


scGEM: Unveiling the Nested Tree-Structured Gene Co-Expressing Modules in Single Cell Transcriptome Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection and Preprocessing
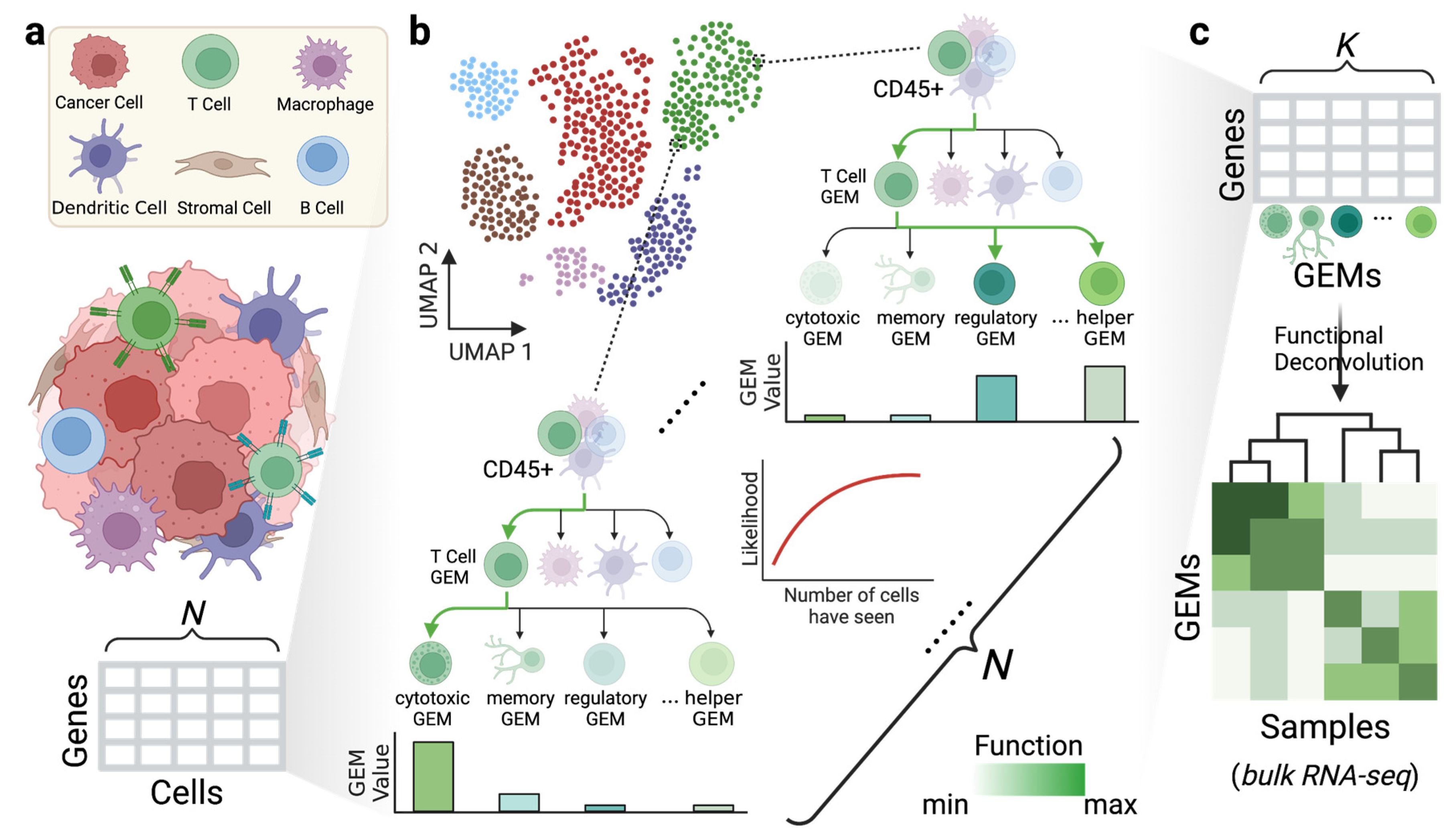
2.2. Tree-Structured Gene Co-Expressing Modules
| Algorithm 1: scGEM Tree initialization |
|
2.3. Model Performance Evaluation
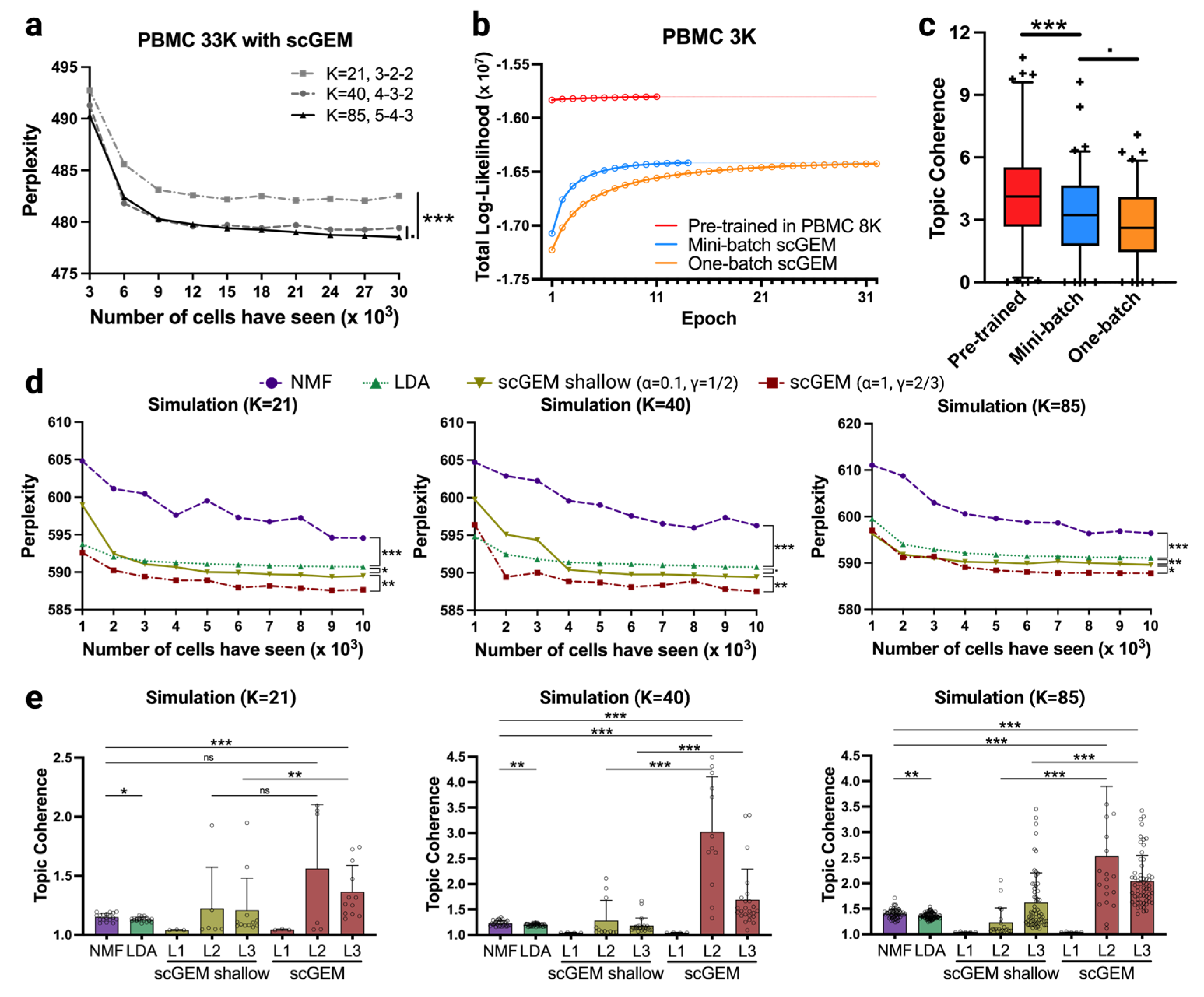
2.3.1. Simulation
2.3.2. Implementation of NMF and LDA
2.3.3. Evaluation Metrics
2.4. Gene Expression Analysis
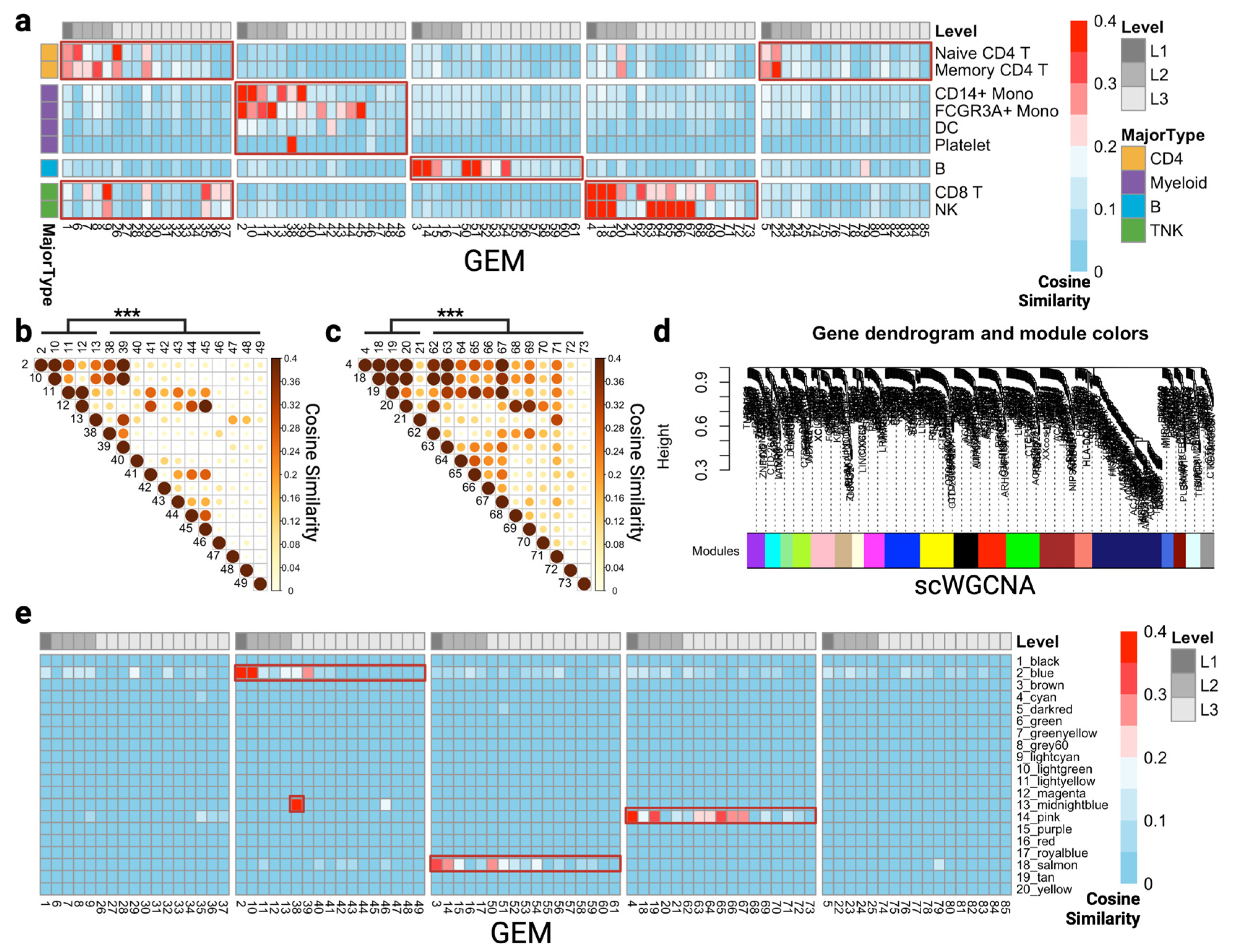
2.5. Cosine Similarity among GEMs and Marker Genes
2.6. Statistical Analysis
3. Results
3.1. scGEM Identifies High-Resolution Gene Co-Expressing Modules in the PBMC Dataset
3.2. Comparison of scGEM and Marker Genes in Cell Type
3.3. Systematic Review of scGEM Model Performance
3.4. scGEM Deconvolutes Function Signals in Bulk Transcriptome Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.-E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Anderson, A.G.; Merullo, D.P.; Konopka, G. Beyond bulk: A review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019, 58, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Grabski, I.N.; Street, K.; Irizarry, R.A. Significance analysis for clustering with single-Cell RNA-sequencing data. Nat. Methods 2023, 20, 1196–1202. [Google Scholar] [CrossRef]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.-A.; Kwok, I.W.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019, 37, 38–44. [Google Scholar] [CrossRef]
- Lahnemann, D.; Koster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven grand challenges in single-cell data science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 2021, 184, 4734–4752.e20. [Google Scholar] [CrossRef]
- DeTomaso, D.; Yosef, N. Hotspot identifies informative gene modules across modalities of single-cell genomics. Cell Syst. 2021, 12, 446–456.e449. [Google Scholar] [CrossRef]
- Longo, D.; Hasty, J. Dynamics of single-cell gene expression. Mol. Syst. Biol. 2006, 2, 64. [Google Scholar] [CrossRef]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Yang, D.; Powell, C.A.; Wang, X. Cell–cell communication: Old mystery and new opportunity. Cell Biol. Toxicol. 2019, 35, 89–93. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Cox Iii, R.S.; Weiss, R.; Arnold, F.H. Programmed population control by cell–cell communication and regulated killing. Nature 2004, 428, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Tang, L.; Dong, W.; Yao, S.; Zhou, W. An overview of topic modeling and its current applications in bioinformatics. Springerplus 2016, 5, 1608. [Google Scholar] [CrossRef]
- Zhu, X.; Ching, T.; Pan, X.; Weissman, S.M.; Garmire, L. Detecting heterogeneity in single-cell RNA-Seq data by non-negative matrix factorization. PeerJ 2017, 5, e2888. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, C.; Quintero, A.; Wu, L.; Yuan, Y.; Wang, M.; Cheng, M.; Leng, L.; Xu, L.; Dong, G. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun. 2019, 10, 470. [Google Scholar] [CrossRef]
- Venkatasubramanian, M.; Chetal, K.; Schnell, D.J.; Atluri, G.; Salomonis, N. Resolving single-cell heterogeneity from hundreds of thousands of cells through sequential hybrid clustering and NMF. Bioinformatics 2020, 36, 3773–3780. [Google Scholar] [CrossRef]
- Shao, C.; Höfer, T. Robust classification of single-cell transcriptome data by nonnegative matrix factorization. Bioinformatics 2017, 33, 235–242. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Z.; Zhou, W.; Wang, P.; Jiang, Q.; Juan, L. An interpretable single-cell RNA sequencing data clustering method based on latent Dirichlet allocation. Brief. Bioinform. 2023, 24, bbad199. [Google Scholar] [CrossRef]
- Gong, B.; Zhou, Y.; Purdom, E. Cobolt: Integrative analysis of multimodal single-cell sequencing data. Genome Biol. 2021, 22, 351. [Google Scholar] [CrossRef] [PubMed]
- duVerle, D.A.; Yotsukura, S.; Nomura, S.; Aburatani, H.; Tsuda, K. CellTree: An R/bioconductor package to infer the hierarchical structure of cell populations from single-cell RNA-seq data. BMC Bioinform. 2016, 17, 363. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, L.; Kürten, C.H.; Jabbari, F.; Vujanovic, L.; Ding, Y.; Lu, B.; Lu, K.; Kulkarni, A.; Tabib, T. An individualized causal framework for learning intercellular communication networks that define microenvironments of individual tumors. PLoS Comput. Biol. 2022, 18, e1010761. [Google Scholar] [CrossRef]
- Liu, M.; Wu, Y.; Jiang, N.; Boot, A.; Rozen, S.G. mSigHdp: Hierarchical Dirichlet process mixture modeling for mutational signature discovery. NAR Genom. Bioinform. 2023, 5, lqad005. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, S.; Koga, Y.; Corbett, S.E.; Shea, C.V.; Johnson, W.E.; Yajima, M.; Campbell, J.D. Celda: A Bayesian model to perform co-clustering of genes into modules and cells into subpopulations using single-cell RNA-seq data. NAR Genom. Bioinform. 2022, 4, lqac066. [Google Scholar] [CrossRef]
- Buntine, W. Variational extensions to EM and multinomial PCA. In Proceedings of the European Conference on Machine Learning, Helsinki, Finland, 19–23 August 2002; pp. 23–34. [Google Scholar]
- Buntine, W.; Jakulin, A. Discrete component analysis. In Proceedings of the International Statistical and Optimization Perspectives Workshop “Subspace, Latent Structure and Feature Selection”, Bohinj, Slovenia, 23–25 February 2005; pp. 1–33. [Google Scholar]
- Blei, D.M. Probabilistic topic models. Commun. ACM 2012, 55, 77–84. [Google Scholar] [CrossRef]
- Nakagawa, H.; Wang, L.; Cantor, H.; Kim, H.-J. New insights into the biology of CD8 regulatory T cells. Adv. Immunol. 2018, 140, 1–20. [Google Scholar]
- Wang, Y.M.; Alexander, S.I. CD8 regulatory T cells: What’s old is now new. Immunol. Cell Biol. 2009, 87, 192. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef]
- Kazanietz, M.G.; Durando, M.; Cooke, M. CXCL13 and its receptor CXCR5 in cancer: Inflammation, immune response, and beyond. Front. Endocrinol. 2019, 10, 471. [Google Scholar] [CrossRef]
- Feregrino, C.; Tschopp, P. Assessing evolutionary and developmental transcriptome dynamics in homologous cell types. Dev. Dyn. 2022, 251, 1472–1489. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Paisley, J.; Wang, C.; Blei, D.M.; Jordan, M.I. Nested Hierarchical Dirichlet Processes. IEEE Trans. Pattern Anal. Mach. Intell. 2015, 37, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Eze, U.C.; Bhaduri, A.; Haeussler, M.; Nowakowski, T.J.; Kriegstein, A.R. Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. 2021, 24, 584–594. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Teh, Y.; Jordan, M.; Beal, M.; Blei, D. Sharing clusters among related groups: Hierarchical Dirichlet processes. In Proceedings of the Advances in Neural Information Processing Systems, Cambridge, MA, USA, 1 June 2004. [Google Scholar]
- Sethuraman, J. A constructive definition of Dirichlet priors. Stat. Sin. 1994, 639–650. [Google Scholar]
- Blei, D.M.; Griffiths, T.L.; Jordan, M.I. The nested chinese restaurant process and bayesian nonparametric inference of topic hierarchies. J. ACM 2010, 57, 1–30. [Google Scholar] [CrossRef]
- Park, S.; Zhao, H. Spectral clustering based on learning similarity matrix. Bioinformatics 2018, 34, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.-N.; Liu, J.-X.; Zheng, C.-H.; Wang, J. SLRRSC: Single-cell type recognition method based on similarity and graph regularization constraints. IEEE J. Biomed. Health Inform. 2022, 26, 3556–3566. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.; Bach, F.; Blei, D. Online learning for latent dirichlet allocation. In Proceedings of the Advances in Neural Information Processing Systems, Vancouver, BC, Canada, 6–9 December 2010. [Google Scholar]
- Paisley, J.; Blei, D.; Jordan, M. Variational Bayesian inference with stochastic search. arXiv 2012, arXiv:1206.6430. [Google Scholar]
- Zappia, L.; Phipson, B.; Oshlack, A. Splatter: Simulation of single-cell RNA sequencing data. Genome Biol. 2017, 18, 174. [Google Scholar] [CrossRef] [PubMed]
- DeBruine, Z.J.; Melcher, K.; Triche Jr, T.J. Fast and robust non-negative matrix factorization for single-cell experiments. bioRxiv 2021. [Google Scholar] [CrossRef]
- Grün, B.; Hornik, K. topicmodels: An R package for fitting topic models. J. Stat. Softw. 2011, 40, 1–30. [Google Scholar] [CrossRef]
- AlSumait, L.; Barbará, D.; Domeniconi, C. On-line lda: Adaptive topic models for mining text streams with applications to topic detection and tracking. In Proceedings of the 2008 Eighth IEEE International Conference on Data Mining, Pisa, Italy, 15–19 December 2008; pp. 3–12. [Google Scholar]
- Newman, D.; Lau, J.H.; Grieser, K.; Baldwin, T. Automatic evaluation of topic coherence. In Proceedings of the Human Language Technologies: The 2010 Annual Conference of the North American Chapter of the Association for Computational Linguistics, Los Angeles, CA, USA, 1–6 June 2010; pp. 100–108. [Google Scholar]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Murakami, H. The power of the modified Wilcoxon rank-sum test for the one-sided alternative. Statistics 2015, 49, 781–794. [Google Scholar] [CrossRef]
- McKight, P.E.; Najab, J. Kruskal-wallis test. Corsini Encycl. Psychol. 2010, 1, 1–10. [Google Scholar]
- Wu, F.; Fan, J.; He, Y.; Xiong, A.; Yu, J.; Li, Y.; Zhang, Y.; Zhao, W.; Zhou, F.; Li, W. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021, 12, 2540. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Hawkins, G.A.; Wudel, L.; Chou, P.C.; Forbes, E.; Pullikuth, A.K.; Liu, L.; Jin, G.; Craddock, L.; Topaloglu, U. Dissecting intratumoral myeloid cell plasticity by single cell RNA-seq. Cancer Med. 2019, 8, 3072–3085. [Google Scholar] [CrossRef]
- Kowalska, M.A.; Rauova, L.; Poncz, M. Role of the platelet chemokine platelet factor 4 (PF4) in hemostasis and thrombosis. Thromb. Res. 2010, 125, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Sampath, P.; Moideen, K.; Ranganathan, U.D.; Bethunaickan, R. Monocyte subsets: Phenotypes and function in tuberculosis infection. Front. Immunol. 2018, 9, 1726. [Google Scholar] [CrossRef]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Su, W.; Tang, H.; Le, W.; Zhang, X.; Zheng, Y.; Liu, X.; Xie, L.; Li, J.; Ye, J. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Jin, S.; Wang, M.; Jiao, Y.; Yang, B.; Lu, X.; Ji, X.; Fei, Y.; Yang, H. Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis. Nat. Commun. 2021, 12, 4977. [Google Scholar] [CrossRef]
- Quintana, F.J. Myeloid cells in the central nervous system: So similar, yet so different. Sci. Immunol. 2019, 4, eaaw2841. [Google Scholar] [CrossRef]
- Luther, S.A.; Cyster, J.G. Chemokines as regulators of T cell differentiation. Nat. Immunol. 2001, 2, 102–107. [Google Scholar] [CrossRef]
- Fu, B.; Tian, Z.; Wei, H. Subsets of human natural killer cells and their regulatory effects. Immunology 2014, 141, 483–489. [Google Scholar] [CrossRef]
- Li, H.; van der Leun, A.M.; Yofe, I.; Lubling, Y.; Gelbard-Solodkin, D.; van Akkooi, A.C.J.; van den Braber, M.; Rozeman, E.A.; Haanen, J.; Blank, C.U.; et al. Dysfunctional CD8 T Cells Form a Proliferative, Dynamically Regulated Compartment within Human Melanoma. Cell 2019, 176, 775–789.e18. [Google Scholar] [CrossRef]
- Joosten, L.A.; Netea, M.G.; Kim, S.-H.; Yoon, D.-Y.; Oppers-Walgreen, B.; Radstake, T.R.; Barrera, P.; van de Loo, F.A.; Dinarello, C.A.; van den Berg, W.B. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2006, 103, 3298–3303. [Google Scholar] [CrossRef]
- Bourque, J.; Kousnetsov, R.; Hawiger, D. Roles of Hopx in the differentiation and functions of immune cells. Eur. J. Cell Biol. 2022, 101, 151242. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Taniuchi, I. CD4 helper and CD8 cytotoxic T cell differentiation. Annu. Rev. Immunol. 2018, 36, 579–601. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Chen, J. A genome-wide regulatory network identifies key transcription factors for memory CD8+ T-cell development. Nat. Commun. 2013, 4, 2830. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, Y.; Wang, D.; Hu, X.; Zhang, Z. Single-cell meta-analyses reveal responses of tumor-reactive CXCL13(+) T cells to immune-checkpoint blockade. Nat. Cancer 2022, 3, 1123–1136. [Google Scholar] [CrossRef]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 2022, 3, 108–121. [Google Scholar] [CrossRef]
- Wan, Z.; Lin, Y.; Zhao, Y.; Qi, H. TFH cells in bystander and cognate interactions with B cells. Immunol. Rev. 2019, 288, 28–36. [Google Scholar] [CrossRef]
- Kim, S.J.; Ryu, K.J.; Hong, M.; Ko, Y.H.; Kim, W.S. The serum CXCL13 level is associated with the Glasgow Prognostic Score in extranodal NK/T-cell lymphoma patients. J. Hematol. Oncol. 2015, 8, 49. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [PubMed]
- Vayansky, I.; Kumar, S.A.P. A review of topic modeling methods. Inf. Syst. 2020, 94, 32. [Google Scholar] [CrossRef]
- Asuncion, A.; Welling, M.; Smyth, P.; Teh, Y.W. On smoothing and inference for topic models. In Proceedings of the Twenty-Fifth Conference on Uncertainty in Artificial Intelligence, Montreal, QC, Canada, 18–21 June 2009; pp. 27–34. [Google Scholar]
- Badia-i-Mompel, P.; Wessels, L.; Müller-Dott, S.; Trimbour, R.; Ramirez Flores, R.O.; Argelaguet, R.; Saez-Rodriguez, J. Gene regulatory network inference in the era of single-cell multi-omics. Nat. Rev. Genet. 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Lu, X.; Lu, B.; Chen, L. scGEM: Unveiling the Nested Tree-Structured Gene Co-Expressing Modules in Single Cell Transcriptome Data. Cancers 2023, 15, 4277. https://doi.org/10.3390/cancers15174277
Zhang H, Lu X, Lu B, Chen L. scGEM: Unveiling the Nested Tree-Structured Gene Co-Expressing Modules in Single Cell Transcriptome Data. Cancers. 2023; 15(17):4277. https://doi.org/10.3390/cancers15174277
Chicago/Turabian StyleZhang, Han, Xinghua Lu, Binfeng Lu, and Lujia Chen. 2023. "scGEM: Unveiling the Nested Tree-Structured Gene Co-Expressing Modules in Single Cell Transcriptome Data" Cancers 15, no. 17: 4277. https://doi.org/10.3390/cancers15174277
APA StyleZhang, H., Lu, X., Lu, B., & Chen, L. (2023). scGEM: Unveiling the Nested Tree-Structured Gene Co-Expressing Modules in Single Cell Transcriptome Data. Cancers, 15(17), 4277. https://doi.org/10.3390/cancers15174277