
Immuno-Contexture and Immune Checkpoint Molecule Expression in Mismatch Repair Proficient Colorectal Carcinoma

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Immunohistochemistry
2.3. Tissue Processing and Flow Cytometry Analysis
2.4. Isolation and Culture of Peripheral Blood Mononuclear Cells and Pleural Fluid Macrophages
2.5. PD-L1 Expression and Modulation via Interferon Gamma (IFN-γ)
2.6. RNAscope
2.7. Statistical Analysis
3. Results
3.1. Clinical and Pathological Features of the CRC Cohort
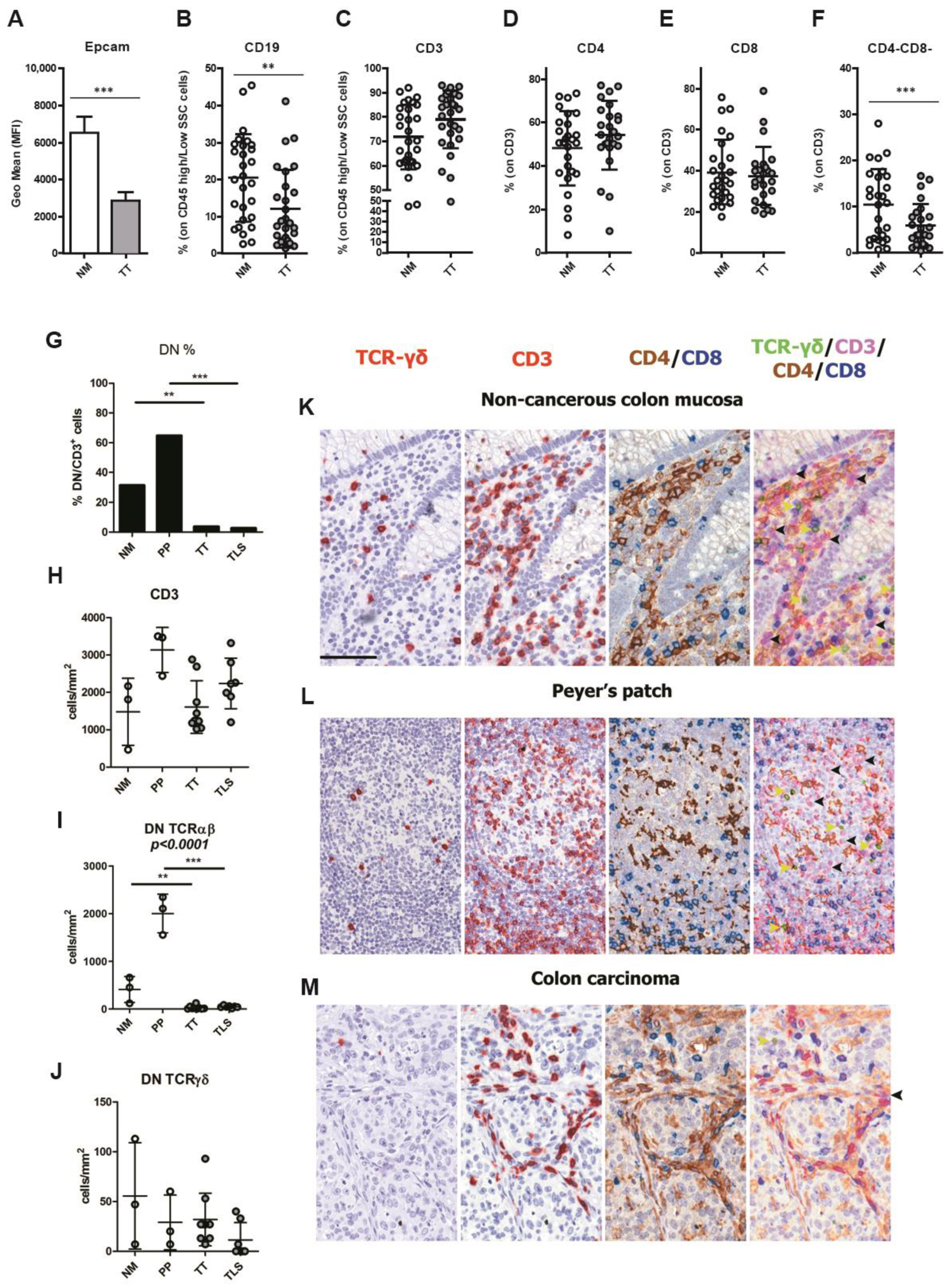
3.2. CRCpMMR Tumor Tissue Displays a Reduced Infiltration of CD19+ B Lymphocytes and CD3+ Double Negative T Lymphocytes (DNTs)
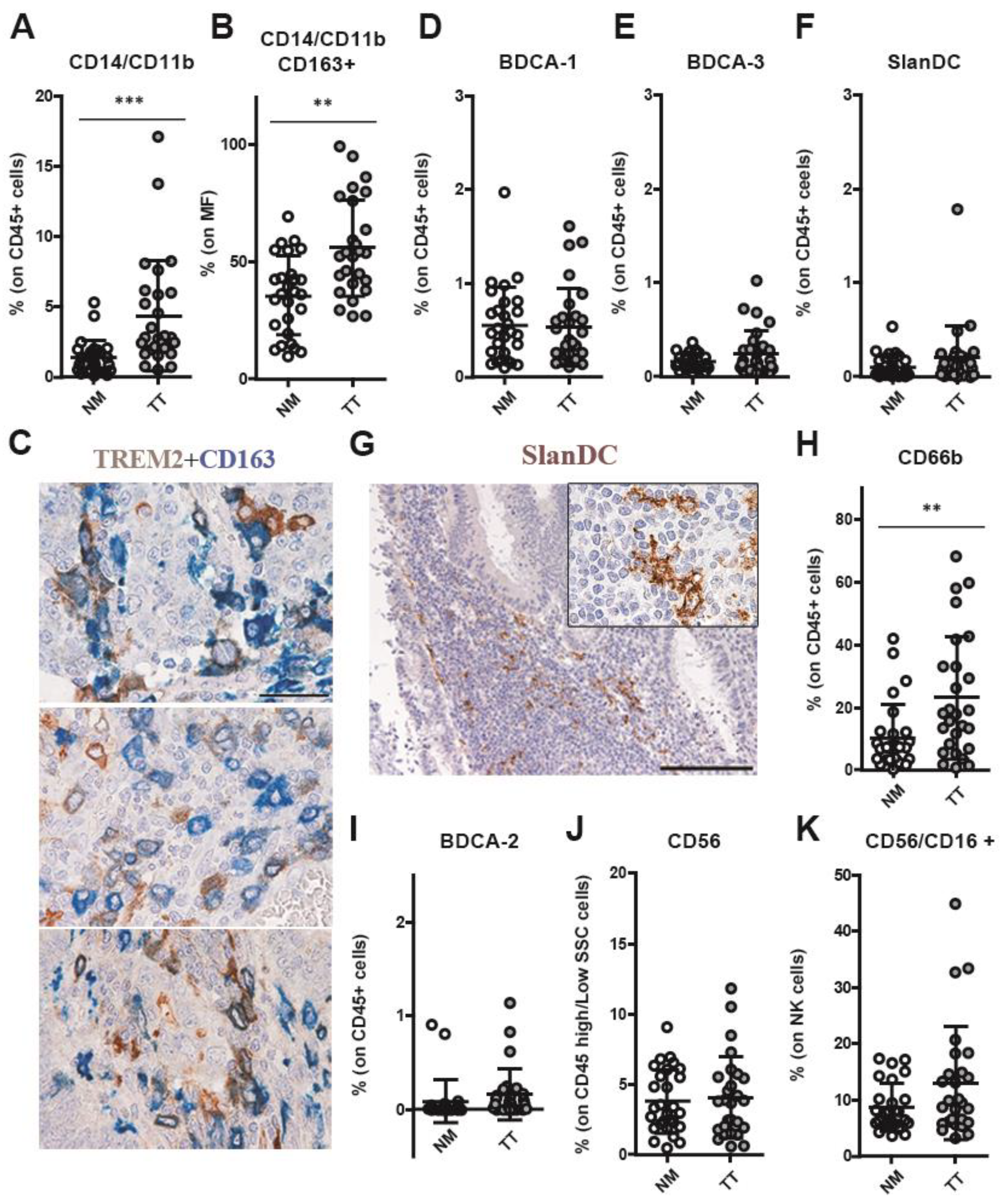
3.3. CRCpMMR Tissue Contains TREM2+ Tumor-Associated Macrophages (TAMs) and Neutrophils (TANs)
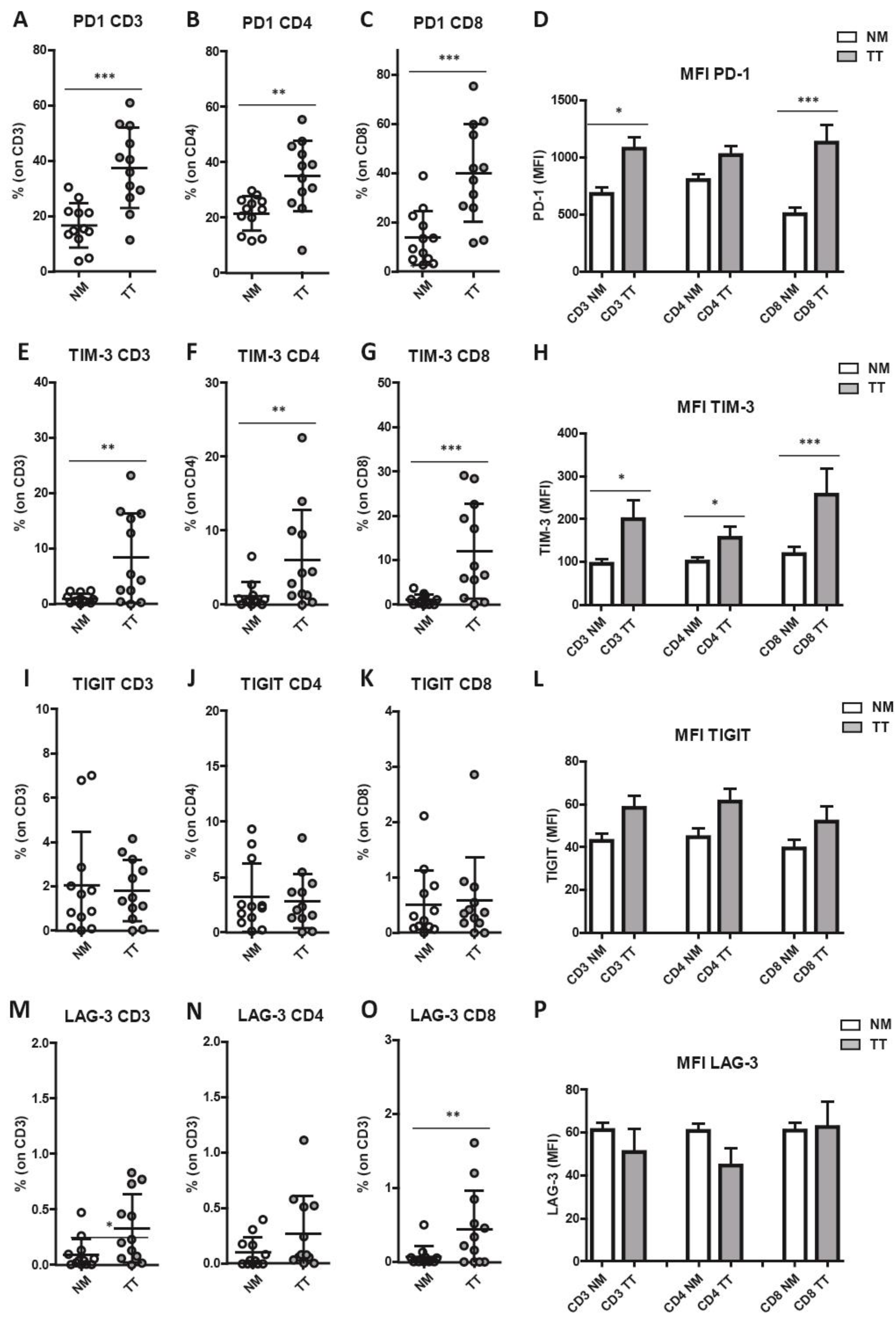
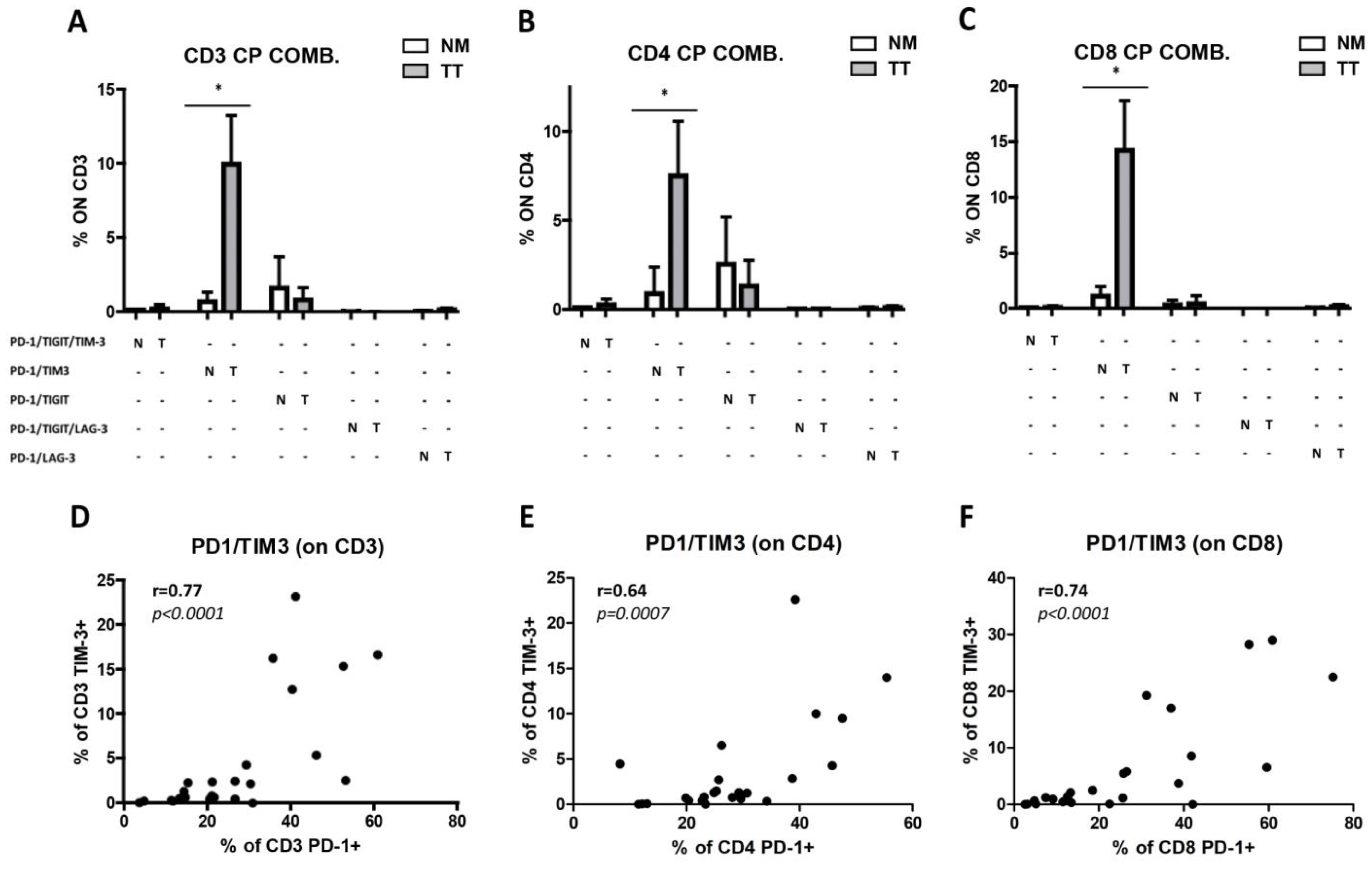
3.4. Increased Combined Expression of PD-1 and TIM-3 in CRCpMMR Tumor-Infiltrating T Cells (TILs)
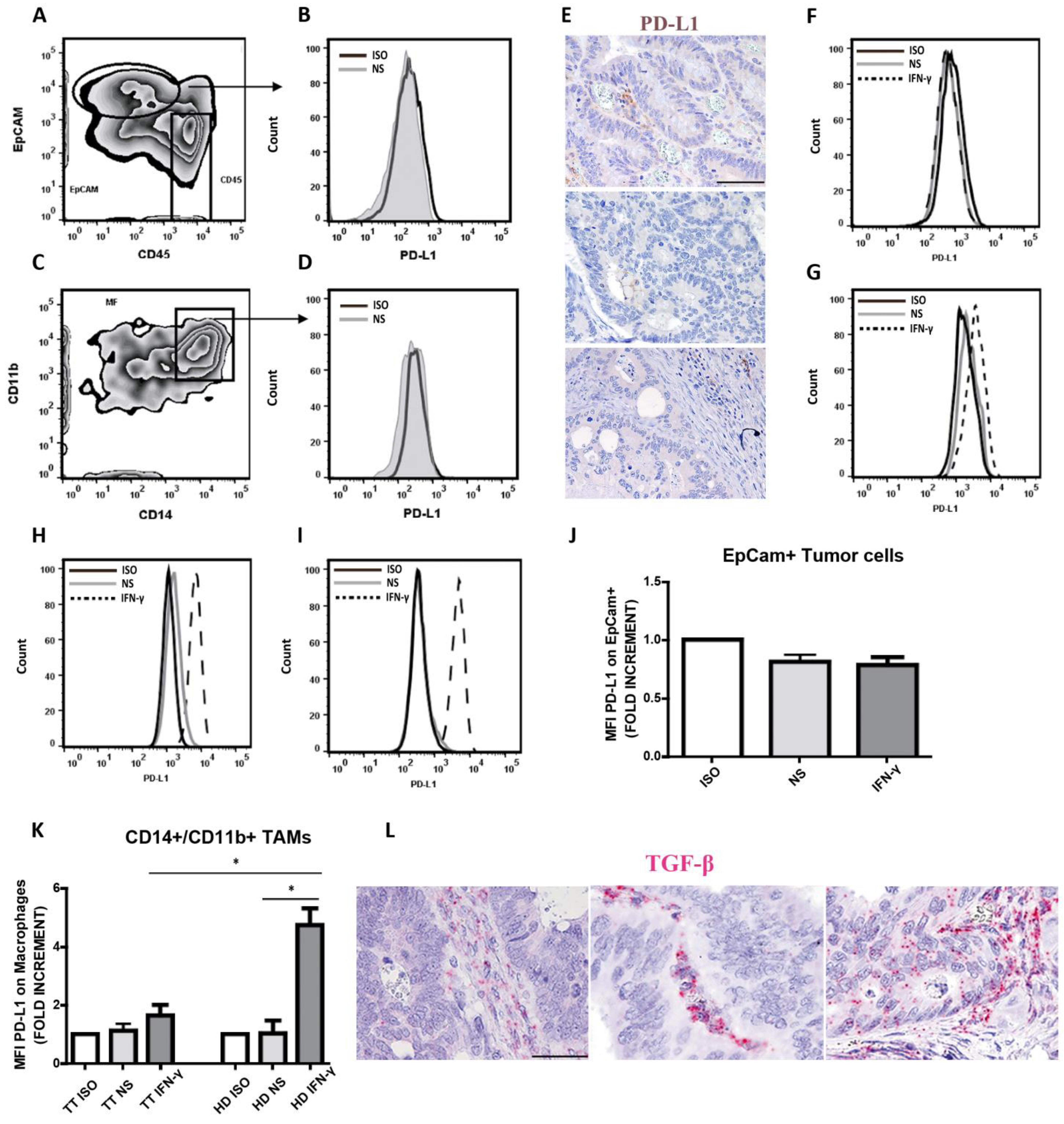
3.5. Defective Modulation of PD-L1 by IFN-γ on CRCpMMR Tumor Cells and Tumor-Associated Macrophages (TAMs)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Verschoor, C.P.; Ma, G.W.; Pawelec, G. Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front. Immunol. 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Na, Y.; Joung, S.Y.; Lee, S.I.; Oh, S.C.; Min, B.W. The significance of microsatellite instability in colorectal cancer after controlling for clinicopathological factors. Medicine 2018, 97, e0019. [Google Scholar] [CrossRef] [PubMed]
- Biller, L.H.; Schrag, D. Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA 2021, 325, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Lo Nigro, C.; Ricci, V.; Vivenza, D.; Granetto, C.; Fabozzi, T.; Miraglio, E.; Merlano, M.C. Prognostic and predictive biomarkers in metastatic colorectal cancer anti-EGFR therapy. World J. Gastroenterol. 2016, 22, 6944–6954. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E-Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Chen, J.; Ye, X.; Pitmon, E.; Lu, M.; Wan, J.; Jellison, E.R.; Adler, A.J.; Vella, A.T.; Wang, K. IL-17 inhibits CXCL9/10-mediated recruitment of CD8+ cytotoxic T cells and regulatory T cells to colorectal tumors. J. Immunother. Cancer 2019, 7, 324. [Google Scholar] [CrossRef]
- Kamal, Y.; Schmit, S.L.; Frost, H.R.; Amos, C.I. The tumor microenvironment of colorectal cancer metastases: Opportunities in cancer immunotherapy. Immunotherapy 2020, 12, 1083–1100. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- Hanna, G.J.; Liu, H.; Jones, R.E.; Bacay, A.F.; Lizotte, P.H.; Ivanova, E.V.; Bittinger, M.A.; Cavanaugh, M.E.; Rode, A.J.; Schoenfeld, J.D.; et al. Defining an inflamed tumor immunophenotype in recurrent, metastatic squamous cell carcinoma of the head and neck. Oral Oncol. 2017, 67, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An immune atlas of clear cell renal cell carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.-A.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Waldner, M.; Obenauf, A.C.; Angell, H.; Fredriksen, T.; Lafontaine, L.; Berger, A.; et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013, 39, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Pohl, M.; Schmiegel, W. Therapeutic Strategies in Diseases of the Digestive Tract—2015 and Beyond Targeted Therapies in Colon Cancer Today and Tomorrow. Dig. Dis. 2016, 34, 574–579. [Google Scholar] [CrossRef]
- Seow, H.F.; Yip, W.K.; Fifis, T. Advances in targeted and immunobased therapies for colorectal cancer in the genomic era. Onco Targets Ther. 2016, 9, 1899–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miko, E.; Meggyes, M.; Doba, K.; Barakonyi, A.; Szereday, L. Immune checkpoint molecules in reproductive immunology. Front. Immunol. 2019, 10, 846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masugi, Y.; Nishihara, R.; Yang, J.; Mima, K.; da Silva, A.; Shi, Y.; Inamura, K.; Cao, Y.; Song, M.; Nowak, J.A.; et al. Tumour CD274 (PD-L1) expression and T cells in colorectal cancer. Gut 2017, 66, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.; Sun, J.; Mao, Y.; Chen, L.-J.; Wu, Y.-Y.; Zhang, X.-G. B7-H1 expression is associated with expansion of regulatory T cells in colorectal carcinoma. World J. Gastroenterol. 2012, 18, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Cruise, M.; Tam, A.; Wicks, E.C.; Hechenbleikner, E.M.; Taube, J.M.; Blosser, R.L.; Fan, H.; Wang, H.; Luber, B.S.; et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015, 5, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Solinas, C.; Garaud, S.; De Silva, P.; Boisson, A.; Van den Eynden, G.; de Wind, A.; Risso, P.; Rodrigues Vitória, J.; Richard, F.; Migliori, E.; et al. Immune Checkpoint Molecules on Tumor-Infiltrating Lymphocytes and Their Association with Tertiary Lymphoid Structures in Human Breast Cancer. Front. Immunol. 2017, 8, 1412. [Google Scholar] [CrossRef] [Green Version]
- Toor, S.M.; Sasidharan Nair, V.; Murshed, K.; Abu Nada, M.; Elkord, E. Tumor-Infiltrating Lymphoid Cells in Colorectal Cancer Patients with Varying Disease Stages and Microsatellite Instability-High/Stable Tumors. Vaccines 2021, 9, 64. [Google Scholar] [CrossRef]
- Angelova, M.; Charoentong, P.; Hackl, H.; Fischer, M.L.; Snajder, R.; Krogsdam, A.M.; Waldner, M.J.; Bindea, G.; Mlecnik, B.; Galon, J.; et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015, 16, 64. [Google Scholar] [CrossRef]
- Jacobs, J.; Smits, E.; Lardon, F.; Pauwels, P.; Deschoolmeester, V. Immune checkpoint modulation in colorectal cancer: What’s new and what to expect. J. Immunol. Res. 2015, 2015, 158038. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Freeman, G.J. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstern, C.R.; Ngu, R.K.; Shalapour, S.; Karin, M. Immunotherapy, inflammation and colorectal cancer. Cells 2020, 9, 618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, A.; Wang, B.; Wang, X.; Nie, Y.; Fan, D.; Zhao, X.; Lu, Y. Immunotherapy in colorectal cancer: Current achievements and future perspective. Int. J. Biol. Sci. 2021, 17, 3837–3849. [Google Scholar] [CrossRef] [PubMed]
- Makaremi, S.; Asadzadeh, Z.; Hemmat, N.; Baghbanzadeh, A.; Sgambato, A.; Ghorbaninezhad, F.; Safarpour, H.; Argentiero, A.; Brunetti, O.; Bernardini, R.; et al. Immune checkpoint inhibitors in colorectal cancer: Challenges and future prospects. Biomedicines 2021, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, J.; Reddy, S.; Verma, N.; Huang, E.H.; Kazmi, S.M. Immune Checkpoint Inhibitors in pMMR/MSS Colorectal Cancer. J. Gastrointest. Cancer 2023. [Google Scholar] [CrossRef] [PubMed]
- Giurisato, E.; Lonardi, S.; Telfer, B.; Lussoso, S.; Risa-Ebrí, B.; Zhang, J.; Russo, I.; Wang, J.; Santucci, A.; Finegan, K.G.; et al. Extracellular-Regulated Protein Kinase 5-Mediated Control of p21 Expression Promotes Macrophage Proliferation Associated with Tumor Growth and Metastasis. Cancer Res. 2020, 80, 3319–3330. [Google Scholar] [CrossRef]
- Garside, P.; Millington, O.; Smith, K.M. The anatomy of mucosal immune responses. Ann. N. Y. Acad. Sci. 2004, 1029, 9–15. [Google Scholar] [CrossRef]
- MacPherson, G.; Milling, S.; Yrlid, U.; Cousins, L.; Turnbull, E.; Huang, F.-P. Uptake of antigens from the intestine by dendritic cells. Ann. N. Y. Acad. Sci. 2004, 1029, 75–82. [Google Scholar] [CrossRef]
- Goossens-Beumer, I.J.; Zeestraten, E.C.M.; Benard, A.; Christen, T.; Reimers, M.S.; Keijzer, R.; Sier, C.F.M.; Liefers, G.J.; Morreau, H.; Putter, H.; et al. Clinical prognostic value of combined analysis of Aldh1, Survivin, and EpCAM expression in colorectal cancer. Br. J. Cancer 2014, 110, 2935–2944. [Google Scholar] [CrossRef]
- Kim, J.H.; Bae, J.M.; Song, Y.S.; Cho, N.-Y.; Lee, H.S.; Kang, G.H. Clinicopathologic, molecular, and prognostic implications of the loss of EPCAM expression in colorectal carcinoma. Oncotarget 2016, 7, 13372–13387. [Google Scholar] [CrossRef]
- Velikkakam, T.; Gollob, K.J.; Dutra, W.O. Double-negative T cells: Setting the stage for disease control or progression. Immunology 2022, 165, 371–385. [Google Scholar] [CrossRef]
- Carrasco, A.; Fernández-Bañares, F.; Pedrosa, E.; Salas, A.; Loras, C.; Rosinach, M.; Aceituno, M.; Andújar, X.; Forné, M.; Zabana, Y.; et al. Regional specialisation of T cell subsets and apoptosis in the human gut mucosa: Differences between ileum and colon in healthy intestine and inflammatory bowel diseases. J. Crohns Colitis 2016, 10, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molgora, M.; Esaulova, E.; Vermi, W.; Hou, J.; Chen, Y.; Luo, J.; Brioschi, S.; Bugatti, M.; Omodei, A.S.; Ricci, B.; et al. TREM2 Modulation Remodels the Tumor Myeloid Landscape Enhancing Anti-PD-1 Immunotherapy. Cell 2020, 182, 886–900.e17. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, L.; Congy-Jolivet, N.; Bolzec, A.; Thibault, G. Gradual Increase of FcγRIIIa/CD16a Expression and Shift toward IFN-γ Secretion during Differentiation of CD56dim Natural Killer Cells. Front. Immunol. 2017, 8, 1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messaoudene, M.; Fregni, G.; Fourmentraux-Neves, E.; Chanal, J.; Maubec, E.; Mazouz-Dorval, S.; Couturaud, B.; Girod, A.; Sastre-Garau, X.; Albert, S.; et al. Mature cytotoxic CD56(bright)/CD16(+) natural killer cells can infiltrate lymph nodes adjacent to metastatic melanoma. Cancer Res. 2014, 74, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, R.E.; Cao, X. Co-stimulatory and co-inhibitory pathways in cancer immunotherapy. Adv. Cancer Res. 2019, 143, 145–194. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef]
- Di Blasi, D.; Boldanova, T.; Mori, L.; Terracciano, L.; Heim, M.H.; De Libero, G. Unique T-Cell Populations Define Immune-Inflamed Hepatocellular Carcinoma. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Stankovic, B.; Bjørhovde, H.A.K.; Skarshaug, R.; Aamodt, H.; Frafjord, A.; Müller, E.; Hammarström, C.; Beraki, K.; Bækkevold, E.S.; Woldbæk, P.R.; et al. Immune Cell Composition in Human Non-small Cell Lung Cancer. Front. Immunol. 2018, 9, 3101. [Google Scholar] [CrossRef] [Green Version]
- Greenplate, A.R.; McClanahan, D.D.; Oberholtzer, B.K.; Doxie, D.B.; Roe, C.E.; Diggins, K.E.; Leelatian, N.; Rasmussen, M.L.; Kelley, M.C.; Gama, V.; et al. Computational Immune Monitoring Reveals Abnormal Double-Negative T Cells Present across Human Tumor Types. Cancer Immunol. Res. 2019, 7, 86–99. [Google Scholar] [CrossRef]
- Wu, Z.; Zheng, Y.; Sheng, J.; Han, Y.; Yang, Y.; Pan, H.; Yao, J. CD3+CD4-CD8- (Double-Negative) T Cells in Inflammation, Immune Disorders and Cancer. Front. Immunol. 2022, 13, 816005. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.; Lombard-Vadnais, F.; Hillhouse, E.E.; Lebel, M.-È.; Chabot-Roy, G.; Melichar, H.J.; Lesage, S. MHC-Independent Thymic Selection of CD4 and CD8 Coreceptor Negative αβ T Cells. J. Immunol. 2020, 205, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, C.L.; Sumaria, N.; Martin, S.; Pennington, D.J. Increased TCR signal strength in DN thymocytes promotes development of gut TCRαβ(+)CD8αα(+) intraepithelial lymphocytes. Sci. Rep. 2017, 7, 10659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crispín, J.C.; Tsokos, G.C. Human TCR-αβ+ CD4− CD8− T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. J. Immunol. 2009, 183, 4675–4681. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, N.; Flores-Mendoza, G.; Apostolidis, S.A.; Rosetti, F.; Tsokos, G.C.; Crispín, J.C. TCR-α/β CD4- CD8- double negative T cells arise from CD8+ T cells. J. Leukoc. Biol. 2020, 108, 851–857. [Google Scholar] [CrossRef]
- Ford, M.S.; Zhang, Z.-X.; Chen, W.; Zhang, L. Double-negative T regulatory cells can develop outside the thymus and do not mature from CD8+ T cell precursors. J. Immunol. 2006, 177, 2803–2809. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Hu, P.; Wu, G.; Zhou, H. Antipancreatic cancer effect of DNT cells and the underlying mechanism. Pancreatology 2019, 19, 105–113. [Google Scholar] [CrossRef]
- Lu, Y.; Hu, P.; Zhou, H.; Yang, Z.; Sun, Y.U.; Hoffman, R.M.; Chen, J. Double-negative T Cells Inhibit Proliferation and Invasion of Human Pancreatic Cancer Cells in Co-culture. Anticancer Res. 2019, 39, 5911–5918. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Ly, D.; Dervovic, D.; Fang, L.; Lee, J.B.; Kang, H.; Wang, Y.-H.; Pham, N.-A.; Pan, H.; Tsao, M.-S.; et al. Human double negative T cells target lung cancer via ligand-dependent mechanisms that can be enhanced by IL-15. J. Immunother. Cancer 2019, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.Q.; Martins, D.S.; Silva-Santos, B. Targeting γδ T lymphocytes for cancer immunotherapy: From novel mechanistic insight to clinical application. Cancer Res. 2010, 70, 10024–10027. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Pagès, F.; Marincola, F.M.; Thurin, M.; Trinchieri, G.; Fox, B.A.; Gajewski, T.F.; Ascierto, P.A. The immune score as a new possible approach for the classification of cancer. J. Transl. Med. 2012, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.S.; McCall, J.L.; Girardin, A.; Munro, F.M.; Black, M.A.; Kemp, R.A. Functional impairment of infiltrating T cells in human colorectal cancer. Oncoimmunology 2016, 5, e1234573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Irahara, N.; Meyerhardt, J.A.; Baba, Y.; Shima, K.; Glickman, J.N.; Ferrone, C.R.; Mino-Kenudson, M.; Tanaka, N.; et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin. Cancer Res. 2009, 15, 6412–6420. [Google Scholar] [CrossRef] [Green Version]
- Meshcheryakova, A.; Tamandl, D.; Bajna, E.; Stift, J.; Mittlboeck, M.; Svoboda, M.; Heiden, D.; Stremitzer, S.; Jensen-Jarolim, E.; Grünberger, T.; et al. B cells and ectopic follicular structures: Novel players in anti-tumor programming with prognostic power for patients with metastatic colorectal cancer. PLoS ONE 2014, 9, e99008. [Google Scholar] [CrossRef]
- Sautès-Fridman, C.; Lawand, M.; Giraldo, N.A.; Kaplon, H.; Germain, C.; Fridman, W.H.; Dieu-Nosjean, M.-C. Tertiary lymphoid structures in cancers: Prognostic value, regulation, and manipulation for therapeutic intervention. Front. Immunol. 2016, 7, 407. [Google Scholar] [CrossRef] [Green Version]
- Di Caro, G.; Bergomas, F.; Grizzi, F.; Doni, A.; Bianchi, P.; Malesci, A.; Laghi, L.; Allavena, P.; Mantovani, A.; Marchesi, F. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. 2014, 20, 2147–2158. [Google Scholar] [CrossRef] [Green Version]
- Bergomas, F.; Grizzi, F.; Doni, A.; Pesce, S.; Laghi, L.; Allavena, P.; Mantovani, A.; Marchesi, F. Tertiary intratumor lymphoid tissue in colo-rectal cancer. Cancers 2011, 4, 1–10. [Google Scholar] [CrossRef]
- Xia, J.; Xie, Z.; Niu, G.; Lu, Z.; Wang, Z.; Xing, Y.; Ren, J.; Hu, Z.; Hong, R.; Cao, Z.; et al. Single-cell landscape and clinical outcomes of infiltrating B cells in colorectal cancer. Immunology 2023, 168, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Sun, X.-F.; Svanvik, J. Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int. J. Cancer 2009, 125, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Elkarim, R.; Sun, X.-F.; Svanvik, J. Macrophage infiltration in tumor stroma is related to tumor cell expression of CD163 in colorectal cancer. Cancer Microenviron. 2014, 7, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabo, I.; Stål, O.; Olsson, H.; Doré, S.; Svanvik, J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Klingen, T.A.; Chen, Y.; Aas, H.; Wik, E.; Akslen, L.A. Tumor-associated macrophages are strongly related to vascular invasion, non-luminal subtypes, and interval breast cancer. Hum. Pathol. 2017, 69, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Nalio Ramos, R.; Missolo-Koussou, Y.; Gerber-Ferder, Y.; Bromley, C.P.; Bugatti, M.; Núñez, N.G.; Tosello Boari, J.; Richer, W.; Menger, L.; Denizeau, J.; et al. Tissue-resident FOLR2+ macrophages associate with CD8+ T cell infiltration in human breast cancer. Cell 2022, 185, 1189–1207.e25. [Google Scholar] [CrossRef]
- Bugatti, M.; Bergamini, M.; Missale, F.; Monti, M.; Ardighieri, L.; Pezzali, I.; Picinoli, S.; Caronni, N.; Missolo-Koussou, Y.; Helft, J.; et al. A population of TIM4+FOLR2+ macrophages localized in tertiary lymphoid structures correlates to an active immune infiltrate across several cancer types. Cancer Immunol. Res. 2022, 10, 1340–1353. [Google Scholar] [CrossRef]
- Ardighieri, L.; Missale, F.; Bugatti, M.; Gatta, L.B.; Pezzali, I.; Monti, M.; Gottardi, S.; Zanotti, L.; Bignotti, E.; Ravaggi, A.; et al. Infiltration by CXCL10 secreting macrophages is associated with antitumor immunity and response to therapy in ovarian cancer subtypes. Front. Immunol. 2021, 12, 690201. [Google Scholar] [CrossRef]
- Cheruku, S.; Rao, V.; Pandey, R.; Rao Chamallamudi, M.; Velayutham, R.; Kumar, N. Tumor-associated macrophages employ immunoediting mechanisms in colorectal tumor progression: Current research in Macrophage repolarization immunotherapy. Int. Immunopharmacol. 2023, 116, 109569. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell. Physiol. Biochem. 2018, 45, 356–365. [Google Scholar] [CrossRef]
- Liu, S.; Gönen, M.; Stadler, Z.K.; Weiser, M.R.; Hechtman, J.F.; Vakiani, E.; Wang, T.; Vyas, M.; Joneja, U.; Al-Bayati, M.; et al. Cellular localization of PD-L1 expression in mismatch-repair-deficient and proficient colorectal carcinomas. Mod. Pathol. 2019, 32, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.-N.; Wiedeman, A.E.; Stevens, A.M. TNF-α and TGF-β counter-regulate PD-L1 expression on monocytes in systemic lupus erythematosus. Sci. Rep. 2012, 2, 295. [Google Scholar] [CrossRef] [Green Version]
- Park, I.-K.; Letterio, J.J.; Gorham, J.D. TGF-beta 1 inhibition of IFN-gamma-induced signaling and Th1 gene expression in CD4+ T cells is Smad3 independent but MAP kinase dependent. Mol. Immunol. 2007, 44, 3283–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Mollinedo, F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [Green Version]
- Hirai, H.; Fujishita, T.; Kurimoto, K.; Miyachi, H.; Kitano, S.; Inamoto, S.; Itatani, Y.; Saitou, M.; Maekawa, T.; Taketo, M.M. CCR1-mediated accumulation of myeloid cells in the liver microenvironment promoting mouse colon cancer metastasis. Clin. Exp. Metastasis 2014, 31, 977–989. [Google Scholar] [CrossRef] [Green Version]
- Pedrazzani, C.; Mantovani, G.; Fernandes, E.; Bagante, F.; Luca Salvagno, G.; Surci, N.; Campagnaro, T.; Ruzzenente, A.; Danese, E.; Lippi, G.; et al. Assessment of neutrophil-to-lymphocyte ratio, platelet-to-lymphocyte ratio and platelet count as predictors of long-term outcome after R0 resection for colorectal cancer. Sci. Rep. 2017, 7, 1494. [Google Scholar] [CrossRef] [Green Version]
- Halazun, K.J.; Aldoori, A.; Malik, H.Z.; Al-Mukhtar, A.; Prasad, K.R.; Toogood, G.J.; Lodge, J.P.A. Elevated preoperative neutrophil to lymphocyte ratio predicts survival following hepatic resection for colorectal liver metastases. Eur. J. Surg. Oncol. 2008, 34, 55–60. [Google Scholar] [CrossRef]
- Yin, C.; Okugawa, Y.; Yamamoto, A.; Kitajima, T.; Shimura, T.; Kawamura, M.; Tsujiura, M.; Okita, Y.; Ohi, M.; Toiyama, Y. Prognostic significance of CD8+ tumor-infiltrating lymphocytes and CD66b+ tumor-associated neutrophils in the invasive margins of stages I-III colorectal cancer. Oncol. Lett. 2022, 24, 212. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Bianchi, P.; Grizzi, F.; Di Caro, G.; Basso, G.; Ponzetta, A.; Bonavita, E.; Barbagallo, M.; Tartari, S.; Polentarutti, N.; et al. Occurrence and significance of tumor-associated neutrophils in patients with colorectal cancer. Int. J. Cancer 2016, 139, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Wu, J.; Peng, Y.; Sun, J.; Cheng, P.; Huang, Q. Tumor-Associated Neutrophils in Colorectal Cancer Development, Progression and Immunotherapy. Cancers 2022, 14, 4755. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhang, H.; Xing, Q.; Cui, J.; Li, J.; Li, Y.; Tan, Y.; Wang, S. PD-1(+) CD8(+) T cells are exhausted in tumours and functional in draining lymph nodes of colorectal cancer patients. Br. J. Cancer 2014, 111, 1391–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, Y.; Saito, H.; Ikeguchi, M. Upregulation of TIM-3 and PD-1 on CD4+ and CD8+ T Cells Associated with Dysfunction of Cell-Mediated Immunity after Colorectal Cancer Operation. Yonago Acta Med. 2012, 55, 1–9. [Google Scholar] [PubMed]
- Gosens, M.J.E.M.; van Kempen, L.C.L.; van de Velde, C.J.H.; van Krieken, J.H.J.M.; Nagtegaal, I.D. Loss of membranous Ep-CAM in budding colorectal carcinoma cells. Mod. Pathol. 2007, 20, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Went, P.; Dirnhofer, S.; Salvisberg, T.; Amin, M.B.; Lim, S.D.; Diener, P.-A.; Moch, H. Expression of epithelial cell adhesion molecule (EpCam) in renal epithelial tumors. Am. J. Surg. Pathol. 2005, 29, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Balzar, M.; Winter, M.J.; de Boer, C.J.; Litvinov, S.V. The biology of the 17-1A antigen (Ep-CAM). J. Mol. Med. 1999, 77, 699–712. [Google Scholar] [CrossRef]
- Van der Gun, B.T.F.; Melchers, L.J.; Ruiters, M.H.J.; de Leij, L.F.M.H.; McLaughlin, P.M.J.; Rots, M.G. EpCAM in carcinogenesis: The good, the bad or the ugly. Carcinogenesis 2010, 31, 1913–1921. [Google Scholar] [CrossRef] [Green Version]
- Maetzel, D.; Denzel, S.; Mack, B.; Canis, M.; Went, P.; Benk, M.; Kieu, C.; Papior, P.; Baeuerle, P.A.; Munz, M.; et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 2009, 11, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.J.; Nagelkerken, B.; Mertens, A.E.E.; Rees-Bakker, H.A.M.; Briaire-de Bruijn, I.H.; Litvinov, S.V. Expression of Ep-CAM shifts the state of cadherin-mediated adhesions from strong to weak. Exp. Cell Res. 2003, 285, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-T.; Osmulski, P.; Wang, Y.; Huang, Y.-W.; Liu, L.; Ruan, J.; Jin, V.X.; Kirma, N.B.; Gaczynska, M.E.; Huang, T.H.-M. EpCAM-Regulated Transcription Exerts Influences on Nanomechanical Properties of Endometrial Cancer Cells That Promote Epithelial-to-Mesenchymal Transition. Cancer Res. 2016, 76, 6171–6182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaves-Pérez, A.; Mack, B.; Maetzel, D.; Kremling, H.; Eggert, C.; Harréus, U.; Gires, O. EpCAM regulates cell cycle progression via control of cyclin D1 expression. Oncogene 2013, 32, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Jachin, S.; Bae, J.S.; Sung, J.J.; Park, H.S.; Jang, K.Y.; Chung, M.J.; Kim, D.G.; Moon, W.S. The role of nuclear EpICD in extrahepatic cholangiocarcinoma: Association with β-catenin. Int. J. Oncol. 2014, 45, 691–698. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pt 1 | Gender | Age | Anatomic District 2 | Istotype 3 | G 4 | N 5 | Neoplastic Emboli 6 | Infl. CT 7 | Infl. IM 8 | Crohn-like 9 | MMR 10 | TNM | AJCC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | M | 42 | R | SR | N1 | L1 | + | + | no | P | pT3N2b | IIIc | |
| #2 | M | 74 | L | NOS | HIGH | N1 | V1 + L1 | + | + | no | P | pT4aN2a | IIIc |
| #3 | M | 43 | R | NOS | LOW | N0 | L1 | + | + | no | P | pT3N0 | IIa |
| #4 | M | 70 | L | NOS | LOW | N1 | L1 | ++ | ++ | yes | P | pT4aN1b | IIIb |
| #5 | M | 76 | L | NOS | LOW | N1 | V1 + L1 | + | + | no | P | pT4aN2bM1a | IVa |
| #6 | F | 77 | L | NOS | LOW | N0 | L1 | ++ | ++ | yes | P | pT3N0 | IIa |
| #7 | M | 76 | R | NOS | LOW | N1 | L1 | + | + | no | P | pT3N1b | IIIb |
| #8 | M | 79 | L | NOS | LOW | N0 | V1 | + | + | no | P | pT3N0 | IIa |
| #9 | M | 60 | L | NOS | LOW | N1 | L1 | + | + | no | P | pT3N1b | IIIb |
| #10 | F | 59 | L | NOS | LOW | N1 | V0-L0 | + | + | yes | P | pT4aN2b | IIIc |
| #11 | M | 69 | R | MUC | LOW | N0 | V1 | + | + | yes | P | pT3N1c | IIIb |
| #12 | F | 80 | R | NOS | LOW | N0 | V1 + L1 | + | + | no | P | pT3N0 | IIa |
| #13 | F | 84 | R | MUC | HIGH | N0 | V0-L0 | + | + | yes | D | pT3N0 | IIa |
| #14 | M | 66 | R | NOS | LOW | N0 | V0-L0 | + | + | yes | P | pT3N0 | IIa |
| #15 | M | 73 | L | MUC | LOW | N1 | L1 | + | + | no | P | pT3N1a | IIIb |
| #16 | M | 77 | R | MUC | LOW | N0 | L1 | + | + | no | P | pT3N0 | IIa |
| #17 | F | 81 | L | MUC | LOW | N0 | L1 | +++ | +++ | no | P | pT4bN0 | IIc |
| #18 | F | 59 | L | NOS | LOW | N0 | V0-L0 | + | + | yes | P | pT3N0 | IIa |
| #19 | F | 75 | R | NOS | LOW | N0 | L1 | + | + | no | P | pT3N0 | IIa |
| #20 | F | 46 | L | NOS | LOW | N0 | L1 | + | + | no | P | pT2N0 | I |
| #21 | M | 78 | L | SR | LOW | N0 | L1 | - | - | no | P | pT2N0 | I |
| #22 | F | 79 | R | MUC | LOW | N0 | L1 | + | + | no | D | pT3N0 (pT2N0) | IIa (I) |
| #23 | M | 77 | R | NOS | LOW | N1 | V1 + L1 | + | + | no | P | pT3N1b | IIIb |
| #24 | M | 72 | L | NOS | LOW | N0 | V0-L0 | + | + | no | P | pT2N0 | I |
| #25 | F | 52 | L | NOS | LOW | N1 | V1 + L1 | + | + | no | P | pT4aN2b | IIIc |
| #26 | M | 80 | R | NOS | LOW | N0 | L1 | + | + | yes | P | pT4bN0 | IIb |
| #27 | M | 95 | L | NOS | LOW | N0 | L1 | + | + | no | P | pT3N0 | IIa |
| #28 | F | 47 | R | NOS | LOW | N0 | L1 | + | + | no | D | pT3N0 | IIa |
| #29 | M | 88 | R | SR | LOW | N0 | V1 + L1 | + | + | no | D | pT4aN0 | IIb |
| #30 | F | 71 | R | NOS | HIGH | N0 | L1 | + | + | no | D | pT4aN1c | IIIb |
| #31 | F | 92 | R | NOS | LOW | N1 | L1 | + | - | no | P | pT4aN1a | IIIb |
| #32 | M | 46 | R | NOS | HIGH | N0 | L1 | - | + | yes | P | pT4aN0 | IIb |
| #33 | M | 76 | R | NOS | LOW | N0 | L1 | - | - | no | D | pT3N0 | IIa |
| #34 | F | 70 | L | NOS | LOW | N1 | V1 + L1 | - | - | no | P | pT3N2a | IIIb |
| #35 | M | 78 | L | NOS | LOW | N0 | V0-L0 | - | + | yes | P | pT1N0 | I |
| #36 | M | 58 | L | NOS | LOW | N1 | V1 + L1 | + | + | no | P | pT3N1b | IIIb |
| #37 | M | 75 | R | NOS | LOW | N1 | L1 | + | + | yes | P | pT3N2a | IIIb |
| #38 | M | 80 | L | NOS | LOW | N1 | L1 | - | - | yes | P | pT3N1b | IIIb |
| #39 | F | 71 | R | MUC | LOW | N0 | L1 | - | - | no | D | pT3n0 | IIa |
| #40 | M | 79 | L | NOS | LOW | N1 | V1 + L1 | + | + | no | P | pT4aN1b | IIIb |
| #41 | M | 79 | R | NOS | LOW | N0 | V0-L0 | + | +++ | no | P | pT3n0 | IIa |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacomelli, M.; Monti, M.; Pezzola, D.C.; Lonardi, S.; Bugatti, M.; Missale, F.; Cioncada, R.; Melocchi, L.; Giustini, V.; Villanacci, V.; et al. Immuno-Contexture and Immune Checkpoint Molecule Expression in Mismatch Repair Proficient Colorectal Carcinoma. Cancers 2023, 15, 3097. https://doi.org/10.3390/cancers15123097
Giacomelli M, Monti M, Pezzola DC, Lonardi S, Bugatti M, Missale F, Cioncada R, Melocchi L, Giustini V, Villanacci V, et al. Immuno-Contexture and Immune Checkpoint Molecule Expression in Mismatch Repair Proficient Colorectal Carcinoma. Cancers. 2023; 15(12):3097. https://doi.org/10.3390/cancers15123097
Chicago/Turabian StyleGiacomelli, Mauro, Matilde Monti, Diego Cesare Pezzola, Silvia Lonardi, Mattia Bugatti, Francesco Missale, Rossella Cioncada, Laura Melocchi, Viviana Giustini, Vincenzo Villanacci, and et al. 2023. "Immuno-Contexture and Immune Checkpoint Molecule Expression in Mismatch Repair Proficient Colorectal Carcinoma" Cancers 15, no. 12: 3097. https://doi.org/10.3390/cancers15123097
APA StyleGiacomelli, M., Monti, M., Pezzola, D. C., Lonardi, S., Bugatti, M., Missale, F., Cioncada, R., Melocchi, L., Giustini, V., Villanacci, V., Baronchelli, C., Manenti, S., Imberti, L., Giurisato, E., & Vermi, W. (2023). Immuno-Contexture and Immune Checkpoint Molecule Expression in Mismatch Repair Proficient Colorectal Carcinoma. Cancers, 15(12), 3097. https://doi.org/10.3390/cancers15123097