
Improving Localized Radiotherapy for Glioblastoma via Small Molecule Inhibition of KIF11
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Animals and In Vivo Studies
2.3. Small Molecule Inhibitor
2.4. Colony Formation Assays
2.5. Hematoxylin and Eosin Staining
2.6. Immunocytochemistry
2.7. Image Analysis
2.8. Statistical Analysis
3. Results
3.1. KIF11 Inhibition Radiosensitized Patient-Derived GBM Cells In Vitro
3.2. The Mitotic Index and Level of Apoptosis Were Increased in Tumors following a Single Treatment with Ispinesib
3.3. Repeated In Vivo Treatment with Ispinesib, with and without Radiotherapy, Led to Increased Mitotic Indexes and Tumor Cell Death
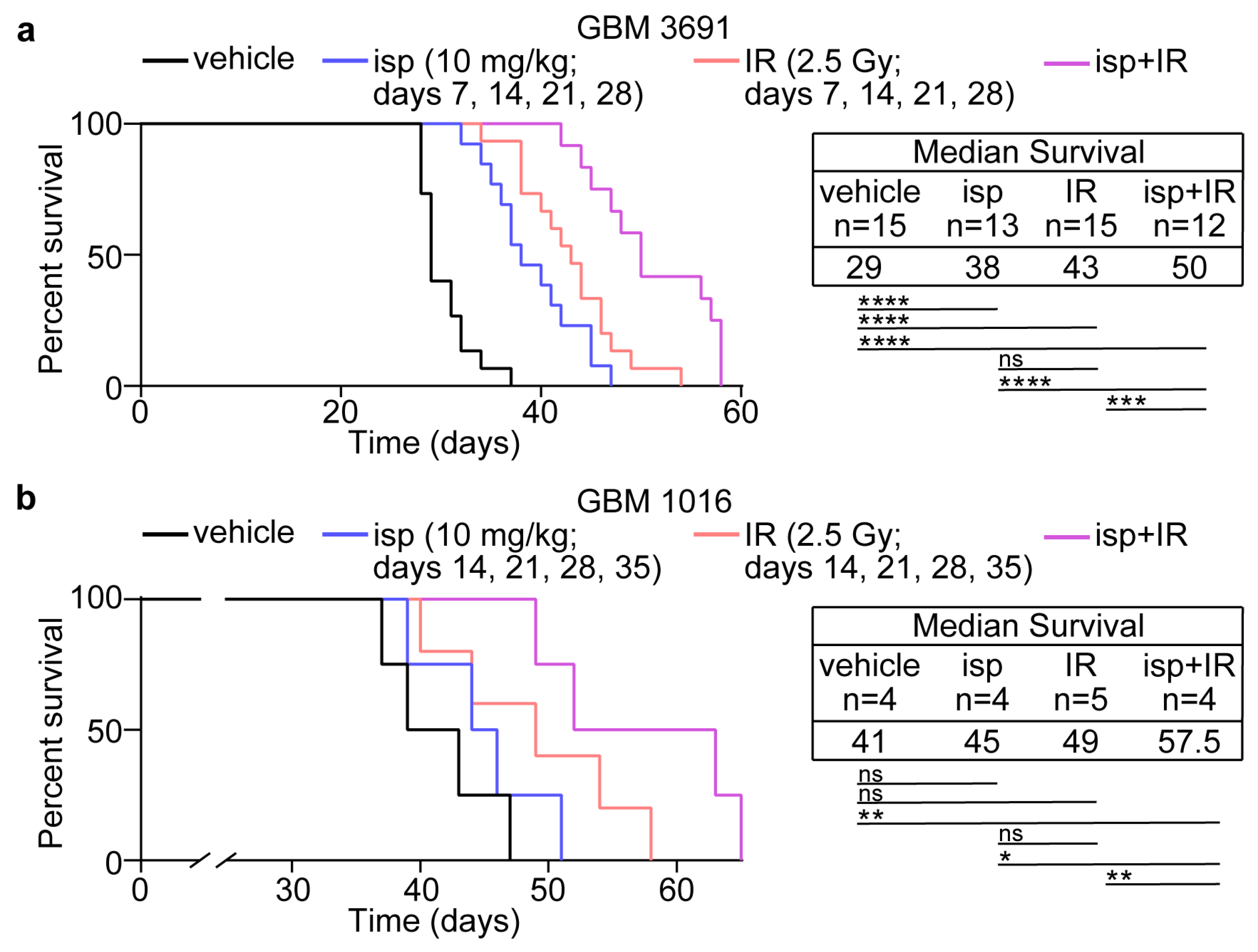
3.4. Combination Treatment with Ispinesib and Radiotherapy Improved Survival in Preclinical Models of GBM
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Squatrito, M.; Holland, E.C. Radiation resistance and stem-like cells in brain tumors. Cancer Cell 2006, 10, 454–456. [Google Scholar] [CrossRef] [Green Version]
- Tallman, M.M.; Zalenski, A.A.; Deighen, A.M.; Schrock, M.S.; Mortach, S.; Grubb, T.M.; Kastury, P.S.; Huntoon, K.; Summers, M.K.; Venere, M. The small molecule drug CBL0137 increases the level of DNA damage and the efficacy of radiotherapy for glioblastoma. Cancer Lett. 2021, 499, 232–242. [Google Scholar] [CrossRef]
- Tamura, K.; Aoyagi, M.; Wakimoto, H.; Ando, N.; Nariai, T.; Yamamoto, M.; Ohno, K. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J. Neurosurg. 2010, 113, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Venere, M.; Hamerlik, P.; Wu, Q.; Rasmussen, R.D.; Song, L.A.; Vasanji, A.; Tenley, N.; Flavahan, W.A.; Hjelmeland, A.B.; Bartek, J.; et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 258–269. [Google Scholar] [CrossRef] [Green Version]
- De, K.; Grubb, T.M.; Zalenski, A.A.; Pfaff, K.E.; Pal, D.; Majumder, S.; Summers, M.K.; Venere, M. Hyperphosphorylation of CDH1 in Glioblastoma Cancer Stem Cells Attenuates APC/C(CDH1) Activity and Pharmacologic Inhibition of APC/C(CDH1/CDC20) Compromises Viability. Mol. Cancer Res. 2019, 17, 1519–1530. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Hubert, C.G.; Herman, J.; Corrin, P.; Toledo, C.M.; Skutt-Kakaria, K.; Vazquez, J.; Basom, R.; Zhang, B.; Risler, J.K.; et al. Cancer-Specific requirement for BUB1B/BUBR1 in human brain tumor isolates and genetically transformed cells. Cancer Discov. 2013, 3, 198–211. [Google Scholar] [CrossRef] [Green Version]
- Godek, K.M.; Venere, M.; Wu, Q.; Mills, K.D.; Hickey, W.F.; Rich, J.N.; Compton, D.A. Chromosomal Instability Affects the Tumorigenicity of Glioblastoma Tumor-Initiating Cells. Cancer Discov. 2016, 6, 532–545. [Google Scholar] [CrossRef] [Green Version]
- Mao, D.D.; Gujar, A.D.; Mahlokozera, T.; Chen, I.; Pan, Y.; Luo, J.; Brost, T.; Thompson, E.A.; Turski, A.; Leuthardt, E.C.; et al. A CDC20-APC/SOX2 Signaling Axis Regulates Human Glioblastoma Stem-like Cells. Cell Rep. 2015, 11, 1809–1821. [Google Scholar] [CrossRef] [Green Version]
- Venere, M.; Horbinski, C.; Crish, J.F.; Jin, X.; Vasanji, A.; Major, J.; Burrows, A.C.; Chang, C.; Prokop, J.; Wu, Q.; et al. The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci. Transl. Med. 2015, 7, 304ra143. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Wu, Q.; Mack, S.C.; Yang, K.; Kim, L.; Hubert, C.G.; Flavahan, W.A.; Chu, C.; Bao, S.; Rich, J.N. CDC20 maintains tumor initiating cells. Oncotarget 2015, 6, 13241–13254. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, W.K. Cyclic x-ray responses in mammalian cells in vitro. Radiat. Res. 1968, 33, 620–643. [Google Scholar] [CrossRef]
- Sinclair, W.K.; Morton, R.A. X-ray sensitivity during the cell generation cycle of cultured Chinese hamster cells. Radiat. Res. 1966, 29, 450–474. [Google Scholar] [CrossRef]
- Terasima, T.; Tolmach, L.J. Variations in several responses of HeLa cells to x-irradiation during the division cycle. Biophys. J. 1963, 3, 11–33. [Google Scholar] [CrossRef] [Green Version]
- Stobbe, C.C.; Park, S.J.; Chapman, J.D. The radiation hypersensitivity of cells at mitosis. Int. J. Radiat. Biol. 2002, 78, 1149–1157. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Kabeche, L.; Compton, D.A.; Powell, S.N.; Bastians, H. Mitotic DNA Damage Response: At the Crossroads of Structural and Numerical Cancer Chromosome Instabilities. Trends Cancer 2017, 3, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.T.; Cesare, A.J.; Fitzpatrick, J.A.; Lazzerini-Denchi, E.; Karlseder, J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat. Struct. Mol. Biol. 2012, 19, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.; Cole, R.W.; Rieder, C.L. DNA damage during mitosis in human cells delays the metaphase/anaphase transition via the spindle-assembly checkpoint. Curr. Biol. 2002, 12, 1797–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audrey, A.; de Haan, L.; van Vugt, M.; de Boer, H.R. Processing DNA lesions during mitosis to prevent genomic instability. Biochem. Soc. Trans. 2022, 50, 1105–1118. [Google Scholar] [CrossRef] [PubMed]
- Giunta, S.; Belotserkovskaya, R.; Jackson, S.P. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 2010, 190, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Leimbacher, P.A.; Jones, S.E.; Shorrocks, A.K.; de Marco Zompit, M.; Day, M.; Blaauwendraad, J.; Bundschuh, D.; Bonham, S.; Fischer, R.; Fink, D.; et al. MDC1 Interacts with TOPBP1 to Maintain Chromosomal Stability during Mitosis. Mol. Cell 2019, 74, 571–583.e8. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Suzuki, K.; Kodama, S.; Watanabe, M. Phosphorylated histone H2AX foci persist on rejoined mitotic chromosomes in normal human diploid cells exposed to ionizing radiation. Radiat. Res. 2006, 165, 269–276. [Google Scholar] [CrossRef]
- Van den Berg, J.; Manjón, A.G.; Kielbassa, K.; Feringa, F.M.; Freire, R.; Medema, R.H. A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res. 2018, 46, 10132–10144. [Google Scholar] [CrossRef]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Deacon, J.; Peckham, M.J.; Steel, G.G. The radioresponsiveness of human tumours and the initial slope of the cell survival curve. Radiother. Oncol. 1984, 2, 317–323. [Google Scholar] [CrossRef]
- Fertil, B.; Dertinger, H.; Courdi, A.; Malaise, E.P. Mean inactivation dose: A useful concept for intercomparison of human cell survival curves. Radiat. Res. 1984, 99, 73–84. [Google Scholar] [CrossRef]
- Fertil, B.; Malaise, E.P. Inherent cellular radiosensitivity as a basic concept for human tumor radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1981, 7, 621–629. [Google Scholar] [CrossRef]
- Fertil, B.; Malaise, E.P. Intrinsic radiosensitivity of human cell lines is correlated with radioresponsiveness of human tumors: Analysis of 101 published survival curves. Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 1699–1707. [Google Scholar] [CrossRef]
- Gampa, G.; Kenchappa, R.S.; Mohammad, A.S.; Parrish, K.E.; Kim, M.; Crish, J.F.; Luu, A.; West, R.; Hinojosa, A.Q.; Sarkaria, J.N.; et al. Enhancing Brain Retention of a KIF11 Inhibitor Significantly Improves its Efficacy in a Mouse Model of Glioblastoma. Sci. Rep. 2020, 10, 6524. [Google Scholar] [CrossRef] [Green Version]
- Aiyappa-Maudsley, R.; Chalmers, A.J.; Parsons, J.L. Factors affecting the radiation response in glioblastoma. Neurooncol. Adv. 2022, 4, vdac156. [Google Scholar] [CrossRef]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef]
- Matsui, J.K.; Perlow, H.K.; Ritter, A.R.; Upadhyay, R.; Raval, R.R.; Thomas, E.M.; Beyer, S.J.; Pillainayagam, C.; Goranovich, J.; Ong, S.; et al. Small Molecules and Immunotherapy Agents for Enhancing Radiotherapy in Glioblastoma. Biomedicines 2022, 10, 1763. [Google Scholar] [CrossRef]
- McAleavey, P.G.; Walls, G.M.; Chalmers, A.J. Radiotherapy-drug combinations in the treatment of glioblastoma: A brief review. CNS Oncol. 2022, 11, CNS86. [Google Scholar] [CrossRef]
- Garcia-Saez, I.; Skoufias, D.A. Eg5 targeting agents: From new anti-mitotic based inhibitor discovery to cancer therapy and resistance. Biochem. Pharmacol. 2021, 184, 114364. [Google Scholar] [CrossRef]
- Jiang, C.; You, Q. Kinesin spindle protein inhibitors in cancer: A patent review (2008–present). Expert Opin. Ther. Pat 2013, 23, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.J.; Garcia-Manero, G.; Borthakur, G.; Kadia, T.; Foudray, M.C.; Arellano, M.; Langston, A.; Bethelmie-Bryan, B.; Rush, S.; Litwiler, K.; et al. A phase 1 dose-escalation study of ARRY-520, a kinesin spindle protein inhibitor, in patients with advanced myeloid leukemias. Cancer 2012, 118, 3556–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Shah, J.J.; Feng, L.; Manasanch, E.E.; Lu, R.; Morphey, A.; Crumpton, B.; Patel, K.K.; Wang, M.L.; Alexanian, R.; et al. A phase 1 study of filanesib, carfilzomib, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Blood Cancer J. 2019, 9, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocio, E.M.; Motllo, C.; Rodriguez-Otero, P.; Martinez-Lopez, J.; Cejalvo, M.J.; Martin-Sanchez, J.; Blade, J.; Garcia-Malo, M.D.; Dourdil, M.V.; Garcia-Mateo, A.; et al. Filanesib in combination with pomalidomide and dexamethasone in refractory MM patients: Safety and efficacy, and association with alpha 1-acid glycoprotein (AAG) levels. Phase Ib/II Pomdefil clinical trial conducted by the Spanish MM group. Br. J. Haematol. 2021, 192, 522–530. [Google Scholar] [CrossRef]
- Masanas, M.; Masia, N.; Suarez-Cabrera, L.; Olivan, M.; Soriano, A.; Majem, B.; Devis-Jauregui, L.; Burgos-Panadero, R.; Jimenez, C.; Rodriguez-Sodupe, P.; et al. The oral KIF11 inhibitor 4SC-205 exhibits antitumor activity and potentiates standard and targeted therapies in primary and metastatic neuroblastoma models. Clin. Transl. Med. 2021, 11, e533. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tallman, M.M.; Zalenski, A.A.; Stabl, I.; Schrock, M.S.; Kollin, L.; de Jong, E.; De, K.; Grubb, T.M.; Summers, M.K.; Venere, M. Improving Localized Radiotherapy for Glioblastoma via Small Molecule Inhibition of KIF11. Cancers 2023, 15, 3173. https://doi.org/10.3390/cancers15123173
Tallman MM, Zalenski AA, Stabl I, Schrock MS, Kollin L, de Jong E, De K, Grubb TM, Summers MK, Venere M. Improving Localized Radiotherapy for Glioblastoma via Small Molecule Inhibition of KIF11. Cancers. 2023; 15(12):3173. https://doi.org/10.3390/cancers15123173
Chicago/Turabian StyleTallman, Miranda M., Abigail A. Zalenski, Ian Stabl, Morgan S. Schrock, Luke Kollin, Eliane de Jong, Kuntal De, Treg M. Grubb, Matthew K. Summers, and Monica Venere. 2023. "Improving Localized Radiotherapy for Glioblastoma via Small Molecule Inhibition of KIF11" Cancers 15, no. 12: 3173. https://doi.org/10.3390/cancers15123173
APA StyleTallman, M. M., Zalenski, A. A., Stabl, I., Schrock, M. S., Kollin, L., de Jong, E., De, K., Grubb, T. M., Summers, M. K., & Venere, M. (2023). Improving Localized Radiotherapy for Glioblastoma via Small Molecule Inhibition of KIF11. Cancers, 15(12), 3173. https://doi.org/10.3390/cancers15123173