectal cancer and the involvement of DNA methylation. Proc. Nutr. Soc. 2012, 71, 592–597. [Google Scholar] [CrossRef]
- Pieroth, R.; Paver, S.; Day, S.; Lammersfeld, C. Folate and Its Impact on Cancer Risk. Curr. Nutr. Rep. 2018, 7, 70–84. [Google Scholar] [CrossRef]
- Hubner, R.A.; Houlston, R.S. Folate and colorectal cancer prevention. Br. J. Cancer 2009, 100, 233–239. [Google Scholar] [CrossRef]
- MacFarlane, A.J.; Behan, N.A.; Matias, F.M.; Green, J.; Caldwell, D.; Brooks, S.P. Dietary folate does not significantly affect the intestinal microbiome, inflammation or tumorigenesis in azoxymethane-dextran sodium sulphate-treated mice. Br. J. Nutr. 2013, 109, 630–638. [Google Scholar] [CrossRef]
- Mahajan, A.; Sapehia, D.; Thakur, S.; Mohanraj, P.S.; Bagga, R.; Kaur, J. Effect of imbalance in folate and vitamin B12 in maternal/parental diet on global methylation and regulatory miRNAs. Sci. Rep. 2019, 9, 17602. [Google Scholar] [CrossRef]
- Zhu, Z.Z.; Sparrow, D.; Hou, L.; Tarantini, L.; Bollati, V.; Litonjua, A.A.; Zanobetti, A.; Vokonas, P.; Wright, R.O.; Baccarelli, A.; et al. Repetitive element hypomethylation in blood leukocyte DNA and cancer incidence, prevalence, and mortality in elderly individuals: The Normative Aging Study. Cancer Causes Control 2011, 22, 437–447. [Google Scholar] [CrossRef]
- Slattery, M.L.; Lundgreen, A.; Wolff, R.K. Dietary influence on MAPK-signaling pathways and risk of colon and rectal cancer. Nutr. Cancer 2013, 65, 729–738. [Google Scholar] [CrossRef]
- Protiva, P.; Mason, J.B.; Liu, Z.; Hopkins, M.E.; Nelson, C.; Marshall, J.R.; Lambrecht, R.W.; Pendyala, S.; Kopelovich, L.; Kim, M.; et al. Altered folate availability modifies the molecular environment of the human colorectum: Implications for colorectal carcinogenesis. Cancer Prev. Res. 2011, 4, 530–543. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A | B | C | D | E | F |
|---|---|---|---|---|---|
| Gene | ND ** FA 8 mg/0 mg | Polyp ζ FA 8 mg/0 mg | 8 mg FA Polyp/ND | 0 mg FA Polyp/ND | Signaling pathways and/or cellular function |
| Tbxt (T, Brachyury) | 11.8 | 0.9 | 4.7 | 61.4 | Wnt target gene [32,33] |
| Slc4a11 | 3.1 | 1.0 | 3.2 | 10.5 | H+(OH×) and NH3-H+ transporter protein; [34] necessary for cell survival [35] |
| Dusp4 (Mkp-2) | 2.1 | 1.4 | 4.7 | 7.0 | Negative regulator of MAPK, overexpressed in CRC [36,37] |
| Spink4 | 2.1 | 1.1 | 0.5 | 0.9 | Marker for intestinal goblet cells, downregulated in CRC [38,39] |
| Thbs1 (Tsp1) | 1.7 | 1.1 | 2.9 | 4.6 | Negative regulator of angiogenesis; [40] increased expression in inflammatory bowel disease [41] |
| Epha2 | 1.8 | 0.9 | 2.1 | 4.1 | Effector of MAPK signaling and overexpressed in CRC [42,43] |
| Zfp703 | 1.7 | 1.3 | 1.5 | 2.0 | Wnt target gene [44] |
| Enc1 | 1.4 | 1.2 | 2.1 | 2.4 | Wnt target gene [45] |
| Atp11a | 1.3 | 1.3 | 2.8 | 2.6 | ATPase with higher expression in CRC [46] |
| Lzts1 (Fez1) | 0.8 | 0.8 | 2.0 | 2.1 | Tumor suppressor gene; regulates M phase during cell cycle [47,48] |
| Tns4 (Cten) | 0.6 | 1.2 | 2.6 | 1.2 | Interacts with β-catenin; overexpressed in CRC [49] |
| Neu1 | 0.6 | 1.1 | 0.8 | 0.4 | Inhibits β-catenin expression [50] |
| Rab11fip4 | 0.6 | 0.9 | 0.5 | 0.3 | HIF-1α target gene and effector of RAB11 [39] |
| Rims4 | 0.5 | 0.3 | 0.2 | 0.1 | Maintains Ca2+ influx [51] |
| Fut9 | 0.3 | 1.1 | 0.8 | 0.3 | Essential in tumor-initiating cells; switched off to enhance the aggressiveness of tumor cells [52] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, W.-C.L.; Ghosh, J.; Cooper, H.S.; Vanderveer, L.; Schultz, B.; Zhou, Y.; Harvey, K.N.; Kaunga, E.; Devarajan, K.; Li, Y.; et al. Folic Acid Supplementation Promotes Hypomethylation in Both the Inflamed Colonic Mucosa and Colitis-Associated Dysplasia. Cancers 2023, 15, 2949. https://doi.org/10.3390/cancers15112949
Chang W-CL, Ghosh J, Cooper HS, Vanderveer L, Schultz B, Zhou Y, Harvey KN, Kaunga E, Devarajan K, Li Y, et al. Folic Acid Supplementation Promotes Hypomethylation in Both the Inflamed Colonic Mucosa and Colitis-Associated Dysplasia. Cancers. 2023; 15(11):2949. https://doi.org/10.3390/cancers15112949
Chicago/Turabian StyleChang, Wen-Chi L., Jayashri Ghosh, Harry S. Cooper, Lisa Vanderveer, Bryant Schultz, Yan Zhou, Kristen N. Harvey, Esther Kaunga, Karthik Devarajan, Yuesheng Li, and et al. 2023. "Folic Acid Supplementation Promotes Hypomethylation in Both the Inflamed Colonic Mucosa and Colitis-Associated Dysplasia" Cancers 15, no. 11: 2949. https://doi.org/10.3390/cancers15112949
APA StyleChang, W. -C. L., Ghosh, J., Cooper, H. S., Vanderveer, L., Schultz, B., Zhou, Y., Harvey, K. N., Kaunga, E., Devarajan, K., Li, Y., Jelinek, J., Fragoso, M. F., Sapienza, C., & Clapper, M. L. (2023). Folic Acid Supplementation Promotes Hypomethylation in Both the Inflamed Colonic Mucosa and Colitis-Associated Dysplasia. Cancers, 15(11), 2949. https://doi.org/10.3390/cancers15112949