
Lactate as Key Metabolite in Prostate Cancer Progression: What Are the Clinical Implications?


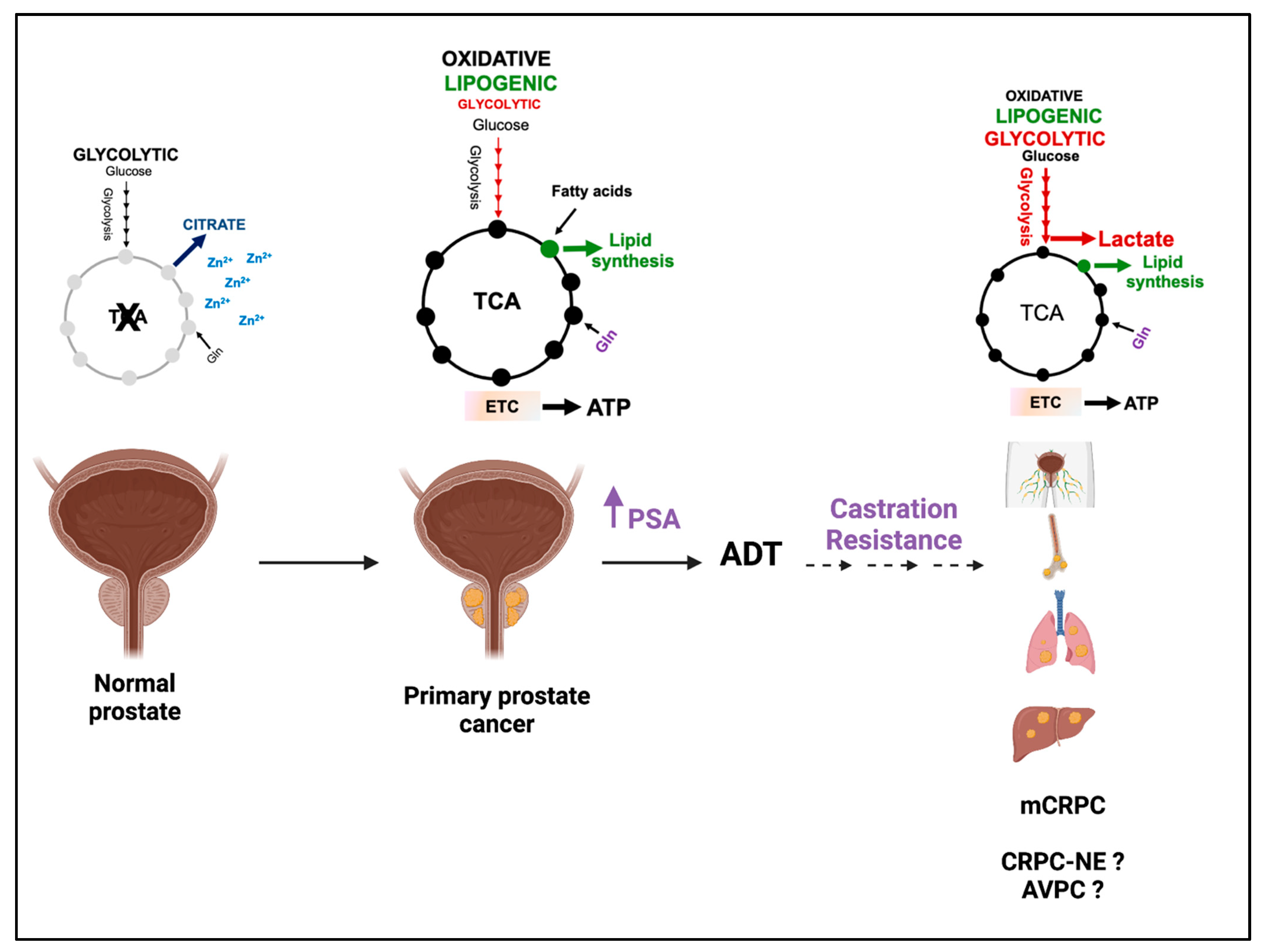{kind=link}
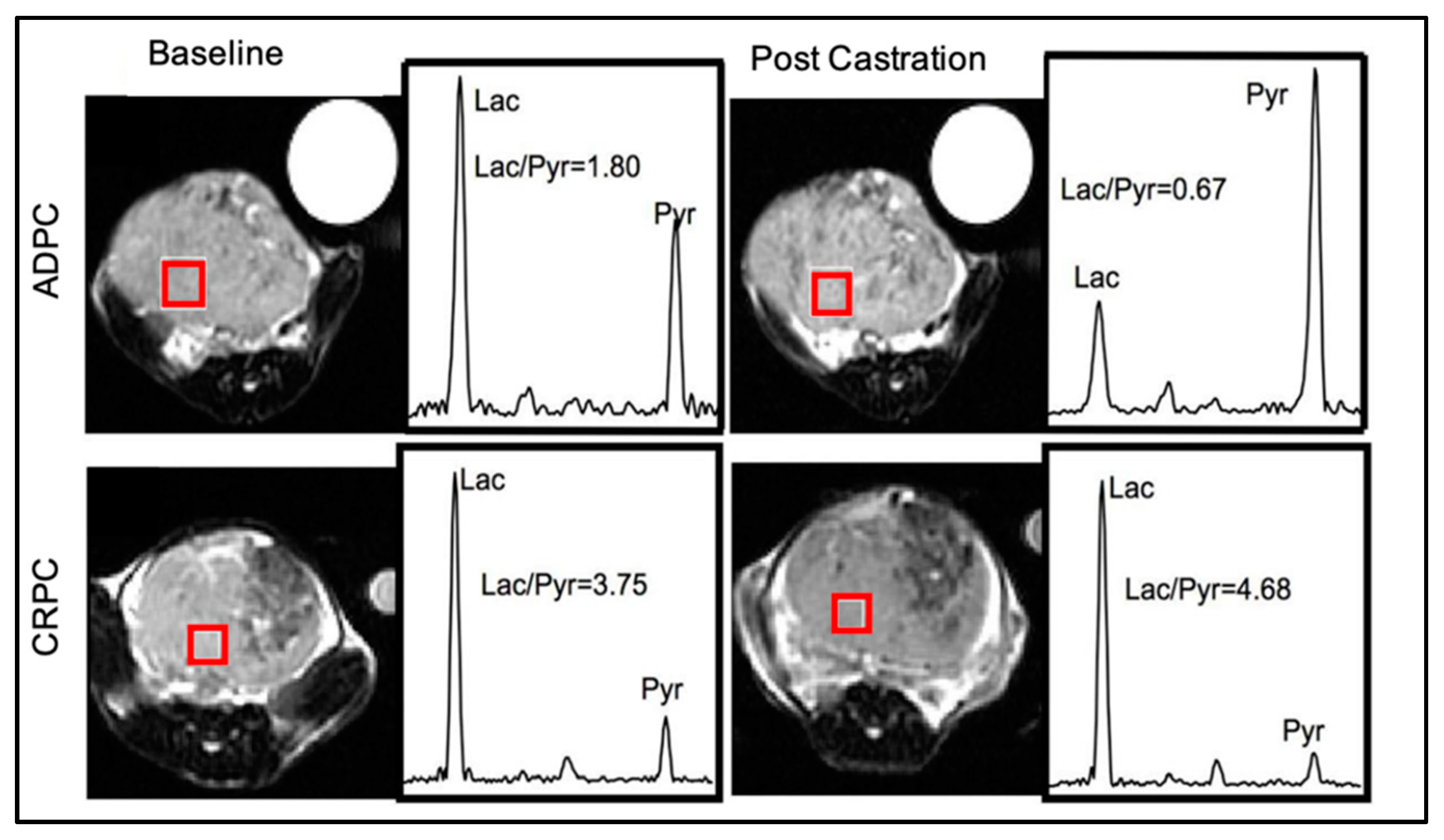{kind=link}
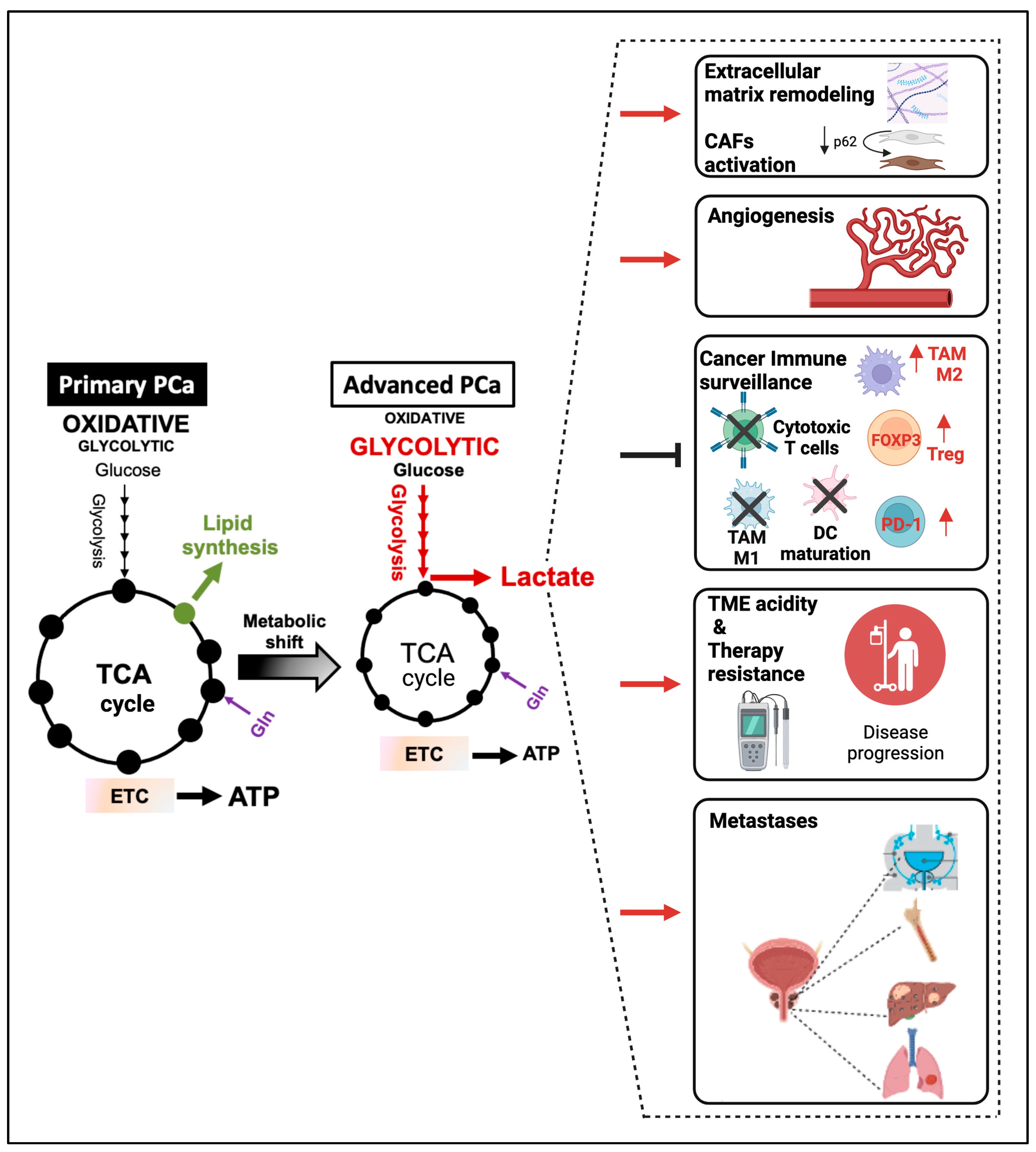{kind=link}
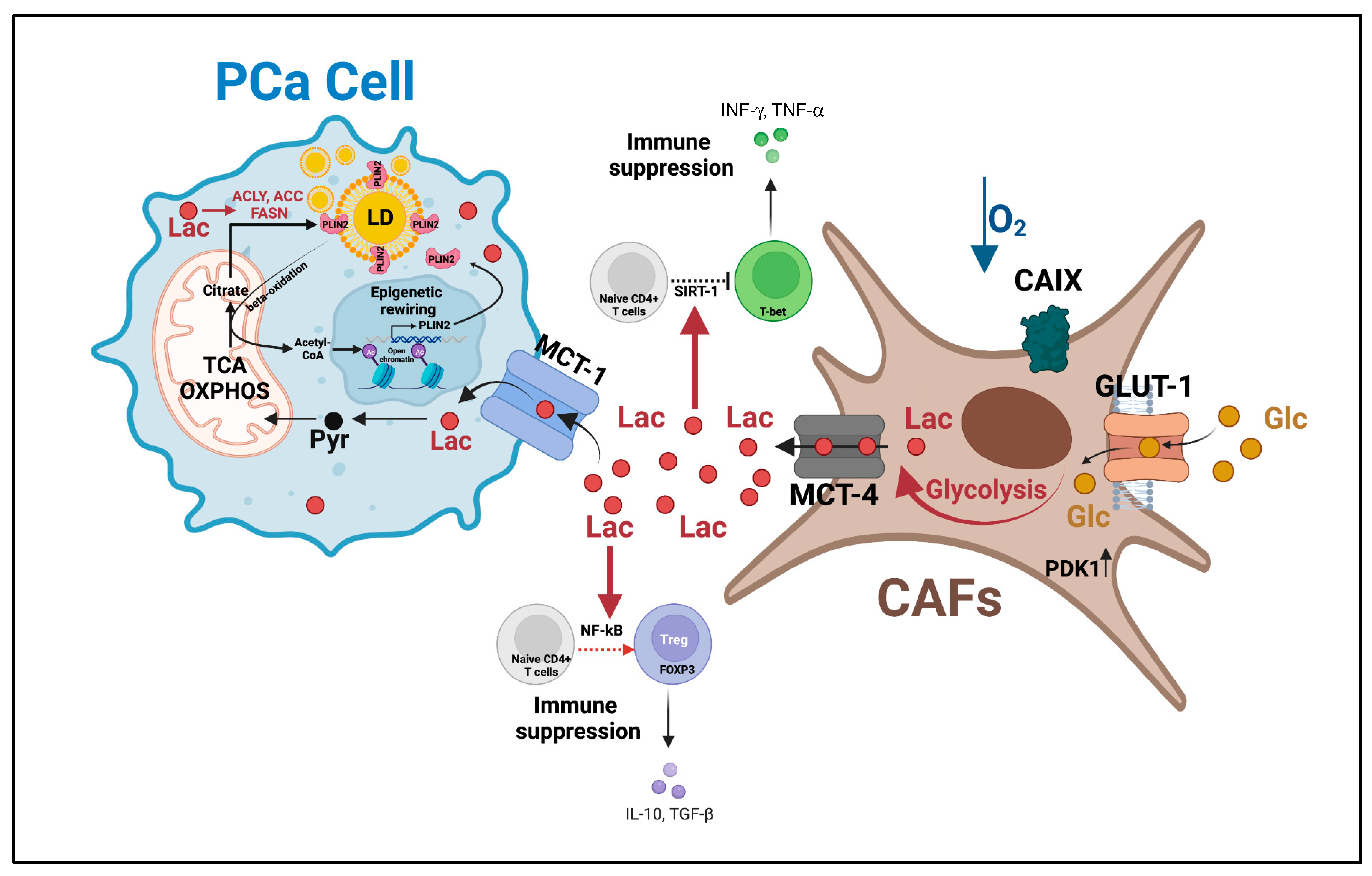{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Lactate Concentration in PCa
3. Lactate Synthesis/Transport and PCa Clinical Outcomes
4. Regulation of Aerobic Glycolysis and Lactate Synthesis during PCa Progression
4.1. Androgen-Receptor Signaling Promotes Aerobic Glycolysis and Lactate Synthesis during PCa Progression
4.2. Alterations in Oncogenes and Tumor Suppressors Modulate Aerobic Glycolysis and Lactate Synthesis in CRPC
5. PCa-Released Lactate Functions as a TME Modulator and Signaling Molecule
5.1. PCa-Derived Lactate Induces an Immunosuppressive and Metastasis-Supportive TME
5.2. Lactate as a Signaling Molecule and Modulator of Protein Activity

6. Metabolic Heterogeneity Supports “Symbiotic” Interactions through Lactate Shuttling to Sustain PCa Progression
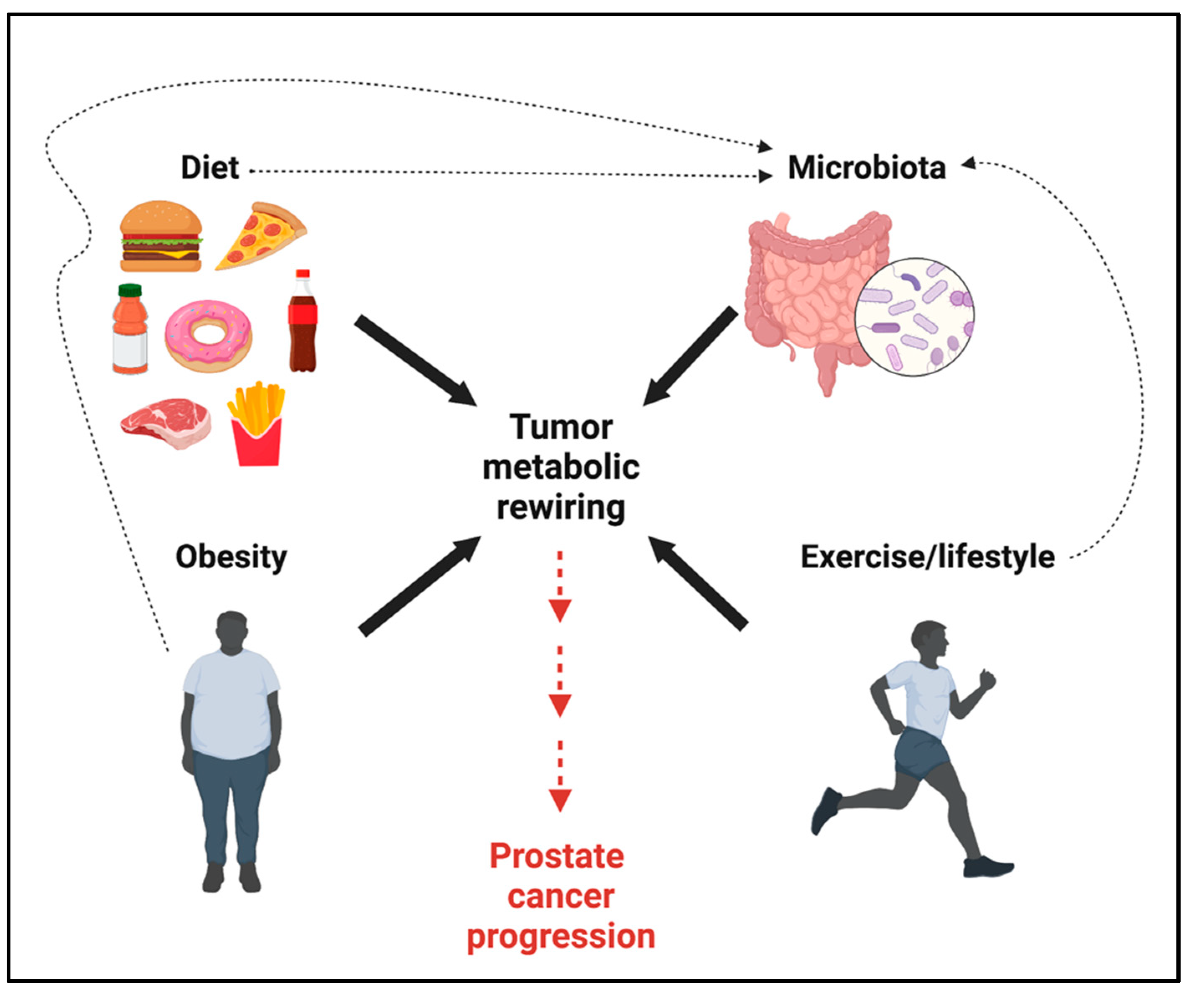
7. Systemic Metabolism and Gut Microbiota Affect Lactate Levels and PCa Progression
8. Imaging Glucose Uptake and Pyruvate-to-Lactate Conversion as Diagnostic, Prognostic, and Predictive Biomarkers
8.1. Preclinical/Ex-Vivo Studies
8.2. Clinical Studies
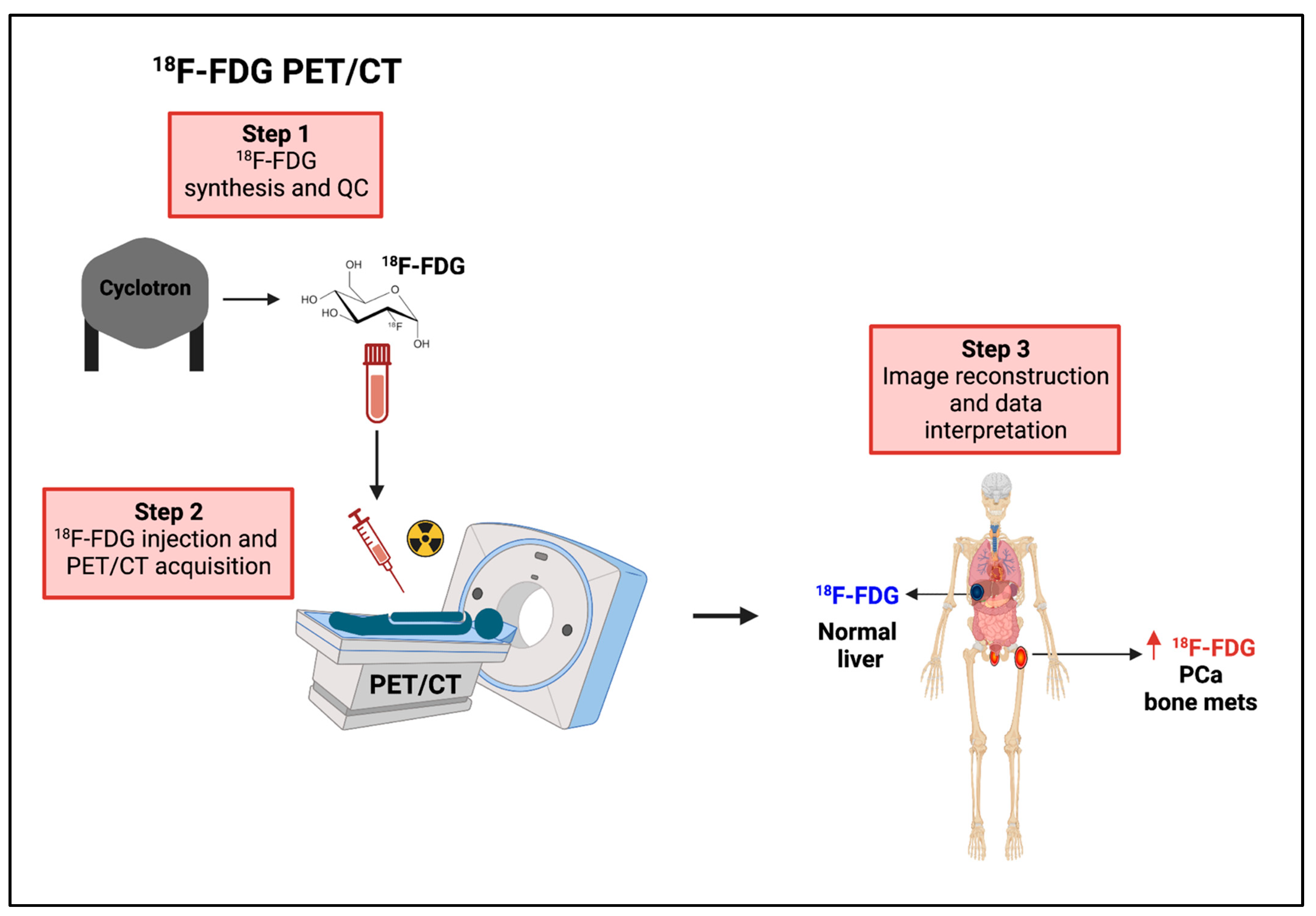
8.2.1. 18F-FDG PET
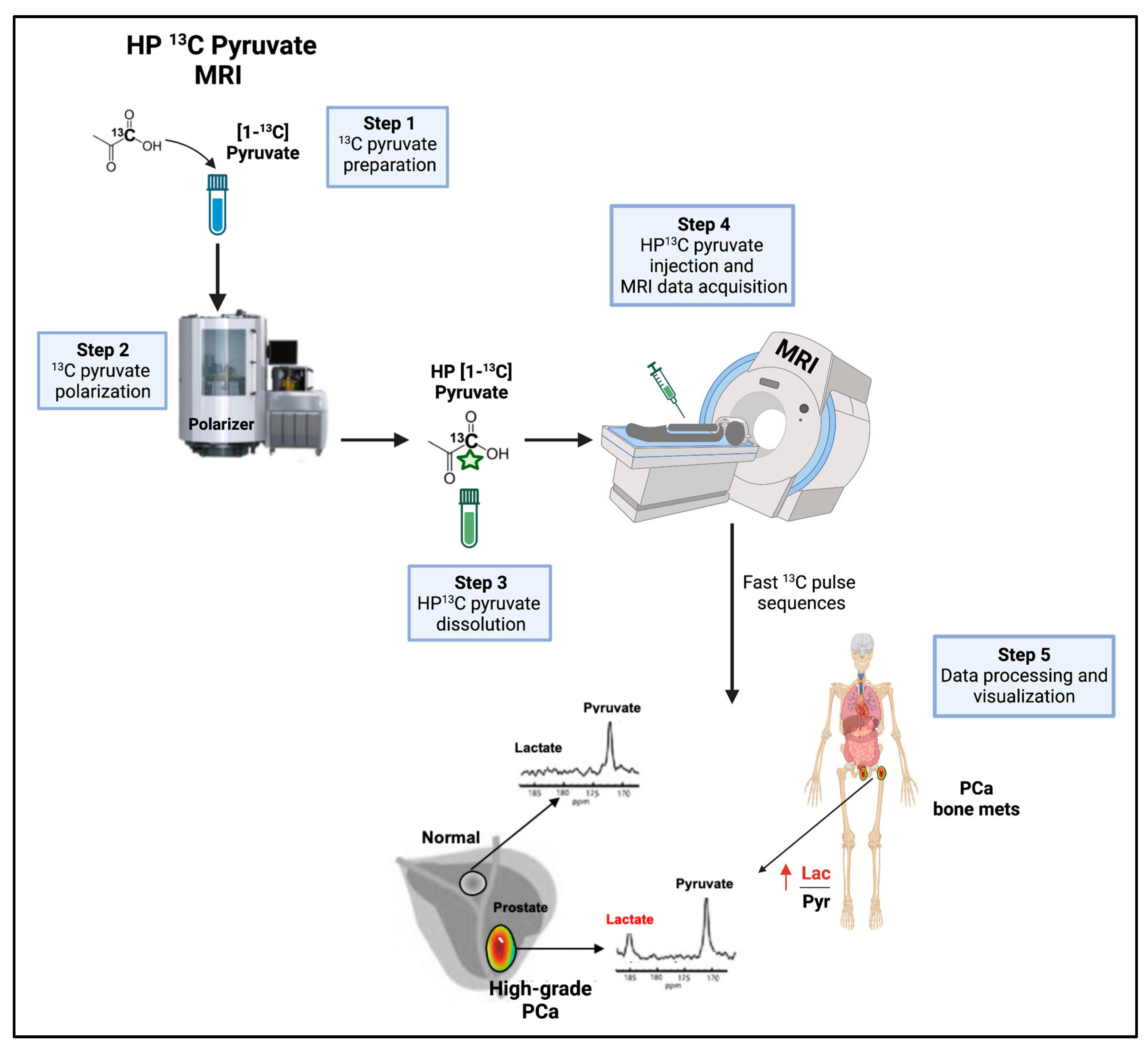
8.2.2. HP 13C Pyruvate MRSI
9. Targeting Lactate Metabolism: New Therapeutic Opportunities
10. Future Prospective
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADT | Androgen deprivation therapy |
| AR | Androgen receptor |
| ATM | Ataxia-telangiectasia mutated |
| ATP | Adenosine triphosphate |
| BID | bis in die (twice a day) |
| BRCA 1 | Breast cancer gene 1 |
| BRCA 2 | Breast cancer gene 2 |
| CAFs | Cancer-associated fibroblasts |
| CaMKK2 | Calcium/calmodulin-dependent kinase kinase 2 |
| CRPC | Castration-resistant prostate cancer |
| CT | Computed tomography |
| DNA-PK | DNA-dependent protein kinase |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| FAs | Fatty acids |
| FASN | Fatty acid synthase |
| 18F-FDG | 18F-fluorodeoxyglucose |
| FGFR1 | Fibroblast growth factor receptor |
| GEMM | Genetically engineered mouse model |
| GLUT1 | Glucose transporter 1 |
| HCAR1 | Hydroxycarboxylic acid receptor 1 |
| HIF-1a | Hypoxia-inducible factor-1 |
| HP | Hyperpolarized |
| HR-MAS | High resolution magic angle spinning |
| ICIs | Immune checkpoint inhibitors |
| LDHA | Lactate dehydrogenase A |
| MALDI-MSI | Matrix-assisted laser desorption/ionization-mass spectrometry imaging |
| MCTs | Monocarboxylate transporters |
| CAFs | Cancer-associated fibroblast |
| MRI | Magnetic resonance imaging |
| MRSI | Magnetic resonance spectroscopic imaging |
| MS | Mass spectrometry |
| NAD | Nicotinamide adenine dinucleotide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NDRG3 | N-MYC downstream-regulated gene 3 |
| NF-KB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK | Natural killer cells |
| NMR | Nuclear magnetic resonance |
| PARP | Poly(ADP-ribose)polymerase |
| PCa | Prostate cancer |
| PDX | Patient-derived xenografts |
| PET | Positron emission tomography |
| PHD2 | HIF prolyl hydroxylase domain-2 |
| PI3K | Phosphoinositide 3-kinase |
| PTEN | Phosphatase and tensin homolog |
| RB1 | RB transcriptional corepressor 1 |
| ROS | Reactive oxygen species |
| SCFAs | Short-chain fatty acids |
| SREBP | Sterol regulatory element-binding proteins |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Malvezzi, M.; Santucci, C.; Boffetta, P.; Collatuzzo, G.; Levi, F.; La Vecchia, C.; Negri, E. European Cancer Mortality Predictions for the Year 2023 with Focus on Lung Cancer. Ann. Oncol. 2023, 34, 410–419. [Google Scholar] [CrossRef]
- Buxton, A.K.; Abbasova, S.; Bevan, C.L.; Leach, D.A. Liver Microenvironment Response to Prostate Cancer Metastasis and Hormonal Therapy. Cancers 2022, 14, 6189. [Google Scholar] [CrossRef] [PubMed]
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Part II-2020 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur. Urol. 2021, 79, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Tilki, D.; Schaeffer, E.M.; Evans, C.P. Understanding Mechanisms of Resistance in Metastatic Castration-resistant Prostate Cancer: The Role of the Androgen Receptor. Eur. Urol. Focus 2016, 2, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Einstein, D.J.; Arai, S.; Balk, S.P. Targeting the androgen receptor and overcoming resistance in prostate cancer. Curr. Opin. Oncol. 2019, 31, 175–182. [Google Scholar] [CrossRef]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. JCO Oncol. Pract. 2022, 18, 45–55. [Google Scholar] [CrossRef]
- Nakazawa, M.; Paller, C.; Kyprianou, N. Mechanisms of Therapeutic Resistance in Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Wang, I.; Song, L.; Wang, B.Y.; Rezazadeh Kalebasty, A.; Uchio, E.; Zi, X. Prostate cancer immunotherapy: A review of recent advancements with novel treatment methods and efficacy. Am. J. Clin. Exp. Urol. 2022, 10, 210–233. [Google Scholar] [PubMed]
- Chen, J.; Shi, M.; Chuen Choi, S.Y.; Wang, Y.; Lin, D.; Zeng, H.; Wang, Y. Genomic alterations in neuroendocrine prostate cancer: A systematic review and meta-analysis. BJUI Compass 2023, 4, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Vlachostergios, P.J.; Beltran, H. Neuroendocrine Differentiation in Prostate Cancer: Emerging Biology, Models, and Therapies. Cold Spring Harb. Perspect. Med. 2019, 9, a030593. [Google Scholar] [CrossRef]
- Uo, T.; Sprenger, C.C.; Plymate, S.R. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front. Oncol. 2020, 10, 580617. [Google Scholar] [CrossRef]
- Chetta, P.; Zadra, G. Metabolic reprogramming as an emerging mechanism of resistance to endocrine therapies in prostate cancer. Cancer Drug Resist. 2021, 4, 143–162. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Citrate metabolism of normal and malignant prostate epithelial cells. Urology 1997, 50, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. The intermediary metabolism of the prostate: A key to understanding the pathogenesis and progression of prostate malignancy. Oncology 2000, 59, 269–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyama, N.; Akino, H.; Kanamaru, H.; Suzuki, Y.; Muramoto, S.; Yonekura, Y.; Okada, K. 11C-acetate PET imaging of prostate cancer. J. Nucl. Med. 2002, 43, 181–186. [Google Scholar]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Mah, C.Y.; Nassar, Z.D.; Swinnen, J.V.; Butler, L.M. Lipogenic effects of androgen signaling in normal and malignant prostate. Asian J. Urol. 2020, 7, 258–270. [Google Scholar] [CrossRef]
- Lin, C.; Salzillo, T.C.; Bader, D.A.; Wilkenfeld, S.R.; Awad, D.; Pulliam, T.L.; Dutta, P.; Pudakalakatti, S.; Titus, M.; McGuire, S.E.; et al. Prostate Cancer Energetics and Biosynthesis. Adv. Exp. Med. Biol. 2019, 1210, 185–237. [Google Scholar] [CrossRef]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 2019, 129, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaglia, N.; Frontini-Lopez, Y.R.; Zadra, G. Prostate Cancer Progression: As a Matter of Fats. Front. Oncol. 2021, 11, 719865. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Gavane, S.; Tarlinton, L.; Fareedy, S.B.; Doran, M.G.; Zelefsky, M.J.; Osborne, J.R. Utility of FDG-PET in clinical neuroendocrine prostate cancer. Prostate 2014, 74, 1153–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.L.; Yin, J.J.; Seng, V.; Casey, O.; Corey, E.; Morrissey, C.; Simpson, R.M.; Kelly, K. Androgen deprivation leads to increased carbohydrate metabolism and hexokinase 2-mediated survival in Pten/Tp53-deficient prostate cancer. Oncogene 2017, 36, 525–533. [Google Scholar] [CrossRef]
- Fox, J.J.; Gavane, S.C.; Blanc-Autran, E.; Nehmeh, S.; Gonen, M.; Beattie, B.; Vargas, H.A.; Schöder, H.; Humm, J.L.; Fine, S.W.; et al. Positron Emission Tomography/Computed Tomography-Based Assessments of Androgen Receptor Expression and Glycolytic Activity as a Prognostic Biomarker for Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 217–224. [Google Scholar] [CrossRef]
- Xiao, H.; Wang, J.; Yan, W.; Cui, Y.; Chen, Z.; Gao, X.; Chen, J. GLUT1 regulates cell glycolysis and proliferation in prostate cancer. Prostate 2018, 78, 86–94. [Google Scholar] [CrossRef]
- Bakht, M.K.; Lovnicki, J.M.; Tubman, J.; Stringer, K.F.; Chiaramonte, J.; Reynolds, M.R.; Porter, L.A. Differential Expression of Glucose Transporters and Hexokinases in Prostate Cancer with a Neuroendocrine Gene Signature: A Mechanistic Perspective for (18)F-FDG Imaging of PSMA-Suppressed Tumors. J. Nucl. Med. 2020, 61, 904–910. [Google Scholar] [CrossRef]
- Meziou, S.; Ringuette Goulet, C.; Hovington, H.; Lefebvre, V.; Lavallee, E.; Bergeron, M.; Brisson, H.; Champagne, A.; Neveu, B.; Lacombe, D.; et al. GLUT1 expression in high-risk prostate cancer: Correlation with (18)F-FDG-PET/CT and clinical outcome. Prostate Cancer Prostatic Dis. 2020, 23, 441–448. [Google Scholar] [CrossRef]
- Zacharias, N.; Lee, J.; Ramachandran, S.; Shanmugavelandy, S.; McHenry, J.; Dutta, P.; Millward, S.; Gammon, S.; Efstathiou, E.; Troncoso, P.; et al. Androgen Receptor Signaling in Castration-Resistant Prostate Cancer Alters Hyperpolarized Pyruvate to Lactate Conversion and Lactate Levels In Vivo. Mol. Imaging Biol. 2019, 21, 86–94. [Google Scholar] [CrossRef]
- Crowell, P.D.; Giafaglione, J.M.; Jones, A.E.; Nunley, N.M.; Hashimoto, T.; Delcourt, A.M.L.; Petcherski, A.; Bernard, M.J.; Huang, R.R.; Low, J.-Y.; et al. Androgen receptor inhibition induces metabolic reprogramming and increased reliance on oxidative mitochondrial metabolism in prostate cancer. bioRxiv 2022, 494200. [Google Scholar] [CrossRef]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.S.; Jin, W.J.; Kwak, J.H.; Kim, H.J.; Yun, M.J.; Kim, J.W.; Park, S.W.; Kim, K.S. Androgen stimulates glycolysis for de novo lipid synthesis by increasing the activities of hexokinase 2 and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 2 in prostate cancer cells. Biochem. J. 2011, 433, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Diedrich, J.D.; Rajagurubandara, E.; Herroon, M.K.; Mahapatra, G.; Huttemann, M.; Podgorski, I. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1alpha activation. Oncotarget 2016, 7, 64854–64877. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wang, J.; Chen, L.; Wang, H.; Liang, C.Z.; Huang, J.; Xu, L.F. The role of glutamine metabolism in castration-resistant prostate cancer. Asian J. Androl. 2023, 25, 192–197. [Google Scholar] [CrossRef]
- Bhowmick, N.; Posadas, E.; Ellis, L.; Freedland, S.J.; Vizio, D.D.; Freeman, M.R.; Theodorescu, D.; Figlin, R.; Gong, J. Targeting Glutamine Metabolism in Prostate Cancer. Front. Biosci. 2023, 15, 2. [Google Scholar] [CrossRef]
- De la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Walenta, S.; Salameh, A.; Lyng, H.; Evensen, J.F.; Mitze, M.; Rofstad, E.K.; Mueller-Klieser, W. Correlation of high lactate levels in head and neck tumors with incidence of metastasis. Am. J. Pathol. 1997, 150, 409–415. [Google Scholar]
- Tessem, M.B.; Swanson, M.G.; Keshari, K.R.; Albers, M.J.; Joun, D.; Tabatabai, Z.L.; Simko, J.P.; Shinohara, K.; Nelson, S.J.; Vigneron, D.B.; et al. Evaluation of lactate and alanine as metabolic biomarkers of prostate cancer using 1H HR-MAS spectroscopy of biopsy tissues. Magn. Reson. Med. 2008, 60, 510–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, R.; Van Criekinge, M.; DeLos Santos, J.; Ahamed, F.; Qin, H.; Nolley, R.; Santos, R.D.; Tabatabai, Z.L.; Bok, R.A.; Keshari, K.R.; et al. Elevated Tumor Lactate and Efflux in High-grade Prostate Cancer demonstrated by Hyperpolarized (13)C Magnetic Resonance Spectroscopy of Prostate Tissue Slice Cultures. Cancers 2020, 12, 537. [Google Scholar] [CrossRef] [Green Version]
- Bancroft Brown, J.; Sriram, R.; VanCriekinge, M.; Delos Santos, R.; Sun, J.; Delos Santos, J.; Tabatabai, Z.L.; Shinohara, K.; Nguyen, H.; Peehl, D.M.; et al. NMR quantification of lactate production and efflux and glutamate fractional enrichment in living human prostate biopsies cultured with [1,6-(13) C(2) ]glucose. Magn. Reason. Med. 2019, 82, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar]
- Sharma, D.; Singh, M.; Rani, R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin. Cancer Biol. 2022, 87, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.; Eddy, E.M.; Duan, C.; Odet, F. LDHC: The ultimate testis-specific gene. J. Androl. 2010, 31, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Felisbino, S.; Massie, C.E.; Vizcaino, J.R.; Coelho, R.; Sandi, C.; Simoes-Sousa, S.; Jurmeister, S.; Ramos-Montoya, A.; Asim, M.; et al. A glycolytic phenotype is associated with prostate cancer progression and aggressiveness: A role for monocarboxylate transporters as metabolic targets for therapy. J. Pathol. 2015, 236, 517–530. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chen, G.; Liu, Z.; Liu, S.; Cai, Z.; You, P.; Ke, Y.; Lai, L.; Huang, Y.; Gao, H.; et al. Aberrant FGFR Tyrosine Kinase Signaling Enhances the Warburg Effect by Reprogramming LDH Isoform Expression and Activity in Prostate Cancer. Cancer Res. 2018, 78, 4459–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Kimura, S.; Parizi, M.K.; Enikeev, D.V.; Glybochko, P.V.; Seebacher, V.; Fajkovic, H.; Mostafaei, H.; Lysenko, I.; Janisch, F.; et al. Prognostic Value of Lactate Dehydrogenase in Metastatic Prostate Cancer: A Systematic Review and Meta-analysis. Clin. Genitourin. Cancer 2019, 17, 409–418. [Google Scholar] [CrossRef]
- Pertega-Gomes, N.; Vizcaino, J.R.; Miranda-Goncalves, V.; Pinheiro, C.; Silva, J.; Pereira, H.; Monteiro, P.; Henrique, R.M.; Reis, R.M.; Lopes, C.; et al. Monocarboxylate transporter 4 (MCT4) and CD147 overexpression is associated with poor prognosis in prostate cancer. BMC Cancer 2011, 11, 312. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.; et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [Green Version]
- Shafi, A.A.; Putluri, V.; Arnold, J.M.; Tsouko, E.; Maity, S.; Roberts, J.M.; Coarfa, C.; Frigo, D.E.; Putluri, N.; Sreekumar, A.; et al. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget 2015, 6, 31997–32012. [Google Scholar] [CrossRef] [Green Version]
- Palanisamy, N.; Yang, J.; Shepherd, P.D.A.; Li-Ning-Tapia, E.M.; Labanca, E.; Manyam, G.C.; Ravoori, M.K.; Kundra, V.; Araujo, J.C.; Efstathiou, E.; et al. The MD Anderson Prostate Cancer Patient-derived Xenograft Series (MDA PCa PDX) Captures the Molecular Landscape of Prostate Cancer and Facilitates Marker-driven Therapy Development. Clin. Cancer Res. 2020, 26, 4933–4946. [Google Scholar] [CrossRef]
- Tzelepi, V.; Zhang, J.; Lu, J.F.; Kleb, B.; Wu, G.; Wan, X.; Hoang, A.; Efstathiou, E.; Sircar, K.; Navone, N.M.; et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin. Cancer Res. 2012, 18, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Bok, R.A.; DeLos Santos, J.; Upadhyay, D.; DeLos Santos, R.; Agarwal, S.; Van Criekinge, M.; Vigneron, D.B.; Aggarwal, R.; Peehl, D.M.; et al. Resistance to Androgen Deprivation Leads to Altered Metabolism in Human and Murine Prostate Cancer Cell and Tumor Models. Metabolites 2021, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Ellwood-Yen, K.; Graeber, T.G.; Wongvipat, J.; Iruela-Arispe, M.L.; Zhang, J.; Matusik, R.; Thomas, G.V.; Sawyers, C.L. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 2003, 4, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xiong, H.; Wu, F.; Zhang, Y.; Wang, J.; Zhao, L.; Guo, X.; Chang, L.J.; Zhang, Y.; You, M.J.; et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 2014, 8, 1461–1474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Chen, W.G.; Li, X.W. MicroRNA-143 acts as a tumor suppressor by targeting hexokinase 2 in human prostate cancer. Am. J. Cancer Res. 2015, 5, 2056–2063. [Google Scholar]
- Priolo, C.; Pyne, S.; Rose, J.; Regan, E.R.; Zadra, G.; Photopoulos, C.; Cacciatore, S.; Schultz, D.; Scaglia, N.; McDunn, J.; et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res. 2014, 74, 7198–7204. [Google Scholar] [CrossRef] [Green Version]
- Labbe, D.P.; Zadra, G.; Yang, M.; Reyes, J.M.; Lin, C.Y.; Cacciatore, S.; Ebot, E.M.; Creech, A.L.; Giunchi, F.; Fiorentino, M.; et al. High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat. Commun. 2019, 10, 4358. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Ma, E.; Zeng, T.; Zhao, R.; Tao, Y.; Chen, X.; Groth, J.; Liang, C.; Hu, H.; Huang, J. ATM deficiency promotes progression of CRPC by enhancing Warburg effect. Endocr. Relat. Cancer 2019, 26, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Perez-Tomas, R.; Perez-Guillen, I. Lactate in the Tumor Microenvironment: An Essential Molecule in Cancer Progression and Treatment. Cancers 2020, 12, 3244. [Google Scholar] [CrossRef] [PubMed]
- Baltazar, F.; Afonso, J.; Costa, M.; Granja, S. Lactate Beyond a Waste Metabolite: Metabolic Affairs and Signaling in Malignancy. Front. Oncol. 2020, 10, 231. [Google Scholar] [CrossRef] [Green Version]
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol. Ther. 2020, 206, 107451. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Peng, W.B.; Zhang, P.; Yang, X.P.; Zhou, Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine 2021, 73, 103627. [Google Scholar] [CrossRef] [PubMed]
- Heuser, C.; Renner, K.; Kreutz, M.; Gattinoni, L. Targeting lactate metabolism for cancer immunotherapy—A matter of precision. Semin. Cancer Biol. 2023, 88, 32–45. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e7. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudagar, K.; Hieromnimon, H.M.; Khurana, R.; Labadie, B.; Hirz, T.; Mei, S.; Hasan, R.; Shafran, J.; Kelley, A.; Apostolov, E.; et al. Reversal of lactate and PD-1-mediated macrophage immunosuppression controls growth of PTEN/p53-deficient prostate cancer. Clin. Cancer Res. 2023, 29, 1952–1968. [Google Scholar] [CrossRef] [PubMed]
- El-Kenawi, A.; Gatenbee, C.; Robertson-Tessi, M.; Bravo, R.; Dhillon, J.; Balagurunathan, Y.; Berglund, A.; Vishvakarma, N.; Ibrahim-Hashim, A.; Choi, J.; et al. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br. J. Cancer 2019, 121, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Cid-Diaz, T.; Duran, A.; Osrodek, M.; Martinez-Ordonez, A.; Reina-Campos, M.; Kuo, H.H.; Elemento, O.; Martin, M.L.; Cordes, T.; et al. The lactate-NAD(+) axis activates cancer-associated fibroblasts by downregulating p62. Cell Rep. 2022, 39, 110792. [Google Scholar] [CrossRef]
- Stern, R.; Shuster, S.; Neudecker, B.A.; Formby, B. Lactate stimulates fibroblast expression of hyaluronan and CD44: The Warburg effect revisited. Exp. Cell Res. 2002, 276, 24–31. [Google Scholar] [CrossRef]
- Vegran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-kappaB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Copetti, T.; De Saedeleer, C.J.; Vegran, F.; Verrax, J.; Kennedy, K.M.; Moon, E.J.; Dhup, S.; Danhier, P.; Frerart, F.; et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE 2012, 7, e33418. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Yang, Z.; Yu, Y.; Zhang, P. HIF1alpha lactylation enhances KIAA1199 transcription to promote angiogenesis and vasculogenic mimicry in prostate cancer. Int. J. Biol. Macromol. 2022, 222 Pt B, 2225–2243. [Google Scholar] [CrossRef]
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W.; Kania, K.D.; Ciszewski, W.M. Stimulation of lactate receptor (HCAR1) affects cellular DNA repair capacity. DNA Repair 2017, 52, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yang, H.; Zhang, Y.; Wei, H.; Zhu, Z.; Zhu, B.; Yang, M.; Cao, W.; Wang, L.; Wu, Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene 2017, 36, 5829–5839. [Google Scholar] [CrossRef] [PubMed]
- De Saedeleer, C.J.; Copetti, T.; Porporato, P.E.; Verrax, J.; Feron, O.; Sonveaux, P. Lactate activates HIF-1 in oxidative but not in Warburg-phenotype human tumor cells. PLoS ONE 2012, 7, e46571. [Google Scholar] [CrossRef] [Green Version]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koltai, T.; Reshkin, S.J.; Baltazar, F.; Fliegel, L. Carbohydrate metabolism in prostate cancer. In Prostate Cancer Metabolism; Academic Press: Cambridge, MA, USA, 2021; pp. 271–294. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [Green Version]
- Sanita, P.; Capulli, M.; Teti, A.; Galatioto, G.P.; Vicentini, C.; Chiarugi, P.; Bologna, M.; Angelucci, A. Tumor-stroma metabolic relationship based on lactate shuttle can sustain prostate cancer progression. BMC Cancer 2014, 14, 154. [Google Scholar] [CrossRef] [Green Version]
- Pertega-Gomes, N.; Vizcaino, J.R.; Attig, J.; Jurmeister, S.; Lopes, C.; Baltazar, F. A lactate shuttle system between tumour and stromal cells is associated with poor prognosis in prostate cancer. BMC Cancer 2014, 14, 352. [Google Scholar] [CrossRef]
- Ippolito, L.; Comito, G.; Parri, M.; Iozzo, M.; Duatti, A.; Virgilio, F.; Lorito, N.; Bacci, M.; Pardella, E.; Sandrini, G.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Montagnani, I.; Raspollini, M.R.; Serni, S.; Simeoni, L. Lactate modulates CD4(+) T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef]
- Ippolito, L.; Comito, G.; Parri, M.; Iozzo, M.; Duatti, A.; Virgilio, F.; Lorito, N.; Bacci, M.; Pardella, E.; Sandrini, G.; et al. Lactate Rewires Lipid Metabolism and Sustains a Metabolic-Epigenetic Axis in Prostate Cancer. Cancer Res. 2022, 82, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Duatti, A. Lactate-Induced COL1A1/DDR1 Axis Promotes Prostate Cancer Aggressiveness and Enhances Metastatic Colonization. Ph.D. Thesis, University of Siena, Siena, Italy, 2023. Available online: https://hdl.handle.net/11365/1227854 (accessed on 31 May 2023).
- Olson, O.C.; Quail, D.F.; Joyce, J.A. Obesity and the tumor microenvironment. Science 2017, 358, 1130–1131. [Google Scholar] [CrossRef]
- Ringel, A.E.; Drijvers, J.M.; Baker, G.J.; Catozzi, A.; Garcia-Canaveras, J.C.; Gassaway, B.M.; Miller, B.C.; Juneja, V.R.; Nguyen, T.H.; Joshi, S.; et al. Obesity Shapes Metabolism in the Tumor Microenvironment to Suppress Anti-Tumor Immunity. Cell 2020, 183, 1848–1866.e26. [Google Scholar] [CrossRef] [PubMed]
- Boufaied, N.; Chetta, P.; Hallal, T.; Cacciatore, S.; Lalli, D.; Luthold, C.; Homsy, K.; Imada, E.L.; Syamala, S.; Photopoulos, C.; et al. High-fat diet fosters c-MYC-driven prostate cancer progression through the accumulation of the oncometabolite lactate and tumor microenvironment remodeling. In Proceedings of the Society for Basic Urologic Research Annual Meeting, Orlando, FL, USA, 10–13 November 2022. [Google Scholar]
- Caffa, I.; Spagnolo, V.; Vernieri, C.; Valdemarin, F.; Becherini, P.; Wei, M.; Brandhorst, S.; Zucal, C.; Driehuis, E.; Ferrando, L.; et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Ligorio, F.; Fuca, G.; Provenzano, L.; Lobefaro, R.; Zanenga, L.; Vingiani, A.; Belfiore, A.; Lorenzoni, A.; Alessi, A.; Pruneri, G.; et al. Exceptional tumour responses to fasting-mimicking diet combined with standard anticancer therapies: A sub-analysis of the NCT03340935 trial. Eur. J. Cancer 2022, 172, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Vernieri, C.; Fuca, G.; Ligorio, F.; Huber, V.; Vingiani, A.; Iannelli, F.; Raimondi, A.; Rinchai, D.; Frige, G.; Belfiore, A.; et al. Fasting-Mimicking Diet Is Safe and Reshapes Metabolism and Antitumor Immunity in Patients with Cancer. Cancer Discov. 2022, 12, 90–107. [Google Scholar] [CrossRef]
- Zhong, W.; Wu, K.; Long, Z.; Zhou, X.; Zhong, C.; Wang, S.; Lai, H.; Guo, Y.; Lv, D.; Lu, J.; et al. Gut dysbiosis promotes prostate cancer progression and docetaxel resistance via activating NF-kappaB-IL6-STAT3 axis. Microbiome 2022, 10, 94. [Google Scholar] [CrossRef]
- Pernigoni, N.; Zagato, E.; Calcinotto, A.; Troiani, M.; Mestre, R.P.; Cali, B.; Attanasio, G.; Troisi, J.; Minini, M.; Mosole, S.; et al. Commensal bacteria promote endocrine resistance in prostate cancer through androgen biosynthesis. Science 2021, 374, 216–224. [Google Scholar] [CrossRef]
- Fujita, K.; Matsushita, M.; Banno, E.; De Velasco, M.A.; Hatano, K.; Nonomura, N.; Uemura, H. Gut microbiome and prostate cancer. Int. J. Urol. 2022, 29, 793–798. [Google Scholar] [CrossRef]
- Reichard, C.A.; Naelitz, B.D.; Wang, Z.; Jia, X.; Li, J.; Stampfer, M.J.; Klein, E.A.; Hazen, S.L.; Sharifi, N. Gut Microbiome-Dependent Metabolic Pathways and Risk of Lethal Prostate Cancer: Prospective Analysis of a PLCO Cancer Screening Trial Cohort. Cancer Epidemiol. Biomark. Prev. 2022, 31, 192–199. [Google Scholar] [CrossRef]
- Louis, P.; Duncan, S.; Sheridan, P.; Walker, A.; Flint, H. Microbial lactate utilisation and the stability of the gut microbiome. Gut Microbiome 2022, 3, E3. [Google Scholar] [CrossRef]
- Matsushita, M.; Fujita, K.; Motooka, D.; Hatano, K.; Fukae, S.; Kawamura, N.; Nonomura, N. The gut microbiota associated with high-Gleason prostate cancer. Cancer Sci. 2021, 112, 3125–3135. [Google Scholar] [CrossRef] [PubMed]
- Liss, M.A.; White, J.R.; Goros, M.; Gelfond, J.; Leach, R.; Johnson-Pais, T.; Lai, Z.; Rourke, E.; Basler, J.; Ankerst, D.; et al. Metabolic Biosynthesis Pathways Identified from Fecal Microbiome Associated with Prostate Cancer. Eur. Urol. 2018, 74, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Fujita, K.; Hayashi, T.; Kayama, H.; Motooka, D.; Hase, H.; Jingushi, K.; Yamamichi, G.; Yumiba, S.; Tomiyama, E.; et al. Gut Microbiota-Derived Short-Chain Fatty Acids Promote Prostate Cancer Growth via IGF1 Signaling. Cancer Res. 2021, 81, 4014–4026. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chi, C.; Fan, L.; Dong, B.; Shao, X.; Xie, S.; Li, M.; Xue, W. The Microbiome of Prostate Fluid Is Associated With Prostate Cancer. Front. Microbiol. 2019, 10, 1664. [Google Scholar] [CrossRef] [Green Version]
- Katongole, P.; Sande, O.J.; Joloba, M.; Reynolds, S.J.; Niyonzima, N. The human microbiome and its link in prostate cancer risk and pathogenesis. Infect. Agents Cancer 2020, 15, 53. [Google Scholar] [CrossRef]
- Miyake, M.; Tatsumi, Y.; Ohnishi, K.; Fujii, T.; Nakai, Y.; Tanaka, N.; Fujimoto, K. Prostate diseases and microbiome in the prostate, gut, and urine. Prostate Int. 2022, 10, 96–107. [Google Scholar] [CrossRef]
- Hurst, R.; Meader, E.; Gihawi, A.; Rallapalli, G.; Clark, J.; Kay, G.L.; Webb, M.; Manley, K.; Curley, H.; Walker, H.; et al. Microbiomes of Urine and the Prostate Are Linked to Human Prostate Cancer Risk Groups. Eur. Urol. Oncol. 2022, 5, 412–419. [Google Scholar] [CrossRef]
- Yu, H.; Meng, H.; Zhou, F.; Ni, X.; Shen, S.; Das, U.N. Urinary microbiota in patients with prostate cancer and benign prostatic hyperplasia. Arch. Med. Sci. 2015, 11, 385–394. [Google Scholar] [CrossRef]
- Nitsch, S.; Hakenberg, O.W.; Heuschkel, M.; Drager, D.; Hildebrandt, G.; Krause, B.J.; Schwarzenbock, S.M. Evaluation of Prostate Cancer with 11C- and 18F-Choline PET/CT: Diagnosis and Initial Staging. J. Nucl. Med. 2016, 57 (Suppl. S3), 38S–42S. [Google Scholar] [CrossRef] [Green Version]
- Emonds, K.M.; Swinnen, J.V.; Lerut, E.; Koole, M.; Mortelmans, L.; Mottaghy, F.M. Evaluation of androgen-induced effects on the uptake of [18F]FDG, [11C]choline and [11C]acetate in an androgen-sensitive and androgen-independent prostate cancer xenograft model. EJNMMI Res. 2013, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Albers, M.J.; Bok, R.; Chen, A.P.; Cunningham, C.H.; Zierhut, M.L.; Zhang, V.Y.; Kurhanewicz, J. Hyperpolarized 13C lactate, pyruvate, and alanine: Noninvasive biomarkers for prostate cancer detection and grading. Cancer Res. 2008, 68, 8607–8615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bok, R.; Lee, J.; Sriram, R.; Keshari, K.; Sukumar, S.; Daneshmandi, S.; Korenchan, D.E.; Flavell, R.R.; Vigneron, D.B.; Kurhanewicz, J.; et al. The Role of Lactate Metabolism in Prostate Cancer Progression and Metastases Revealed by Dual-Agent Hyperpolarized (13)C MRSI. Cancers 2019, 11, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, C.S.; Venkatesh, H.S.; Chaumeil, M.M.; Brandes, A.H.; Vancriekinge, M.; Dafni, H.; Sukumar, S.; Nelson, S.J.; Vigneron, D.B.; Kurhanewicz, J.; et al. Noninvasive detection of target modulation following phosphatidylinositol 3-kinase inhibition using hyperpolarized 13C magnetic resonance spectroscopy. Cancer Res. 2010, 70, 1296–1305. [Google Scholar] [CrossRef] [Green Version]
- Tee, S.S.; Suster, I.; Truong, S.; Jeong, S.; Eskandari, R.; DiGialleonardo, V.; Alvarez, J.A.; Aldeborgh, H.N.; Keshari, K.R. Targeted AKT Inhibition in Prostate Cancer Cells and Spheroids Reduces Aerobic Glycolysis and Generation of Hyperpolarized [1-(13)C] Lactate. Mol. Cancer Res. 2018, 16, 453–460. [Google Scholar] [CrossRef] [Green Version]
- Dafni, H.; Larson, P.E.; Hu, S.; Yoshihara, H.A.; Ward, C.S.; Venkatesh, H.S.; Ronen, S.M. Hyperpolarized 13C spectroscopic imaging informs on hypoxia-inducible factor-1 and myc activity downstream of platelet-derived growth factor receptor. Cancer Res. 2010, 70, 7400–7410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scroggins, B.T.; Matsuo, M.; White, A.O.; Saito, K.; Munasinghe, J.P.; Sourbier, C.; Yamamoto, K.; Diaz, V.; Takakusagi, Y.; Ichikawa, K.; et al. Hyperpolarized [1-13C]-Pyruvate Magnetic Resonance Spectroscopic Imaging of Prostate Cancer In Vivo Predicts Efficacy of Targeting the Warburg Effect. Clin. Cancer Res. 2018, 24, 3137–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Zhang, V.; Bok, R.A.; Santos, R.D.; Cunha, J.A.; Hsu, I.C.; Santos Bs, J.D.; Lee, J.E.; Sukumar, S.; Larson, P.E.Z.; et al. Simultaneous Metabolic and Perfusion Imaging Using Hyperpolarized (13)C MRI Can Evaluate Early and Dose-Dependent Response to Radiation Therapy in a Prostate Cancer Mouse Model. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Liu, B.; Zhou, X.; Ji, Y.; Chen, L.; Wang, Q.; Xue, W. The Evolving Role of (18)F-FDG PET/CT in Diagnosis and Prognosis Prediction in Progressive Prostate Cancer. Front. Oncol. 2021, 11, 683793. [Google Scholar] [CrossRef]
- Chen, R.; Wang, Y.; Zhu, Y.; Shi, Y.; Xu, L.; Huang, G.; Liu, J. The added value of (18)F-FDG PET/CT compared to (68)Ga-PSMA PET/CT in patients with castration-resistant prostate cancer. J. Nucl. Med. 2021, 64, 1–28. [Google Scholar] [CrossRef]
- Afshar-Oromieh, A.; Zechmann, C.M.; Malcher, A.; Eder, M.; Eisenhut, M.; Linhart, H.G.; Holland-Letz, T.; Hadaschik, B.A.; Giesel, F.L.; Debus, J.; et al. Comparison of PET imaging with a (68)Ga-labelled PSMA ligand and (18)F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasikumar, A. Specificity of 68Ga-PSMA PET/CT for Prostate Cancer—Myths and Reality. Indian J. Nucl. Med. 2017, 32, 11–12. [Google Scholar] [CrossRef] [Green Version]
- Perera, M.; Papa, N.; Roberts, M.; Williams, M.; Udovicich, C.; Vela, I.; Christidis, D.; Bolton, D.; Hofman, M.S.; Lawrentschuk, N.; et al. Gallium-68 Prostate-specific Membrane Antigen Positron Emission Tomography in Advanced Prostate Cancer-Updated Diagnostic Utility, Sensitivity, Specificity, and Distribution of Prostate-specific Membrane Antigen-avid Lesions: A Systematic Review and Meta-analysis. Eur. Urol. 2020, 77, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Oyama, N.; Hasegawa, Y.; Kiyono, Y.; Kobayashi, M.; Fujibayashi, Y.; Ponde, D.E.; Dence, C.; Welch, M.J.; Yokoyama, O. Early response assessment in prostate carcinoma by (1)(8)F-fluorothymidine following anticancer therapy with docetaxel using preclinical tumour models. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Lavallee, E.; Bergeron, M.; Buteau, F.A.; Blouin, A.C.; Duchesnay, N.; Dujardin, T.; Tiguert, R.; Lacombe, L.; Fradet, V.; Makao-Nguile, M.; et al. Increased Prostate Cancer Glucose Metabolism Detected by (18)F-fluorodeoxyglucose Positron Emission Tomography/Computed Tomography in Localised Gleason 8-10 Prostate Cancers Identifies Very High-risk Patients for Early Recurrence and Resistance to Castration. Eur. Urol. Focus 2019, 5, 998–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadvar, H.; Velez, E.M.; Desai, B.; Ji, L.; Colletti, P.M.; Quinn, D.I. Prediction of Time to Hormonal Treatment Failure in Metastatic Castration-Sensitive Prostate Cancer with (18)F-FDG PET/CT. J. Nucl. Med. 2019, 60, 1524–1530. [Google Scholar] [CrossRef]
- Bauckneht, M.; Bertagna, F.; Donegani, M.I.; Durmo, R.; Miceli, A.; De Biasi, V.; Laudicella, R.; Fornarini, G.; Berruti, A.; Baldari, S.; et al. The prognostic power of 18F-FDG PET/CT extends to estimating systemic treatment response duration in metastatic castration-resistant prostate cancer (mCRPC) patients. Prostate Cancer Prostatic Dis. 2021, 24, 1198–1207. [Google Scholar] [CrossRef]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.; Harzstark, A.L.; Ferrone, M.; van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-(1)(3)C]pyruvate. Sci. Transl. Med. 2013, 5, 198ra108. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Aggarwal, R.; Bok, R.A.; Ohliger, M.A.; Zhu, Z.; Lee, P.; Gordon, J.W.; van Criekinge, M.; Carvajal, L.; Slater, J.B.; et al. Hyperpolarized (13)C-pyruvate MRI detects real-time metabolic flux in prostate cancer metastases to bone and liver: A clinical feasibility study. Prostate Cancer Prostatic Dis. 2020, 23, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Granlund, K.L.; Tee, S.S.; Vargas, H.A.; Lyashchenko, S.K.; Reznik, E.; Fine, S.; Laudone, V.; Eastham, J.A.; Touijer, K.A.; Reuter, V.E.; et al. Hyperpolarized MRI of Human Prostate Cancer Reveals Increased Lactate with Tumor Grade Driven by Monocarboxylate Transporter 1. Cell Metab. 2020, 31, 105–114.e3. [Google Scholar] [CrossRef]
- Sushentsev, N.; McLean, M.A.; Warren, A.Y.; Benjamin, A.J.V.; Brodie, C.; Frary, A.; Gill, A.B.; Jones, J.; Kaggie, J.D.; Lamb, B.W.; et al. Hyperpolarised (13)C-MRI identifies the emergence of a glycolytic cell population within intermediate-risk human prostate cancer. Nat. Commun. 2022, 13, 466. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Vigneron, D.B.; Kurhanewicz, J. Hyperpolarized 1-[(13)C]-Pyruvate Magnetic Resonance Imaging Detects an Early Metabolic Response to Androgen Ablation Therapy in Prostate Cancer. Eur. Urol. 2017, 72, 1028–1029. [Google Scholar] [CrossRef] [PubMed]
- De Kouchkovsky, I.; Chen, H.Y.; Ohliger, M.A.; Wang, Z.J.; Bok, R.A.; Gordon, J.W.; Larson, P.E.Z.; Frost, M.; Okamoto, K.; Cooperberg, M.R.; et al. Hyperpolarized 1-[(13)C]-Pyruvate Magnetic Resonance Imaging Detects an Early Metabolic Response to Immune Checkpoint Inhibitor Therapy in Prostate Cancer. Eur. Urol. 2022, 81, 219–221. [Google Scholar] [CrossRef]
- Chen, H.Y.; Bok, R.A.; Cooperberg, M.R.; Nguyen, H.G.; Shinohara, K.; Westphalen, A.C.; Wang, Z.J.; Ohliger, M.A.; Gebrezgiabhier, D.; Carvajal, L.; et al. Improving multiparametric MR-transrectal ultrasound guided fusion prostate biopsies with hyperpolarized (13) C pyruvate metabolic imaging: A technical development study. Magn. Reson. Med. 2022, 88, 2609–2620. [Google Scholar] [CrossRef]
- Xian, Z.Y.; Liu, J.M.; Chen, Q.K.; Chen, H.Z.; Ye, C.J.; Xue, J.; Yang, H.Q.; Li, J.L.; Liu, X.F.; Kuang, S.J. Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumour Biol. 2015, 36, 8093–8100. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Nunes, A.; Simoes-Sousa, S.; Pinheiro, C.; Miranda-Goncalves, V.; Granja, S.; Baltazar, F. Targeting lactate production and efflux in prostate cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165894. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Panteliadou, M.; Pouliliou, S.E.; Chondrou, P.S.; Mavropoulou, S.; Sivridis, E. Lactate dehydrogenase 5 isoenzyme overexpression defines resistance of prostate cancer to radiotherapy. Br. J. Cancer 2014, 110, 2217–2223. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, H.; Sumitomo, M.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Watanabe, M.; Zennami, K.; Kanao, K.; et al. Targeting lactate dehydrogenase-A promotes docetaxel-induced cytotoxicity predominantly in castration-resistant prostate cancer cells. Oncol. Rep. 2019, 42, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.N.; Kauer, N.; Blazquez, R.; Hacker, L.; et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef] [Green Version]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e4. [Google Scholar] [CrossRef] [PubMed]
- Renner, O.; Mayer, M.; Leischner, C.; Burkard, M.; Berger, A.; Lauer, U.M.; Venturelli, S.; Bischoff, S.C. Systematic Review of Gossypol/AT-101 in Cancer Clinical Trials. Pharmaceuticals 2022, 15, 144. [Google Scholar] [CrossRef]
- Halford, S.E.R.; Jones, P.; Wedge, S.; Hirschberg, S.; Katugampola, S.; Veal, G.; Payne, G.; Bacon, C.; Potter, S.; Griffin, M.; et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin Cancer Res. 2023, 29, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.D.; Fyrestam, J.; Lanekoff, I. Advances in mass spectrometry based single-cell metabolomics. Analyst 2019, 144, 782–793. [Google Scholar] [CrossRef] [Green Version]
- Lanekoff, I.; Sharma, V.V.; Marques, C. Single-cell metabolomics: Where are we and where are we going? Curr. Opin. Biotechnol. 2022, 75, 102693. [Google Scholar] [CrossRef]
- Hsieh, W.C.; Budiarto, B.R.; Wang, Y.F.; Lin, C.Y.; Gwo, M.C.; So, D.K.; Tzeng, Y.S.; Chen, S. Spatial multi-omics analyses of the tumor immune microenvironment. J. Biomed. Sci. 2022, 29, 96. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chetta, P.; Sriram, R.; Zadra, G. Lactate as Key Metabolite in Prostate Cancer Progression: What Are the Clinical Implications? Cancers 2023, 15, 3473. https://doi.org/10.3390/cancers15133473
Chetta P, Sriram R, Zadra G. Lactate as Key Metabolite in Prostate Cancer Progression: What Are the Clinical Implications? Cancers. 2023; 15(13):3473. https://doi.org/10.3390/cancers15133473
Chicago/Turabian StyleChetta, Paolo, Renuka Sriram, and Giorgia Zadra. 2023. "Lactate as Key Metabolite in Prostate Cancer Progression: What Are the Clinical Implications?" Cancers 15, no. 13: 3473. https://doi.org/10.3390/cancers15133473
APA StyleChetta, P., Sriram, R., & Zadra, G. (2023). Lactate as Key Metabolite in Prostate Cancer Progression: What Are the Clinical Implications? Cancers, 15(13), 3473. https://doi.org/10.3390/cancers15133473